1
ФЕДЕРАЛЬНОЕ ГОСУДАРСТВЕННОЕ БЮДЖЕТНОЕ ОБРАЗОВАТЕЛЬНОЕ УЧРЕЖДЕНИЕ ВЫСШЕГО ПРОФЕССИОНАЛЬНОГО ОБРАЗОВАНИЯ
Нижегородский государственный университет им. Н.И. Лобачевского Национально-исследовательский университет
Химический факультет
Кафедра неорганической химии
ТЕХНИКА ПРОВЕДЕНИЯ ЛАБОРАТОРНЫХ РАБОТ
Методическая разработка к лабораторной работе
по неорганической химии
Для студентов ННГУ, обучающихся по направлению подготовки 020100 “Химия” и специальностям 020101 “Химия”, 020801 “Экология”, 240306 “Химическая технология монокристаллов, материалов и изделий электронной техники”
Нижний Новгород
2013 г.
2
УДК 542 (075.8)
ББК
Т-38
ТЕХНИКА ПРОВЕДЕНИЯ ЛАБОРАТОРНЫХ РАБОТ. Часть 1. Оборудование химических лабораторий. Составители: О.А.Замятин, А.А.Сибиркин / Методические разработки к лабораторным работам по неорганической химии – Нижний Новгород: Нижегородский госуниверситет, 2013. – с.
Рецензент:
В учебно-методическом пособии описано классическое лабораторное оборудование и стандартная химическая посуда. Освещены физикохимические основы важнейших лабораторных операций и изложена техника их выполнения.
Учебно-методическое пособие предназначено для студентов ННГУ, обучающихся по направлению подготовки 020100 “Химия” и специальностям 020101 “Химия”, 020801 “Экология”, 240306 “Химическая технология монокристаллов, материалов и изделий электронной техники”, изучающих курс неорганической химии.
Ответственный за выпуск:
УДК 542
|
3 |
|
|
Содержание |
|
|
1. Оборудование химических лабораторий ……………………………. |
4 |
|
1.1. Лабораторная стеклянная посуда общего назначения …….. |
4 |
1.2.Мерная посуда ………………………………………………… 16
1.3.Фарфоровая посуда …………………………………………… 18
1.4.Химическая тара ……………………………………………… 21
|
2. Основные лабораторные операции ………………………………….. |
22 |
|
2.1. Взвешивание ………………………………………………….. |
22 |
|
2.2. Фильтрование ………………………………………………… |
33 |
2.3.Титрование ……………………………………………………. 37
2.4.Определение плотности веществ ……………………………. 39
|
2.5. Нагревание ……………….…………………………………… |
41 |
|
2.6. Охлаждение …………………………………………………… |
50 |
|
3. Практическая часть …………………………………………………… |
51 |
3.1.Взвешивание ………………………………………………….. 51
3.2.Фильтрование и декантация …………………………………. 56
3.3. Фильтрование под уменьшенным давлением ……………… 59
3.4.Перекристаллизация …………………………………………. 61
3.5.Перегонка ……………………………………………………… 63
3.6.Возгонка ………………………………………………………. 65
3.7.Титрование ……………………………………………………. 66
3.8.Определение плотности жидкости ………………………….. 71
|
3.9. Определение температуры плавления ……………………… |
73 |
|
3.10. Определение температуры кипения жидкости …………… |
75 |
|
Список использованных источников …………………………………… |
77 |
4
1.Оборудование химических лабораторий
1.1.Лабораторная стеклянная посуда общего назначения
Проведение химического эксперимента традиционно осуществляется в стандартной химической посуде. При его выполнении используются различные виды емкостей от самых простых, например стаканов и пробирок, до изделий высокой сложности, изготовленных по индивидуальному чертежу. Сбор и монтаж установки предполагает использование различных соединительных и запорных устройств – трубок, шлангов, пробок, шлифов, кранов. Почти всегда в работе в химической лаборатории необходимы фильтры и теплоизолирующие материалы, чистая вода. Выбирая посуду для проведения эксперимента необходимо знать ее термическую и химическую устойчивость по отношению к веществам, с которыми предстоит работать.
Преобладающая часть лабораторных работ осуществляется в посуде и приборах из специального тонкостенного или толстостенного прозрачного стекла. Благодаря своей коррозионной стойкости, твердости, прозрачности и сравнительно небольшому коэффициенту линейного теплового расширения стекло является ценным конструктивным материалом для изготовления лабораторной посуды, приборов и аппаратов. Прозрачность стекла позволяет непосредственно визуально следить за ходом процесса в реакционном сосуде, а гладкость поверхности стекла облегчает мытье посуды.
Недостатки стекла — его хрупкость, относительно малая устойчивость к резким перепадам температуры, недостаточная стойкость в отношении некоторых агрессивных химических веществ, например, концентрированные щелочи, фосфорная и фтористоводородная кислоты.
Используемая в лабораториях химическая посуда может быть разделена на ряд групп. По назначению посуду можно разделить на посуду общего назначения, специального назначения и мерную. По материалу — на посуду из простого стекла, специального стекла, из кварцевого стекла.
Кгруппе общего назначения относятся те предметы, которые всегда должны быть в лаборатории и которые требуются для большинства работ. Это пробирки, воронки простые и делительные, стаканы, колбы, кристаллизаторы, холодильники, реторты, тройники, краны.
Кгруппе специального назначения относятся те предметы, которые употребляются для какой-либо одной цели, например, аппарат Киппа, дефлегматоры, склянки Вульфа, склянки Тищенко, пикнометры, ареометры, склянки Дрекселя.
Кмерной посуде относятся мерные цилиндры и мензурки, пипетки, бюретки и мерные колбы.
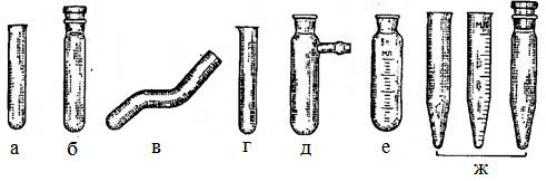
5
Пробирки
Пробирки – это наиболее простая химическая посуда для проведения испытаний химических свойств веществ (в том числе качественных реакций) и выполнения препаративных работ с использованием малых количеств веществ. Они представляют собой изготовленные из стекла узкие сосуды цилиндрической формы различного диаметра с закругленным дном (рис. 1). Различают простые, градуированные, конические пробирки для центрифугирования, пробирки с боковыми отводами.
Круглодонные пробирки с боковым отводом и взаимозаменяемым конусом предназначаются для фильтрования под вакуумом небольших объемов жидкостей.
Рис. 1. Пробирки: простые (а), с пришлифованной пробкой (б), изогнутые (в), с носиком (г), для вакуумного фильтрования (д), градуированные (е) и центрифужные (ж).
Для хранения пробирок применяют специальные деревянные, пластмассовые или металлические штативы. Для разложения твердых веществ, состав и свойства которых мало известны, применяют изогнутые пробирки.
При работе с пробирками следует придерживаться следующих правил.
1.При проведении реакции не следует применять слишком большое количество реактивов. Достаточно бывает одной четверти или менее объема пробирки.
2.Недопустимо, чтобы пробирка была наполнена до краев.
3.Недопустимо закрывать пробирку пальцем и встряхивать ее в таком виде. При этом содержимое обычно загрязняется бытовыми примесями. При работе с едкими веществами можно повредить кожу пальца или получить ожог.
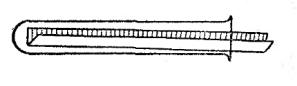
6
4.Для перемешивания налитых реактивов пробирку держат большим
иуказательным пальцами левой руки за верхний конец и поддерживают ее средним пальцем, а указательным пальцем правой руки ударяют косым ударом по низу пробирки. Если пробирка наполнена жидкостью больше чем на половину, содержимое перемешивают стеклянной палочкой.
5.Для внесения твердых веществ (порошков, кристаллов) используют полоску бумаги шириной немного меньше диаметра пробирки (рис. 2). Ее складывают вдвое по длине и в полученный совочек насыпают нужное количество твердого вещества. Пробирку держат в левой руке, расположив ее горизонтально, и вводят в нее совочек с веществом почти до дна. Затем пробирку ставят вертикально и слегка ударяют по ней. Когда все твердое вещество высыплется, бумажный совочек вынимают.
Рис. 2. Внесение в пробирку порошкообразных веществ.
6. Если пробирку нужно нагреть, ее следует зажать в держателе. Нагревание нужно проводить осторожно. Вначале нужно обогреть всю пробирку, после чего приступить к нагреванию ее нижней части. Если нагреваемая жидкость начинает закипать (появляются пузырьки пара), пробирку следует переместить дальше от источника тепла и продолжать нагревание горячим воздухом. При нагревании открытый конец пробирки должен быть обращен в сторону от работающего и от соседей по столу или лаборатории. Когда не требуется сильного нагрева, пробирку с нагреваемым веществом можно поместить в горячую воду или разогретый песок.
Химические стаканы
Лабораторные химические стаканы представляют собой стеклянные тонкостенные низкие или высокие плоскодонные или круглые цилиндры с носиком или без него различной вместимости (рис. 3). Бывают также простые и мерные стаканы. Изготавливают их из разных сортов стекла и фарфора, а также полимерных материалов.
Применяются стаканы для проведения простейших химических операций и в качестве вспомогательных сосудов. Нагревать химические стаканы следует на водяной или песчаной бане или применять электрические плитки с керамическим верхом. Запрещается осуществлять нагревание лабораторных стаканов на открытом пламени газовой горелки или на электрической плитке с открытой спиралью.
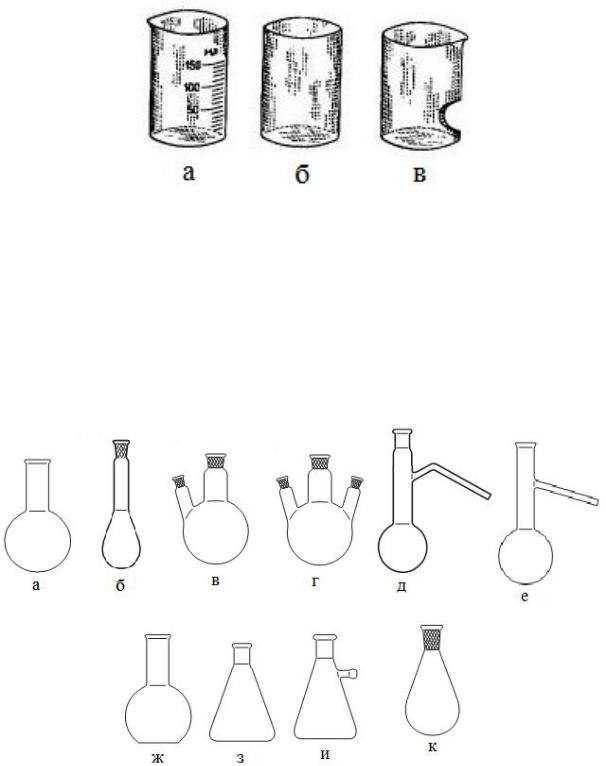
7
Рис. 3. Химические стаканы: мерный с носиком (а), с отшлифованной верхней кромкой (б) и стакан для промывки осадков декантацией (в).
Колбы
Колбы имеют общелабораторное назначение и предсталяют собой круглодонные, конические, плоскодонные, грушевидные, остродонные сосуды, с взаимозаменяемыми конусными и сферическими шлифами или без них (рис. 4).
Рис. 4. Колбы: круглодонная с коротким горлом (а), с длинным горлом или колба Кьельдаля (б), с двумя горловинами или двугорлая (в), с тремя горловинами или трехгорлая (г), с боковым отростком или колба Вюрца (д), с боковым отростком или колба Энглера (е), плоскодонная (ж), коническая колба Эрленмейера (з), толстостенные с тубусом или колба Бунзена (и), грушевидная (к).
8
Круглодонные колбы с длинным и коротким горлом с взаимозаменяемыми конусами и без них применяют для нагревания и перегонки жидкостей, проведения различных препаративных и аналитических работ, в качестве приемников при простой и вакуумной перегонке. Колбы Кьельдаля применяют также для перегонки веществ с водяным паром и для определения содержания азота.
Круглодонные колбы с несколькими горловинами с взаимозаменяемыми конусами и без них служат для перегонки под вакуумом и проведения, например, препаративных работ, требующих применения мешалки, холодильника, термометра, капельной воронки, кранами для соединения с вакуумной системой или для подачи газа.
Круглодонная колба с боковым отростком – наиболее простая и удобная колба для перегонки жидкости. К боковому отростку колбы присоединяют холодильник для конденсации паров. При работе с высококипящими жидкостями отросток должен быть расположен ближе к шарообразной части колбы. Легкокипящие жидкости перегоняют в колбах Вюрца с отростком, расположенным ближе к открытому концу горла.
Плоскодонные колбы предназначаются для аналитических работ, простейших операций при атмосферном давлении, иногда для хранения жидких веществ. Используют в качестве приемников при простой перегонке.
Конические колбы благодаря своей форме обеспечивают малую поверхность испарения, вследствие чего их используют для кристаллизации.
Для закрывания горла колбы используют часовое стекло соответствующего размера или стеклянную крышку. Нагревать колбы следует подобно стаканам, а именно, на бане или плитке с закрытой спиралью.
Основная область применения колб Эрленмейера малого объема – проведение титрования.
Толстостенные колбы с тубусом (колбы Бунзена) обычно применяют в качестве приемников при фильтровании при уменьшенном давлении. Они изготавливаются из стекла толщиной до 8 мм, что позволяет выдерживать атмосферное давление снаружи. При работе с колбами Бунзена рекомендуется для страховки закрывать колбу полотенцем или помещать в ящик из толстого картона или жести. Для защиты от разлетающихся осколков стекла (при возможном взрыве) можно также наклеивать на наружную стенку колбы липкую прозрачную ленту, накладывая слой на слой так, чтобы каждый виток закрывал около половины предыдущего. Внимание ! Толстостенные колбы Бунзена запрещается использовать в работах, предполагающих нагревание. Их нельзя помещать на плитку или баню или допускать попадание в них горячих жидкостей или твердых веществ во избежание разрушения.
Грушевидные колбы используются в качестве приемников при вакуумной перегонке и в случае, когда при перегонке жидкости пар не
9
должен перегреваться в конце процесса. Обогреваемая поверхность такой колбы не уменьшается при понижении зеркала жидкости.
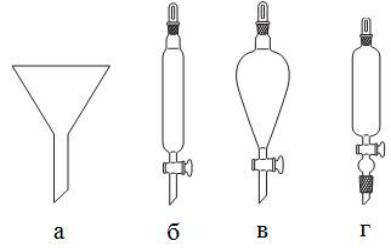
Воронки
Стеклянные воронки (рис. 5) общего назначения подразделяются на лабораторные, делительные, капельные и для фильтрования.
Рис. 5. Воронки: лабораторная (а), делительная (б), делительная шарообразная (в), капельная (г).
Лабораторные воронки, часто называемые химическими или простыми воронками, имеют конусообразную форму (угол конуса 60°), со срезанным длинным концом. Они служат для переливания жидкостей из сосуда в сосуд, для фильтрования при помощи вкладного фильтра, для переноса твердых веществ в колбы. Простые воронки имеют ровную внутреннюю стенку, но для ускоренного фильтрования иногда применяют воронки с ребристой внутренней поверхностью.
При наливании жидкости через воронку ее не следует наполнять до краев. Если воронка плотно прилегает к горлу сосуда, в который переливают жидкость, то переливание затрудняется, так как внутри сосуда создается повышенное давление. Поэтому воронку время от времени нужно приподнимать или обеспечить между воронкой и горлом сосуда щель, вложив между ними, например, кусочек бумаги.
Делительные воронки предназначаются для разделения несмешивающихся жидкостей при экстрагировании. Они бывают цилиндрические, конические, грушевидные и шаровидные, неградуированные, градуированные и с боковой трубкой для выравнивания давления. Все они снабжены притертым стеклянным спускным краном.
Перед началом работы герметичность крана следует проверить наливанием воды или другой более текучей жидкости в воронку. При недостаточной герметичности воронкой не пользуются. Ремонт воронки
10
заключается в притирке крана. При заполнении воронки объем разделяемых жидкостей не должен превышать двух третей ее общей вместимости.
Капельные воронки отличаются от делительных тем, что они более легкие, тонкостенные и в большинстве случаев с длинным концом. Они предназначены для приливания жидкости в реакционный сосуд небольшими порциями или по каплям. Поэтому они обычно составляют часть прибора. Эти воронки укрепляют в горле колбы на шлифе или при помощи корковой либо резиновой пробки. Перед работой на шлиф крана и шлиф керна наносят небольшое количество вакуумной смазки, так чтобы смазка не попадала в трубку воронки или внутрь отверстия крана. Смазанный кран должен поворачиваться легко и без усилия.
Воронки для фильтрования характеризуются удлиненным срезанным концом сливной трубки, внутренний диаметр которого в верхней части меньше, чем в нижней, что приводит к ускорению процесса фильтрования. Они предназначаются в основном для аналитических целей.
Реторты
Реторты (рис. 6) представляют собой химическую посуду, напоминающую колбу видоизмененной формы. Из применяемой в настоящее время стандартной химической посуды реторта является, пожалуй, наиболее древним лабораторным прибором.
Рис. 6. Реторта.
Реторты применяют для перегонки жидкостей с высокой температурой кипения. Выходящие из сферической части пары конденсируются в удлиненном носике благодаря контакту с окружающим воздухом. Изготавливают реторты двух видов: без тубуса и с тубусом. Вместимость реторт составляет в среднем 2-3 литра.
Кристаллизаторы
Кристаллизаторы (рис. 7) представляют собой стеклянные плоскодонные сосуды различных диаметров и емкости. Их применяют для кристаллизации веществ из раствора. Процесс протекает за счет
Соседние файлы в предмете [НЕСОРТИРОВАННОЕ]
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
Handbuch
der Laboratoriumsdestillation
Mit einer Einfiihrung in die Pilotdestillation
von Erich Kreil
3.,bearbeitete und erweiterte Auflage
VEB Deutscher Verlag der Wissenschaften
Berlin 1976
Э.Крель
РУКОВОДСТВО ПО ЛАБОРАТОРНОЙ ПЕРЕГОНКЕ
Перевод с немецкого канд. техн, наук В.И.Чернышева канд. техн, наук А.В. Шафрановского
Под редакцией д ра техн, наук В.М.Олевского
Москва «Химия» 1980
УДК 542.48
Крель Э.
Руководство по лабораторной перегонке — Пер. с нем./Под ред. В. М. Олевского — М.:Химия, 1980.— 520 с., ил.
Книга представляет собой руководство по технике лабораторной перегонки. В ней изложены физические основы процессов дистилляции и ректификации, описаны различные методы перегонки и соответствующая аппаратура, а также контрольно-измерительные устройства. Один из разделов книги посвящен вопросам ректификации на пилотных (опытнопромышленных) установках.
Книга содержит графики, таблицы, номограммы, облегчающие математическую обработку данных по лабораторной перегонке и расчет применяемой аппаратуры.
Книга предназначена для научных сотрудников, инженеров н техников, занимающихся проведением дистилляции и ректификации в лабораторных и промышленных условиях. Она может быть полезна для учащихся высших и средних технических учебных заведений, а также аспирантов н преподавателей.
520 с.; 70 табл.; 434 рнс.; список литературы 918 ссылок.
20507-003
К050 (01)-80
3.80.1801000000
© VEB Deutscher Verlag der Wissenschaften, Berlin 1976
© Перевод на русский язык. Издательство «Химия», 1980 г.
СОДЕРЖАНИЕ
Предисловие редактора ........................................... Ю
Предисловие автора к третьему немецкому изданию ................. И
Основные обозначения и сокращения .............................. 13
1. Введение.................................................... 15
2. Из истории лабораторной перегонки........................... 19
3. Стандартизация в технике перегонки. Способы выражения концентрации ................................. 28
3.1. Стандартизация методов перегонки и применяемых приборов ... 28
3.2. Определение основных понятий .............................. 32
3.3. Единицы измерения ......................................... 33
3.4. Способы выражения и пересчета концентрации................. 34
4. Физические основы процесса разделения .... 38
4.1. Принципы дистилляции и ректификации ....................... 38
4.2. Особенности движения жидкой фазы в колоннах с орошаемой насадкой ......................................................... 41
4.2.1. Распределение жидкости по насадке ....................... 41
4.2.2. Пленочное течение жидкости .......................... . 48
4.2.3. Динамические характеристики колонн ...................... 49
4.3. Разделение смесей взаимно растворимых компонентов.......... 50
4.4. Зависимость между давлением пара и температурой р—t........ 53
4.4.1. Экспериментальное определение давления насыщенных паров 54
4.4.2. Расчет и способы представления данных по давлению насыщенных паров .......................................................... 60
4.5. Уравнение состояния и фазовая диаграмма р—х................ 70
4.6. Фазовая диаграмма t—х—у и кривая равновесия у*—х .......... 74
4.6.1. Расчет кривой равновесия ................................ 76
4.6.2. Летучесть, относительная летучесть и коэффициент активности 81
4.6.3. Экспериментальное определение кривых равновесия.......... 86
4.7. Число теоретических ступеней разделения (число теоретических тарелок) ....................................................... 93
5
VI '
4.7.1. Расчет числа теоретических ступеней разделения по методу Мак-Кэба и Тиле для периодической ректификации .......................... 1W-
4.7.2. Расчет числа теоретических ступеней разделения по методу Мак-
Кэба и Тиле для непрерывной ректификации....................... 103
4.7.3. Определение числа теоретических ступеней разделения по методу Мак-Кэба и Тиле для смесей с кривыми равновесия, имеющими
точку перегиба и азеотропную точку .......................... 107
4.7.4. Определение числа теоретических ступеней разделения для смесей со слабо выпуклыми кривыми равновесия и при небольшом расстоянии между кривой равновесия и рабочей линией .... 108
4.7.5. Аналитические методы определения числа теоретических ступеней разделения для периодической ректификации ........................... 111
4.7.5.1. Определение числа теоретических ступеней разделения по разности температур кипения ........................................... 111
4.7.5.2. Расчет числа теоретических ступеней разделения с помощью фактора обогащения и формул Роуза .................................. 112
4.7.5.3. Расчет минимального числа теоретических ступеней разделения по уравнению Фенске для идеальных смесей при v = со . . . 115
4.7.5.4. Другие методы расчета числа теоретических ступеней разделения и их сравнительная оценка . .................................... 117
4.8. Теория процессов, протекающих в насадочной колонне.............. 118
4.8.1. Процесс разделения в насадочной колонне....................... 118
4.8.2. Определение числа единиц переноса ............................ 122
4.8.3. Интенсивность противоточного массообмена и время, необходимое для разгонки .................................................... 127
4.9. Определение числа теоретических ступеней разделения при периодической и непрерывной ректификации многокомпонентных смесей 131
4.10. Аппаратурно-технологические параметры тарельчатых и насадочных колонн. Испытания колонн ........................................ 135
4.10.1. Диаметр колонны . .......................................... 137
4.10.2. Рабочая высота колонны и способы укладки насадочных тел . .. . 138
4.10.3. Эталонная смесь и состав загруженной жидкости............... 139
4.10.4. Флегмовое число и количество орошающей жидкости............ 145
4.10.5. Общая, статическая и динамическая удерживающая способность колонны ............................................................ 149
4.10.6. Рабочее давление ........................................... 151
4.10.7. Нагрузка и скорость потока паров ........................... 153
4.10.8. Проведение испытаний ....................................... 155
4.10.9. Некоторые данные по разделяющей способности насадочных и тарельчатых колонн ................................................. 161
4.11. Гидравлическое сопротивление, предельная скорость паров и расчет основных размеров колонны . ................................. 164
4.12. Тепловые расчеты ......................................... 175
4.13. Характеристика разделяемых веществ и получение кривых разгонки ............................................................... 179
4.14. Примерная последовательность расчета процесса разделения . . . 186
4.14.1. Периодическая ректификация при атмосферном давлении . . . 186
4.14.2. Непрерывная ректификация ................................. 190
4.14.3. Вакуумная перегонка ...................................... 190
4.15. Использование ЭВМ при расчете колонн ....................... 190
6
5. Обычные методы перегонки..................... 195
5.1. Классификация методов перегонки п« количеству перерабатываемого продукта .................................................... 195
5.1.1. Микро- и полумикроперегонка ............................... 195
5.1.2. Аналитическая ректификация . ........................ 202
5.1.3. Препаративная и наработочная перегонка..................... 207
5.1.3.1. Полупромышленные установки , . .......................... 208
5.1.3.2. Пилотные установки ...................................... 211
5.1.3.3. Получение дистиллированной воды ......................... 216
5.1.4. Разделение изотопов ....................................... 219
5.1.4.1. Разделение изотопов низкотемпературной ректификацией .... 222
5.1.4.2. Получение D и 18О путем ректификации воды................ 224
5.1.4.3. Получение других изотопов .............................. 233
5.2. Классификация методов перегонки по способу проведения процесса 234
5.2.1. Периодическая и полунепрерывная перегонка.................. 234
5.2.2. Непрерывная ректификация. , . ,. , ........................ 235
5.2.2.1. Области применения непрерывной ректификации.............. 237
5.2.2.2. Теоретические расчеты ................................... 239
5.2.2.3. Аппаратура для непрерывной ректификации.................. 240
5.2.2.4. Примеры непрерывной лабораторной ректификации............ 243
5.2.2.5. Выведение ректификационных установок на стационарный режим 244
5.2.3. Разделение методом парциальной конденсации ................ 245
5.3. Классификация методов перегонки по температуре процесса .... 249
5.3.1. Низкотемпературная ректификация ........................... 250
5.3.2. Высокотемпературная и изотермическая перегонка............. 257
5.4. Классификация методов перегонки по рабочему давлению......... 262
5.4.1. Вакуумная дистилляция и ректификация ...................... 263
5.4.2. Расширительная перегонка .................................. 269
5.4.3. Пленочная перегонка (пленочное испарение).................. 272
5.4.4. Молекулярная дистилляция .................................. 280
5.4.5. Ректификация под давлением ................................ 292
6. Селективные методы перегонки ......... 294
6.1. Перегонка в парах носителя ............................... 294
6.2. Азеотропная и экстрактивная перегонка .................... 299
6.2.1. Азеотропная перегонка .................................. 304
6.2.2. Экстрактивная перегонка ................................ 314
6.3. Солевая перегонка. . ................................... 321
7
7. Материалы и аппаратура..................................... 324
7.1. Материалы для изготовления ректификационной аппаратуры . . . 324
7.2. Стандартные и сборные приборы ............................. 326
7.2.1. Краны и клапаны ......................................... 332
7.3. Ректификационные колонны .................................. 335
7.3.1. Безнасадочные колонны ................................... 335
7.3.2. Насадочные колонны ...................................... 344
7.3.3. Тарельчатые колонны ..................................... 345
7.3.4. Колонны с неподвижными контактными устройствами.......... 352
7.3.5. Колонны с вращающимися контактными устройствами ......... 359
7.4. Холодильники и дефлегматоры ............................... 369
7.5. Форштосы, приставки для дистилляционных аппаратов и головки колонн ......................................................... 376
7.5.1. Форштосы ................................................ 376
7.5.2. Приставки для дистилляционных аппаратов.................. 376
7.5.3. Головки колонн .......................................... 378
7.6. Кубы, приемники и сборники фракций дистиллята.............. 387
7.6.1. Кубы .................................................... 387
7.6.2. Приемники и сборники фракций дистиллята.................. 390
7.7. Нагревательные приборы и теплоизоляция .................... 393
7.7.1. Обогрев кубов ........................................... 394
7.7.2. Нагревание исходной смеси и кубовой жидкости при непрерывной ректификации ................................................... 398
7.7.3. Теплоизоляция............................................ 401
7.8. Насадки ................................................... 407
7.8.1. Формы выполнения элементов насадок ...................... 407
7.8.2. Влияние материала насадки на эффективность разделения . . . 415
8. Автоматические установки, измерительные и регулирующие устройства........................................417
8.1. Автоматические установки .................................. 417
8.1.1. Автоматизированные установки для стандартного дистилляционного анализа ................................................... 419
8.1.2. Полностью автоматизированные установки для фракционной перегонки ...................................................... 422
8.2. Измерение и регулирование температуры ..................... 428
8.2.1. Измерение температуры ................................... 428
8.2.2. Регулирование температуры ............................... 434
8.3. Измерение и регулирование давления ........................ 438
8.3.1. Измерение и регулирование давления в областях выше 760 и от 760 до 1 мм рт. ст.............................................. 438
8
8.3.1.1. Методы частичного вакуумирования ......................... 443
8.3.2. Измерение и регулирование давления в интервале от 1 до 10“8 мм рт. ст..................................................... 446
8.3.2.1. Компрессионный манометр Мак-Леода ........................ 448
8.3.2.2. Регулирование давления в интервале от 100 до 10"в мм рт. ст. 451
8.4. Регулирование флегмового числа и скорости выкипания........... 453
8.4.1. Реле времени для регулирования флегмового числа............. 453
8.4.2. Регулирование скорости выкипания............................ 455
8.5. Измерение физико-химических свойств дистиллята в процессе ректификации ......................................................... 458
8.5.1. Измерение температуры затвердевания ..................... 458
8.5.2. Измерение показателя преломления ..................... 459
8.5.3. Измерение диэлектрической проницаемости..................... 461
8.5.4. Измерение других физико-химических свойств ................. 462
8.6. Измерение расхода и дозирование газов и жидкостей............. 463
9. Оборудование лаборатории. Ввод в эксплуатацию ректификационных установок ................. 469
9.1. Планировка помещения ................................... 469
9.2. Стенды и коммуникации .................................. 472
9.3. Сборка установок ....................................... 474
9.4. Герметизация установок ................................. 477
9.5. Ввод ректификационных установок в эксплуатацию ......... 480
9.6. Техника безопасности ................................... 481
Литература .................................................. 483
Предметный указатель ........................................ 512
ПРЕДИСЛОВИЕ РЕДАКТОРА
Первое издание монографии Э. Креля «Руководство по лабораторной перегонке», опубликованное Издательством иностранной литературы в 1960 г. (перевод с 1-го немецкого издания 1958 г.), получило широкое одобрение советских специалистов.
За прошедшие 20 лет литература по лабораторной перегонке существенно обогатилась, расширилась область применения ректификации, в связи с чем выпуск перевода третьего немецкого издания книги Э. Креля «Руководство по лабораторной перегонке» является своевременным.
При подготовке третьего немецкого издания автор в значительной мере учел недочеты, имевшиеся в первых двух изданиях. Как особо положительный момент следует отметить включение раздела, в котором рассматривается ректификация на укрупненных (пилотных) установках, что представляет большой интерес в качестве ступени для перехода к промышленным агрегатам.
Полагаю, что настоящее издание монографии Э. Креля послужит дальнейшему прогрессу в области техники перегонки, приобретающей особое значение в борьбе за повышение качества продукции на предприятиях.
Шафрановский А. В. перевел главы 1, 2, 3, 4, 6 и начало главы 5 по раздел 5.1.4.1. Чернышев В. И. перевел главы 7, 8, 9 и конец главы 5, начиная с раздела 5.1.4.2.
В. ОЛЕВСКИЙ
ПРЕДИСЛОВИЕ АВТОРА К ТРЕТЬЕМУ НЕМЕЦКОМУ .-ИЗДАНИЮ
Первое издание этой книги, вышедшее в 1958 г., разошлось сравнительно быстро и уже в 1960 г. возникла необходимость во втором издании. Книга нашла признание и была переведена на русский, английский и венгерский языки.
Цель третьего издания (как и предыдущих) — дать возможно более полное введение в технику лабораторной перегонки; в нем представлены новейшие научные данные и достижения; приведена специальная литература по 1975 г. включительно. При составлении книги автор придерживался мнения, что излагать следует только общепризнанные факты, поскольку на многие вопросы дистилляции и ректификации до сих пор еще нет ясного и окончательного ответа. Однако с целью стимулирования дальнейших исследований в книге отражены также и тенденции, наметившиеся в развитии техники перегонки.
Разумеется, ввиду наличия обширного материала к его отбору необходимо было отнестись критически. Автор стремился просто и понятно описать уравнения и расчетные методы, облегчающие практическую работу в лаборатории. Читатели, интересующиеся вопросами математической обработки и другими специальными вопросами и желающие углубить приобретенные знания, найдут в конце книги обширные списки рекомендуемой литературы.
Расположение материала, принятое в первом издании, в основном сохранено. Дополнительно рассмотрены вопросы ректификации на пилотных * установках (разд. 5.1.3.2). Содержанием раздела 4.2 в третьем издании является гидродинамика потоков в насадочных колоннах. Гл. 8 значительно сокращена ввиду того, что стандартные детали дистилляционных и ректификационных „ приборов и соответствующие контрольно-измерительные приборы уже нашли достаточно широкое применение в лабораторной практике. Таблицы, ранее приводившиеся в приложении, в третьем издании включены в текст. Литературные ссылки распределены по главам и дополнены новыми важнейшими работами; в списки литературы включено по возможности больше обзорных статей.
Книга рассчитана, главным образом, на физиков, химиков и инженеров, занятых в химической промышленности и научно
* Под пилотными установками автор подразумевает малопроизводительные . опытно-промышленные установки. — Прим. ред.
11
исследовательских центрах, в задачу которых входит проведение дистилляции и ректификации в лабораторных и промышленных условиях. Учащиеся высших и средних технических учебных заведений, химики-техники и лаборанты также найдут в этой книге ответы на многие вопросы, связанные с практикой лабораторной перегонки и с расчетами процессов разделения.
Автор надеется, что данное руководство послужит советчиком и путеводителем при разработке более совершенных и экономичных методов разделения для всех, кому приходится заниматься перегонкой в лабораторных условиях.
Автор выражает свою особую благодарность профессору доктору наук Кл. Ветцелю за его благосклонное внимание к этой книге, за его ценные указания и постоянную поддержку. Автор приносит благодарность коллегам доктору Г. Штаге и Е. Гибе-леру за многочисленные замечания, а также благодарит фирмы, выпускающие лабораторные приборы и изделия из стекла, за любезное представление каталогов и другой технической документации. Автор выражает также благодарность издательству за образцовое оформление книги и особенно отделу химии за тщательное редактирование рукописи.
Автор надеется, что и третье издание этой книги окажет содействие дальнейшему развитию техники перегонки на лабораторных и пилотных установках, а также послужит учебным и рабочим пособием при решении задач разделения путем дистилляции и ректификации в лабораториях и в опытных производствах химической промышленности и научно-исследовательских центров, а также в высших и средних учебных заведениях.
БЕРЛИН, ЛЕТО 1975 г.
Доктор технических наук ЭРИХ КРЕЛЬ
ОСНОВНЫЕ ОБОЗНАЧЕНИЯ И СОКРАЩЕНИЯ
А — количество (расход) кубового продукта при непрерывной ректификации а — полная поверхность насадки
— смоченная поверхность насадки
ае — активная поверхность насадки
В — количество жидкости в кубе
ср — теплоемкость при постоянном давлении
си — теплоемкость при постоянном объеме
D — количество паров (суммарное количество дистиллята и флегмы)
dK — диаметр колонны
dH — диаметр насадочного тела
Е — количество (расход) дистиллята
Нк — рабочая высота колонны
НуС — общая удерживающая способность колонны по жидкости
h — высота слоя насадки
I — фактор интенсивности
L — объемная плотность орошения, средняя (по поперечному сечению колонны)
L — объемная плотность орошения в определенной точке т — масса
mq — степень перемешивания
п — число теоретических ступеней разделения
пмнн — число теоретических ступеней разделения, соответствующее минимальному флегмовому числу vMHH
Пуд — число теоретических ступеней разделения, приходящееся на Гм рабочей высоты колонны
р — общее давление паров смеси, рабочее давление процесса перегонки Р° — давление паров чистого вещества
р{ — парциальное давление /-го компонента
Q — количество тепла
Т — термодинамическая (абсолютная) температура
t — температура в градусах Цельсия
V — объем
v — флегмовое число
vMHH — минимальное флегмовое число
vpa6 — рабочее флегмовое число
wn — скорость потока паров
х — концентрация легколетучего компонента в растворе (мольная доля) хд — концентрация легколетучего компонента в кубовой жидкости хд — мольная доля легколетучего компонента в кубовой жидкости
Х£ — мольная доля легколетучего компонента в дистилляте
xz — концентрация легколетучего компонента в питающей жидкости (исходной смеси)
у — концентрация легколетучего компонента в парах
у* — равновесная концентрация легколетучего компонента в парах
Z — количество (расход) питающей жидкости при непрерывной ректификации
13
a — относительная летучесть (отношение давлений паров чистых компонентов)
у — коэффициент активности
е — доля свободного объема (насадки)
6 — характеристическая температура (приведенная температура)
X — коэффициент теплопроводности
|х — динамическая вязкость
л — приведенное давление
р — плотность
о — поверхностное натяжение
т — время
Ф — приведенный объем
Ф — краевой угол смачивания
фю, фс — доля смоченной или активной поверхности насадки
ВЭТС — высота, эквивалентная теоретической ступени разделения
ВЕП — высота единицы переноса
УС — удерживающая способность колонны по жидкости
ЧЕП — число единиц переноса
Введение
Дистилляция и ректификация как методы физического разделения нашли весьма широкое применение в химической промышленности, и в том числе в заводских и исследовательских лабораториях. Однако следует отметить, для лабораторной перегонки часто еще используют малоэффективную с современной точки зрения аппаратуру. К тому же в большинстве случаев не проводят расчетов процесса разделения, а работают чисто эмпирически, руководствуясь опытными данными.
За последние 30 лет проведена большая исследовательская работа по усовершенствованию техники лабораторной перегонки. Теперь в нашем распоряжении имеются современные приборы, изготовленные из стандартных деталей, а также полностью автоматизированные и высоковакуумные установки; разработаны методы расчетов процесса перегонки; лабораторные способы разделения включают разнообразные методы перегонки: от микроректификации с загрузкой менее 1 г до непрерывных процессов с пропускной способностью до 5 л/ч, от низкотемпературной ректификации сжиженных газов до высокотемпературной разгонки смол, от перегонки при атмосферном давлении до молекулярной дистилляции при остаточном давлении ниже 10-4 мм рт. ст. Усовершенствованы селективные методы разделения; путем изменения соотношения парциальных давлений компонентов в парах удается разделять такие смеси, которые до сих пор не поддавались разделению обычными методами.
Классические работы по лабораторной перегонке Рехенберга [1] и Янга [2] содержат превосходные обзоры по теории и практике перегонки, включая описание промышленных установок, однако оии уже во многих отношениях устарели; это особенно касается технологических расчетов и аппаратуры. В монографиях Торманна [3], а также Беджера и Мак-Кэба [4], изданных к началу тридцатых годов, описаны до сих пор еще применяемые графические методы расчета по. Мак-Кэбу и Тиле [5]. Эти монографии представляют особую ценность благодаря ясности изложения. Однако они посвящены исключительно процессам дистилляции и ректификации в промышленных условиях и не содержат рекомендаций по лабораторной перегонке. Работы Янцена и его учеников послужили своеобразным стимулом к дальнейшим исследованиям в области лабораторной перегонки. В монографии [6], выпущенной в 1932 г. Германским обществом химического машиностроения (Dechema), Янцен подробно изложил сформулированные нм еще в 1923 г. основные требования, которые следует предъявлять к ректификационной колонне. Эти требования сохраняют свое значение еще и сегодня.
Работы Киршбаума [7], Гиюла [8] и Якобса [9] посвящены проблемам промышленной ректификации, как н монография Робинсона и Джиллиленда [10], в которой подробно изложены теоретические основы перегонки и рассмотрены сложные проблемы разделения многокомпонентных смесей, а также методы азеотропной и экстрактивной ректификации. Справочник Перри для инженеров-химиков [10 а] включает главу «Перегонка» с многочисленными примерами, таблицами н номограммами для расчета промышленных установок. В справочнике почти нет сведений о лабораторной перегонке.
15
Указанные выше книги предполагают наличие у читателя теоретических знаний и практического опыта в области ректификации. Те же требования предъявляются к читателю в более обширном труде Шульце и Штаге [11] по проблемам ректификации, в брошюре по обмену опытом «Методы работы и приборы для дистилляции и ректификации» Германского общества химического машиностроения, подготовленной Торманном [ 12 ] и охватывающей лабораторные методы перегонки, а также в брошюре «Ректификация в промышленности» [13], в основу которой положены производственные данные.
О темпах развития техники перегонки в период 1920—1944 г. дает представление литературный обзор Штаге и Шульце [14], который содержит ссылки на 2300 важнейших работ по теории и методам дистилляции и ректификации, а также применяемой аппаратуре. Всю мировую литературу по технике перегонки за 1941—1945 г. и 1946—1952 г. А. и Е. Роуз [15] отразили в кратких рефератах, которые за первый период времени охватывают 1000 публикаций, а за второй 5000 публикаций. Обзор за два года приведен в сборнике «Успехи технологии» [16] и, в частности, в его разделах «Дистилляция и ректификация», «Ректификация при низких температурах», «Массопередача», «Данные по равновесию для одно- и многокомпонентных смесей», а также в разделе «Вакуумная техника и методы работы под вакуумом». Уэльш [17] ежегодно публикует критические обзоры технической литературы под рубрикой «Unit Operations Review» *. Ценные указания о константах индивидуальных веществ и методах их расчета можно найти в пособии Орличека и Пёлля [18] для инженеров-нефтяников, в частности в специальном разделе книги приводятся данные по температурам кипения смесей и фазовым равновесиям. Современный уровень перегонки как в лабораторных, так и в промышленных условиях отражен Зигвартом [19] в превосходном обзоре, составленном для Ульмановской энциклопедии технической химии. Теория перегонки и экстракции с позиций термодинамики изложена в монографиях Кортюма и Бухгольц-Майзенхаймера [20] и Бошняковича [21].
Известные каждому изучающему химию книги Гаттерманна и Виланда [22] и Виттенбергера [23] дают практические сведения для химиков-органиков, однако по практике перегонки в них содержатся, к сожалению, лишь очень краткие обзоры. В книгах Вейганда [24] и Бернхауэра [25] по методам лабораторной техники кратко описаны также и современные приборы и методы; однако до сих пор не было исчерпывающего изложения проблем лабораторной перегонки, в том числе и методов расчета. Лишь в работах Карни [26] и Розенгарта [27], а также Роуза с сотр. [28] систематически изложены специфические вопросы лабораторной перегонки. Работа Вебера [29] посвящена получению эфирных масел с помощью перегонки, в частности перегонке с водяным паром. Для четвертого издания книги Губен-Вейля Зигварт [30] написал главу «Дистилляция и ректификация»- В этой главе дан превосходный обзор с многочисленными примерами из лабораторной практики и приведены 13 задач разделения с намеренно усложненными условиями процесса. Вышли в свет небольшие книги Цуидервега [31] и Коулсона и Херингтона [32], в которых кратко изложены основные принципы перегонки. В монографии Букала, Маевского и Родзинского [33] подробно рассматривается применение в лаборатории фракционной дистилляции и ректификации.
Начиная с 1960 г. резко возросло число публикаций по теоретическим основам процессов дистилляции и ректификации. Поскольку объем реферативных сборников постоянно растет, информацию теперь обрабатывают, широко применяя вычислительную технику. Тем не менее в ряде журналов еще появляются периодические литературные обзоры по вопросам перегонки [34]. Экспресс-информацию содержат реферативные карточки и доклады, обобщающие научные достижения [35, 16]. Назрела необходимость в издании для студентов обстоятельных руководств по технике лабораторных работ, включающих также разделы по методам дистилляции и ректификации [36], поскольку в большинстве имеющихся пособий методы разделения освещены недостаточно [37].
В ряде монографий обсуждаются специальные вопросы перегонки. Так, книга Рёка [38] является превосходным введением в экстрактивную и азеотроп
* «Обзор элементарных процессов». —Прим. ред.
16
ную ректификацию, а в книгах Штаге с сотр. [39] в доступной форме изложена методика экспериментального исследования фазового равновесия в системе пар— жидкость на примере жирных кислот, а также приведены сведения о соотношениях нагрузок в насадочных колоннах- Опубликованный в 1960 г, классический труд Хала с сотр. [40] по вопросам теории и экспериментального исследования равновесия в системе жидкость—пар был дополнен обширным сборником равновесных данных тех же авторов [41]. Переизданы таблицы Когана и Фридмана, обобщающие данные по равновесию для более чем 2000 бинарных систем. Кроме того, те же авторы подготовили справочник по азеотропным смесям (21069 систем, содержащих от 2 до 5 компонентов) [42]. В двух монографиях Биллет подробно и поэтапно излагает основы разделения жидких смесей путем перегонки [43] и принципы оптимизации в технике ректификации (особенно вакуумной ректификации) [44]. Материал монографий-относится к промышленной ректификации, вместе с тем приведенные многочисленные расчетные методы пригодны также для лабораторной и полупромышленной ректификации. В двух солидных трудах — Оливера [45] «Диффузионные процессы разделения» и Пратта [46] «Противоточные процессы разделения» — отчетливо проявляется тенденция рассматривать все методы разделения под одним и тем же углом зрения. С теоретической точки зрения это дает большие преимущества. Поэтому Крель с сотр. [47] в учебном пособии для студентов-химиков «Введение в методы разделения» попытался дать общее представление о методах разделения и их возможных комбинациях.
В последние годы появились новые монографии, трактующие специальные проблемы ректификации, например «Основы моделирования тарельчатых колонн» Хоппе и Миттельштрасса [48], «Низкие температуры в технологии» Юнгникеля и Отто [49] и «Ректификация как метод очистки» Франка и Куче [50]. В то время как последняя работа в основном охватывает лабораторные методы, монография Олевского и Ручинского [51] ориентирована на дистилляцию и ректификацию термически нестойких продуктов в промышленных условиях*. В монографии Холло с сотр. [52] рассмотрены вопросы применения молекулярной дистилляции в лаборатории, а также в пилотных и в промышленных установках; книга содержит многочисленные литературные ссылки и обширный справочный и иллюстративный материал.
В первой части своего двухтомного труда [53] «Термодинамические основы ректификации и экстракции» Шуберт обсуждает принципы термодинамики много-«азных систем, а также вопросы классификации и разделения бинарных смесей, торая часть посвящена тройным системам, экстракционному разделению двух компонентов с помощью одного или нескольких растворителей, специальным методам селективной ректификации, а также проблемам ректификации и экстракции многокомпонентных смесей. Приведенные в этой книге частные теоретические положения, справедливые для процессов ректификации, логически следуют из строгих термодинамических принципов.
Брауэром [54] в общей форме изложены теоретические основы процессов массообмена и разделения одно-и многофазных систем. При этом рассмотрен массо-перенос в неподвижных и движущихся средах. Для изучающих ректификацию особенный интерес представляют разделы «Массопередача в неподвижных и подвижных слоях насадки», «Массоперенос через границу раздела в простых двухфазных системах» и «Массоперенос в двухфазных потоках промышленных аппаратов». Холланд [55] подробно обсуждает вопросы многокомпонентной ректификации. В своей монографии [43а] Биллет освещает вопросы применения ректификации в промышленности.
На международном симпозиуме по перегонке, состоявшемся в Брайтоне (Англия) в 1969 г., особенно четко выявилась необходимость в дальнейшей разработке теоретических основ ректификации с ориентацией на применение электронной вычислительной техники при расчетах процесса разделения [56]. На
* В позднее опубликованной монографии В. М. Олевского и В. Р. Ручинского «Роторно-пленочные тепло-и массообменные аппараты» (М., Химия, 1977 г.) описаны также лабораторные и пилотные установки для дистилляции и ректификации термически нестойких продуктов под вакуумом. —Прим. ред.
17
симпозиуме были обсуждены всевозможные исследования и разработки в области дистилляции и ректификации. Вместе с тем отчетливо было показано, что в многоплановой области перегонки по-прежнему остается еще множество нерешенных проблем. К такому выводу приходит и Биллет [57], который освещает современный уровень и перспективы развития техники дистилляции и ректификации, проводя сравнение с другими методами разделения, основанными на тепловом воздействии на смесь веществ. Он подчеркивает, что ввиду ограниченности наших знаний о процессах массообмена и пропускной способности колонн затрудняется расчет оптимальных параметров и поэтому необходимы дальнейшие обширные и детальные исследования экспериментального и теоретического характера.
В результате бурного развития техники лабораторной перегонки за последние 30 лет и углубления специализации назрела необходимость в создании руководства по перегонке для лабораторий и опытных производств, рассчитанного на читателя, не имеющего специальных знаний в этой области. Такое руководство должно включать, методику определения давления паров и кривых равновесия, а также подробное описание непрерывных и селективных методов перегонки и рекомендации по применению контрольно-измерительной и регулирующей аппаратуры. Подобный обобщающий труд должен способствовать устранению многих неверных представлений о данном процессе разделения, подверженном влиянию многочисленных факторов, и послужить обстоятельным руководством при практическом проведении различных, в том числе и специальных, видов перегонки
Вторая глава настоящей книги «Из истории лабораторной перегонки» одновременно знакомит читателей с общими принципами перегонки. В третьей главе уточняются основные понятия, вводятся единицы измерения и условные обозначения, при этом особое внимание уделяется стандартизации, которая дает возможность за счет унификации определенных приборов и методик получать сопоставимые результаты, служащие фундаментом для дальнейших научных исследований. В главах 4—6 сначала изложены физические основы процесса перегонки и приведена классификация разделяемых смесей, после чего разносторонне рассмотрены обычные и селективные методы перегонки, с помощью которых можно решать самые разнообразные задачи разделения. В главах 7 и 8 описываются необходимые для проведения перегонки приборы и установки, включая вспомогательное оборудование, а также контрольно-измерительную и регулирующую аппаратуру. Наконец, девятая глава касается вопросов, которые следует принимать во внимание при оборудовании лабораторий дистилляции и ректификации и при вводе установок в эксплуатацию.
При изложении материала особое значение придавалось тому, чтобы процессы лабораторной и полупромышленной дистилляции и ректификации рассматривались во взаимосвязи с задачами промышленной перегонки, по отношению к которой эти процессы часто являются предварительным этапом. Ранее в лабораториях нередко разрабатывали методы дистилляции и ректификации, не имеющие никакой взаимосвязи с промышленностью. В итоге при переносе данных лабораторных исследований в промышленные и даже полупромышленные условия часто возникали значительные трудности. Если же при разработке новых способов, представляющих интерес для промышленности, с самого начала рассчитывать на их промышленное внедрение, то можно сэкономить значительные средства и много времени; при этом лабораторные данные почти без коорректи-ровки найдут применение при расчете промышленных процессов. Такой подход не должен, конечно, исключать тех случаев, когда в лаборатории намеренно подбирают условия, совершенно не экономичные для промышленной ректификации (например, при аналитических разгонках). Только глубокое знание закономерностей процесса ректификации обеспечит подбор оптимальных условий при решении любой задачи разделения, и данная книга должна способствовать приобретению этих знаний. '
Условные обозначения физико-химических величин и единицы измерения, использованные в прежнем издании, остались в основном без изменения. Однако сделана попытка привести условные обозначения в соответствие с международной практикой. Библиографические данные упорядочены по главам и приведены в списке литературы. Рисунки, формулы и таблицы пронумерованы по порядку с начала до конца книги,
18
2
Из истории лабораторной перегонки
Проследить, как на протяжении веков развивалась какая-либо химическая отрасль, интересно не только с исторической точки зрения; сравнение с современным уровнем развития часто приводило к ценным начинаниям в научно-исследовательской деятельности сегодняшнего дня. Исторический обзор особенно полезен при ознакомлении с такой специальной отраслью химической технологии, какой является перегонка. При этом мы становимся свидетелями того, как человеческая мысль на основе уже достигнутого постоянно находит все новые пути к повышению экономичности технологических процессов и производительности применяемой аппаратуры.
Шеленц [1], Гильдемайстер [2] и Рехенберг [3] проанализировали многочисленные исторические сведения, позволяющие проследить развитие методов перегонки на примере получения эфирных масел. Андервуд [4] дает краткий обзор развития техники перегонки вплоть до 19 века. В своей превосходной книге Фобе [5] подробно излагает историю этого своего рода искусства с самого его зарождения и вплоть до смерти Селлье-Блюменталя. Этот французский инженер умер в Брюсселе в 1840 г. Он был одним из самых талантливых конструкторов ректификационных колонн начала 19 века. Другие исторические сведения можно найти в разрозненном виде в многочисленных работах по истории алхимии и химии. Из книг, вышедших за последние десятилетия, следует отметить двухтомный труд Фобса и Дийкстеруиза [6] по истории естественных и технических наук, в котором общедоступно излагаются этапы развития техники, астрономии, математики, химии и физики, причем особое внимание уделено физике.
В. Шнайдер [7] составил словарь алхимических и фармацевтических терминов, который облегчает изучение старинных трудов.
В настоящем обзоре автор не стремился дать возможно более полную хронологию событий и лингвистически объяснить происхождение специальных терминов, он скорее пытался точно проследить этапы развития лабораторных методов и аппаратов и сопоставить их с современным уровнем техники. При этом внимание уделялось также и полупромышленным установкам.
Перегонка — это своего рода искусство, к которому еще до новой эры приобщились древние египтяне. Жрецы совершенствовали и оберегали это искусство в храмах как тайную науку. Индусы, персы и китайцы также, по-видимому, уже с древнейших времен использовали дистилляционные приборы. По мнению Шеленца [1], изобретение дистилляции следует приписать персам, которые возможно сначала использовали ее при получении розовой воды. Кроме того, он полагает, что в принципе дистилляция аналогична сухой перегонке дерева, с чем согласен также и Липпман [8—10], который сообщает, что сведения о перегонке с нисходящим'потоком газовой фазы можно найти еще в папирусе Эберса («Papyrus Ebers»), относящемся примерно к 1500 г. до нашей эры. Поэтому можно считать, что история перегонки насчитывает почти 3500 лет. Следует учесть, что в те времена слово «дистилляция» применяли в более широком смысле для обозначения всех известных тогда методов разделения. Это слово можно перевести как «разделение по каплям»*. На языке алхимиков это означало выде-
* Это слово (от лат. distillatio) можно также перевести как «стекание по ('каплям», что точнее соответствует специфике мелкомасштабных процессов разделения, когда целевой продукт собирают по каплям в приемные сосуды. — Прим. ред.
19
Рис. 1.
«Шлем» (alembic) на печи в окружении магических знаков.
Из трактата египетского алхимика Клеопатры «Приготовление золота» (II век н. э.).
ление более или менее ценных веществ. Поэтому термином «дистилляция» обозначали также фильтрацию, кристаллизацию, экстракцию, а также отжимание масла.
Мы будем рассматривать историю дистилляции (перегонки) только в современном значении этого слова, подразумевая под дистилляцией метод разделения веществ, основанный на испарении жидкости и конденсации образующихся паров. Разумеется, по данному определению дистилляция с нисходящим потоком паров представляет собой разновидность обычной дистилляции в современном ее понимании.
Примерами самого древнего применения перегонки являются получение розового и других эфирных масел, дистиллированной воды для мореплавателей (Аристотель сообщает о получении пресной воды из морской), а также приготовление алхимиками большого числа микстур и напитков. На рис. 1 показан «шлем» (alembic), установленный на печи; «шлем» окружен магическими знаками, которым при проведении перегонки в древности приписывалась особая роль. Типичный дистилляционный прибор того времени показан на рис. 2а. Он представляет собой стеклянный прибор с песочной или водяной баней. Приборы подобного устройства применяют еще и сегодня, что иллюстрирует рис. 26, на котором представлен прибор для перегонки ртути. Четыре основных элемента прибора: баня, куб (curcurbita, колба), головка (alembicus, шлем) и приемник дистиллята (receptacula) до настоящего времени сохранились практически в первоздан-
Рис. 2.
Простые перегонные приборы типовых конструкций:
а — стеклянный дистилляционный прибор с песочной или водяной баней (II век н. э.); б — прибор для вакуумной дистилляции ртути с расширением в виде кармана для сбора и отвода дистиллята (XX век).
20
Рис. 3.
б
1952 г.) снова применен этот
Приборы для перегонки по методу «per descensum» (с нисходящим потоком паров):
а — прибор для сухой перегонки коры и кореньев пряных растений (1300 г.); б — аппарат Блоума для дистилляции воды (XX век).
ном виде. Интересно отметить, что и расширение в виде кольцевого кармана для сбора и отвода дистиллята до сих пор еще используют в различных Дистилляционных приборах. В качестве конструкционных материалов для приборов в древние времена и в средние века кроме меди применяли в основном стекло и керамику-
Известные до 1300 г. методы перегонки в основном можно подразделить на дистилляцию с восходящим потоком паров (per ascensum) и на дистилляцию с нисходящим потоком паров (per descensum). Второй способ (per descensum) после 1880 г. был временно забыт, хотя он и представляет собой оптимальное решение для ряда случаев разделения (рис. За). В одном из аппаратов для дистилляции воды (выпуск принцип, как обеспечивающий экономное ис
пользование тепла (рис. 36)
Вскоре после изобретения книгопечатания появились книги по дистилляции,
в которых весьма наглядно отражен уровень развития техники перегонки в средние века. Имеются в виду главным образом следующие труды: Шрик «Указатель перегнанных вод» (1483 г.); Брунсвиг «Книга об истинном искусстве дистилляции
Рис. 4.
Установки с обогревательной системой, расположенной вне куба:
а — деревянный перегонный аппарат со змеевиковым холодильником и вынесенной топкой (XVII век); б — промышленная ректификационная установка непрерывного действия с вынесенным кипятильником (XX век).
21
Рис. 5.
Террасообразная печь для перегонки.
Внутри конического корпуса установлена топка, а по бокам расположены газоходы, в которых поддерживается рабочая температура. Керамические полки служат для размещения перегонных кубов (XVI век).
отдельных веществ» (1500 г.); Брунсвиг «Книга о подлинном искусстве дистилляции» (1507 г.); Ульштад«Сое1ит philosophorum» (1528 г.); Рифф «Новая большая книга по дистилляции», излагающая хорошо обоснованные методы искусной дистилляции (1536) г.
Начиная с XVI века наблюдается большее
разнообразие применяемых систем обогрева перегонных кубов. Кубы обогревали] с помощью воздушной, песочной или зольной бани, для обогрева применяли также и восковые свечи. Чтобы обеспгчить непрерывный режим работы, печи оснащались шахтными топками. Иногда применяли и весьма причудливые средства, например кислое тесто или баню с отжатой фруктовой мякотью, выделяющими тепло при брожении. В жарких странах для нагревания иногда применяли фокусирующие зеркала. В связи с этим интересно отметить, что в 1943 г. был выдан патент на устройство для обогрева перегонных аппаратов с помощью фокусирующего зеркала в форме параллелепипеда.
В промышленных установках часто требуется отделять нагреватель от собственно куба (рис. 46). Такое исполнение было предложено еще Глаубером (1604—1668 г.) (рис. 4а). При переработке значительных количеств дистиллята еще в XVI веке было принято проводить перегонку одновременно на нескольких Дистилляционных приборах, устанавливаемых в одной террасообразной печи (рис. 5).
Первые попытки применить теплоизоляцию, а именно шерсть животных, замешанную на глине, принадлежат еще Люлли (1415 г.). В XVI веке были установлены преимущества ступенчатого обогрева и уже тогда стали применять для обогрева тепло конденсирующегося водяного пара. Применение водяного пара в качестве теплоносителя в широких масштабах началось только после 1800 г.
Примерно до 1300 г. из способов конденсации паров был известен только прием, основанный на воздушном охлаждении. Для лучшего охлаждения паровую
Рис. 6.
Охлаждающая труба в форме змеевика: в полупромышленном н лабораторном дистилляционных аппаратах (а), в качестве конденсаторов с водяным и с воздушным охлаждением (XVI век) н в холодильнике Димрота (б) со стандартными шлифами (XX век).
22
Рис. 7.
Перегонные аппараты с удлиненной паровой трубой:
а — дистилляционные приборы, применявшиеся в химико-фармацевтической отрасли [большой аппарат с головкой в форме кулича и малый аппарат со «шлемом» установлены наГпечах и снабжены зигзагообразными паровыми трубками с воздушным охлаждением; приемник дистиллята закреплен на передвижном штативе (1709 г.)]; б — ректификационная колонна Янцеиа с корпусом в виде спиральной трубки (XX век).
трубу удлиняли, а позднее ее стали пропускать через бочку или чан с водой. Позже обнаружили, что наиболее выгодной формой охлаждаемой трубы является змеевик (рис. 6а), который и сейчас используется в конденсаторе Димрота (рис. 66). В XVI веке широко использовался непрерывный способ водяного охлаждения. Бы-
ло установлено обогащающее действие удлиненной паровой трубы, не имеющей тепловой изоляции (1709 г.), и частичной конденсации* (1648 г) (рис. 7, а). Аналогичный современный прибор показан на рис. 76. В 1770 г. Писсонье ввел принцип противотока при охлаждении, который и теперь еще используется, например в известном холодильнике Либиха (рис. 8, 6), применявшемся еще Дарио (1533—1594 г.) (рис. 8а).
Развитие техники перегонки в период с XVI по XIX век шло по пути совершенствования аппаратурного оформления. Еще в середине XVI века было установлено, что металлические кубы легко корродируют, и поэтому предпочтение стали оказывать приборам из стекла и керамики. Благодаря Кункелю (1638— 1703 г.) стекло стало преобладающим материалом в лаборатории и осталось таковым до настоящего времени. На рис. 9 представлены разнообразные дистилляционные головки («шлемы»), применявшиеся в то время. Уже тогда в употреблении были и приборы в миниатюрном исполнении (см. рис. 9 п). О том, как выглядела лаборатория алхимика примерно в 1700 г., дает представление
* Т. е. дефлегмации, когда часть паров конденсируется еще до ввода в конденсатор, а образовавшийся при этом конденсат возвращается в перегонную колбу.—Прим. ред.
Рис. 8.
Противоточные холодильники:
а — холодильник Дарио с непрерывным охлаждением (XVI век); б — холодильник Либиха (XIX век).
23
Рис. 9.
Головин («шлемы») дистилляционных приборов по данным Андреаса Ли-бау (около 1600 г.):
а — с длинным носиком; б — с коротким носиком; в, г, з — глухне; д — из олова; е, ж — сублимационные; а, у — глухне, с тубусом; к — колоколообразная; л — в виде тиары; м. — оловянная с оттянутым иосиком; н — с патрубком; о — с охлаждаемым устройством; п — в миниатюрном исполнении; р — глухая с носиком; с — тройная глухая; т — тройная с ио-снкамн.
Рис. 10.
Лаборатория аптеки при монастыре капуцинов в Париже (около 1700 г,).
рис. 10, изображающий лабораторию аптеки при монастыре капуцинов в Париже. По обилию дистилляционных приборов можно догадаться, что дистилляция занимала большое место в лабораторной практике. До XVIII века типичные лабораторные приборы практически ничем не отличались от самых древних образцов, но уже в середине XVIII века химики, как например Бом и Вульф, начали создавать новые модификации приборов. Систематические исследования процесса перегонки впервые были начаты английским физиком Бойлем (1627—1691 г.), который уже ставил опыты с дистилляцией под вакуумом и при давлении выше атмосферного (рис. 11). В середине XVIII века был создан ряд типовых установок, которые нашли применение во многих странах (рис. 12). Перегонные кубы имели диаметр 45—75 см и высоту 90—120 см.
Спиртовая промышленность Франции стимулировала бурное развитие техники перегонки, начавшееся в XIX веке при активном участии французских инженеров. В результате ряда усовершенствований (Адам, Берар, Перье) Селлье-Блюменталю в 1813 г. был выдан патент на ректификационную установку непрерывного действия, которая в принципе вполне соответствует современным аппаратам (рис. 13). Аппараты для вакуумной перегонки сконструировал в 1828 г. Триттон. Следующим достижением была колонна с ситчатыми тарелками, предложенная Коффи в 1830 г. Принцип барботажных {тарелок использован Шам-поннуа в промышленных аппаратах (рис. 14а). Этот принцип ' -----х лежит в основе современной промышленной ректификации
наряду с принципом орошаемой насадки, предложенной Игле-сом. В 1873 г. ои впервые применил шары в качестве насадки.
Рис. И.
Прибор Бойля для простой вакуумной перегонки (1627-—1691 г.).
24
Рис. 12.
Полупромышленные Дистилляционные установки XIX века, применявшиеся: в Германии (а); в Китае (б) и в Болгарии (в).
На основании изложенного выше можно прийти к заключению, что в древности и в средние века, несмотря на простоту применявшихся аппаратов, уже были известны основные принципы перегонки. До середины XIX века лабораторные и полупромышленные аппараты различались только размерами. Лишь в период бурного развития органической химии в середине XIX века были созданы дистилляционные и ректификационные приборы, специально предназначенные для экспериментальных исследований в химических лабораториях. Такие выдающиеся химики этого периода (по 1900 г.), как Кляйзен, Димрот, Глинский, Хемпель, Либих, Мичерлих, Мор и Вурц, создали ряд приборов для лабораторной перегонки. В качестве куба и приемника стали применять реторту, а от дистилляционной колбы с боковой трубкой для воздушного охлаждения через колбу Кляйзена и приставку с шариковым дефлегматором Вурца пришли к барботажной колонне. Приставки Линнеманна, Глинского, а также Ле-Беля и Хеннингера являются прототипами колпачковой колонны [11] (рис. 146 и 15). Колонна с насадкой из стеклянных шаров была введена в лабораторную практику Хем-пелем в 1881 г.
В конце XIX века назрела необходимость сравнить по разделяющей способности уже имевшиеся в большом числе дефлегматоры, выполняемые в виде приставок к дистилляционным кубам. Крайс, Янг и Фридрихе [12] дополнительно исследовали также эффективность холо-
Рис. 13.
Непрерывно действующая ректификационная __ . - .- ................. установка Селлье—Блюменталя (1813 г.).
25
Рис. 14.
Колпачковые колонны:
а — колонна Шампониуа (1854 г.); б — стеклянная колонна Брууна с 20 та* релками и вакуумированной рубашкой (1931 г.).
Рис. 15.
Этапы развития колодны с колпачковыми тарелками:
а ~ перегонная колба с боковой трубкой для отвода паров; б — колба Кляй* зека; в — дефлегматоры с шарообразными расширениями и без расширения; г — шариковый дефлегматор Вюрца (1854 г.); д — дефлегматор Линнеманна с ситчатыми тарелками (1871 г.); е — дефлегматор Глинского с ситчатыми тарелками (1873 г.); тс — барботажная колонна Ле-Беля— Хеннингера (1875 г.); з — ректификационная колонна Янга—Томаса (1889 г.); и — тарельчатая колонна Баума (1910 г.); к — колпачковая колонна Брууна (1931 г.).
Рис. 16.
Перегонные аппараты образца 1920 г.:
а — вакуумная ректификационная установка Рехенберга с колонной Хемпеля и приемником дистиллята Бертрана; б — ректификационная колонна Эльснерса для работы при атмосферном давлении и под вакуумом.
26
дильников для конденсации паров. Наряду с холодильником Либиха приобретает известность холодильник Димрота с охлаждаемым змеевиком (см. рис. 66), и уже применяется эффективный холодильник Мичерлиха для низкоки-пящих веществ, послуживший прототипом для ряда аналогичных устройств.
На период с 1900 по 1920 г. приходятся многочисленные изобретения, применяемые и сегодня, например колонна Янцена (см. рис. 76), насадочные кольца Рашига (1916 г.) и насадочные кольца «Прим» (1919 г.). Был усовершенствован обогрев аппаратов и создан регулятор давления для вакуумной перегонки. На рис. 16а показана установка для вакуумной ректификации, которой пользовался известный специалист по перегонке Рехенберг [3] в период с 1900 по 1920 г. Эта установка, как и ректификационная колонна Эльснерса (рис. 166), демонстрирует уровень развития техники перегонки в 1920 г.
Теоретические труды Янцена [13] и его школы, проводивших систематическую научно-исследовательскую работу по изучению физических основ процесса ректификации, послужили фундаментом Для бурного развития этой отрасли техники после 1920 г. Период с 1920 г. и по настоящее время еще слишком мал, чтобы ему можно было дать историческую оценку. Однако мы видим, что корни современной техники перегонки с ее сложными аппаратами и методами уходят глубоко в прошлое, когда уже были известны ее основные принципы [14].
3
Стандартизация
в технике перегонки.
Способы выражения концентрации
3.1. Стандартизация методов перегонки и применяемых приборов
Научное и технико-экономическое значение стандартизации настолько общеизвестно, что, по-видимому, нет нужды еще раз это подробно обосновывать. Однако следует постоянно пропагандировать применение стандартных приборов. Во всех странах прослеживается тенденция к созданию стандартных приборов для различных процессов разделения. Благодаря серийному производству такие приборы стоят сравнительно дешево, они всегда имеются в наличии и позволяют получать сопоставимые результаты. Начало стандартизации было положено известными приборами Эиглера и Американского общества по испытанию материалов [ASTMJ. Аналогичный прибор можно собрать из стандартных деталей, например, для работы под вакуумом (рис. 17). В работах [1, 2] описаны стандартизованные приборы, их детали, а также специальные методы перегонки.
Основное преимущество метода блочной сборки состоит в том. что он позволяет надежно и быстро собирать нужные установки и обходиться при возможности многих комбинаций без применения специальной аппаратуры. При этом дополнительно появляется возможность в мельчайших деталях копировать конструкцию промышленных установок. При ремонте прибора в этом случае требуется лишь заменить вышедшую из строя деталь.
В наше время в любой лаборатории органической химии и в том числе в любой лаборатории, занимающейся процессами перегонки, применяют почти исключительно аппаратуру со стеклянными шлифами (табл. 1). Телле [3] подготовил указатель аппаратуры для химических лабораторий в соответствии с требованиями стандарта TGL, действующего в ГДР.
С начала XX века в качестве взаимозаменяемых соединений нашли применение главным образом конические шлифы (рис. 18); сферические шлифы (рис. 19) используют пока в тех приборах, в которых использование конических шлифов давало бы слишком жесткое соединение. В ГДР сферические шлифы изготавливают в соответствии со стандартами TGL 20678 и DIN 12244. Прецизионные сферические шлифы с номинальными диаметрами от 25 до 150 мм фирмы Бюхи (г. Флавиль, Швейцария) обеспечивают герметичное вакуумное уплотнение без применения специальной смазки.
В стандарте DIN 12242 дается подробное описание различных типов шлифов и возможных областей их применения для лабораторных установок. Размеры взаимозаменяемых стандартных шлифов с конусностью 1 : 10 регламентированы стандартами
28
TGL 14972 (лист 2) и DlN 12242. Для лабораторной перегонки интерес представляют шлифы рядов 1 и 2, из которых преимущественно используют шлифы NS 14,5/23, 29/32 и 45/40. Для микро- и полумикроприборов находят применение также шлифы NS 7,5/16, 10/19 и 19/26. Целесообразно было бы ограничиться применением в лабораторных приборах общего назначения шлифов NS 14,5/23, приспособленных также для термометров (TGL 40—339, DlN 12784), и шлифов NS 29/32. Тем самым значительно улучшилась бы взаимозаменяемость стандартных деталей и расширилась бы возможность их комбинирования. В приборах «Дестинорм», разработанных автором (см. гл. 7), этот принцип выдержан и отлично себя оправдал. Специально для тех случаев, когда требуется облегченная разборка шлифового
Таблица 1.
Размеры применяемых в ГДР стандартных шлифов для взаимозаменяемых соединений.
А — конические шлифы NS с конусностью 1:10 (тип Кб) согласно стандартам TGL 14972 (ноябрь 1971 г.) н DIN 12242 (по рекомендациям Международной комиссии по стандартам, ISO)
Кр аткое обозначение шлифа Больший диаметр конуса, мм Длина конуса, мм Краткое обозначение шлифа Больший диаметр конуса, мм Длина конуса, мм
NS 5/13 5 13 29/32 * 29,2 32
7/16 7,5 16 34/35 34,5 35
10/19 10 19 45/40 * 45 40
12/21 12,5 21 60/46 60 46
14/23 * 14.5 23 71/51 71 51
19/26* 18,8 26 85/55 85 55
24/29 24 29 100/60 100 60
Предпочтительный размер.
Б — сферические шлифы S согласно стандартам TGL 20678 (ноябрь 1971 г.) и DIN 12244 (июль 1963 г., шаровые шлифы)
Краткое обозначение шлифа Диаметр сферического шлифа, мм Краткое обозначение шлифа Диаметр сферического шлифа, мм
TGL DIN • TGL DIN*
S 7 7,144 38 ** 40/25 38,100
13,2 13/2 12,700 41 41/25 41,275
13 13/5 12 51 51/30 50,800
19 * 19/9 19,050 64 ** 64/40 63,500
29 29/15 28,575 76 76,200
35 35/20 34,925 102 101,600
* Второе число равно внутреннему диаметру примыкающей стеклянной трубки (в мм).
** Предпочтительный размер.
29
Рис. 17.
Прибор для вакуумной дистилляции, собранный из стандартных деталей:
1 — Трехгорлая колба NS2‘9 с двумя боковыми тубусами NS14.5; 2 — термометры со стандартным шлифом NS14.5; 3 — капилляр для подсоса воздуха; 4 — угловой переходник со стандартными шлифами NS29 и 14,5;
5 — холодильник Димрота; 6 — вакуумный форштос; 7 — короткогорлая круглодонная колба.
соединения, например при перегонке высококипящих смесей и особенно при работе с высоким вакуумом, стандартами TGL 14972/3 и DIN 12243 регламентированы стандартные шлифы 1 : 5. Кроме того, главным образом в крупногаба-
с конусностью
ритных приборах применяют линзовые и плоские шлифы. Разработаны проекты стандартов на детали аппаратов и на трубопроводы промышленного назначения, выполненные из стекла [7].
Фридрихе [4] и Флиднер [5] дают обзоры способов испыта-
ния и ухода за стандартными шлифами.
Известно много традиционных методов анализа определенных смесей путем перегонки, например метод, заключающийся в исследовании процесса выкипания сырой смеси фенолсодержащих кислот. Куэнханссом с coTp.Jfl6] разработан метод экспресс-анализа бензина с температурой кипения до 180 °C, который позволяет уловить имеющиеся примеси парафинов, циклопарафинов (с 5—6-звенной углеродной цепью) и ароматических веществ. Используемый прибор комплектуется стандартной головкой (см. разд. 7.5, рис. 312). В настоящее время стандартизованы методы определения температурных пределов кипения (табл. 2); регламентированы также и основные размеры приборов.
Так как стандартные шлифы нуждаются в смазке, то предпри-
нимались неоднократные попытки создать принципиально новые соединительные элементы для стеклянных приборов. Фирма «Научно-технические стеклянные приборы» (Вертхайм) разработала бесшлифовое конусное соединение, в котором для надежной герметизации стыка вместо шлифов применено вакуумное лабиринтное уплотнение. Благодаря двум уплотнительным кольцам из тефлона полностью отпадает необходимость в смазке соединения. Сферические шлифы фирмы Бюхи также не нуждаются в каких-либо специальных уплотнениях или смазках.
Высокая точность изготовления шлифов гарантирует герметичность соединения как при высоковакууме, так и при избыточном давлении. Подобные сферические шлифы выпускаются с номинальными диаметрами 25, 50, 80 100 и 150 мм. Французский завод лабораторного стекла «Совирель» освоил выпуск разъемного соединения, в котором применены штуцеры нового типа. Гермети-
30
|S'; Рис. 18.
Стандартный конический шлиф (конусность ! : 10): / — конус шлифа; 2 — муфта шлифа.
К р“с-
'Стандартный сферический шлиф с зажимом.
Ж'Рис- 20-
«В Разъемное соединение «Совирель»:
ЯК' 1 — штуцеры С резьбой; 2 — фланцы с виуТрейней резьбой; 3 — Защитная прокладка из тефлона; 4 — промежуточное кольцо для фиксации фланцев; зЯ|г 5 — кольцо из силиконовой резины.
Изготовитель: фирма «Совирель», Левалуа, Франция.
д» Таблица 2.
нет Стандартизованные методы перегонки
Стандарт Назначение метода перегонки
TGL21120 Испытание минеральных масел, жидких топлив и аналогичных
21125 продуктов; определение температурных пределов кипения Испытание жидких топлив; определение давления паров по Рейду
0-51761 Испытание технических углеводородов бензольного ряда; определение температурных пределов кипения по Кремеру—Шпиль-
0-51786 керу Испытание жидких топлив; определение содержания неотстаи-
fDlN 51751 вающейся воды в мазутах перегонкой по ксилольному методу Определение температурных пределов кипения моторных топлив для двигателей внутреннего сгорания с посторонним зажиганием, а также бензинов
? 51752 Испытание дизельных топлив и аналогичных материалов по тем-
51761 пературным пределам кипения Проверка температурных пределов кипения по методу Кремера—
51767 Испытание нефтей, высококипящих нефтяных фракций и кубовых остатков при перегонке нефти; дробная перегонка по методу
| 51611 Г россе—Етрингхауса Низкотемпературная ректификация газов
31
чность соединения обеспечивается химически стойкой эластичной прокладкой, зажимаемой между двумя обезжиренными стеклянными поверхностями (рис. 20). Подобные соединения сохраняют герметичность как под вакуумом, так и при избыточном давлении; их термическая стойкость определяется свойствами прокладочного материала.
Крамер [16] изложил историю разработки современных уплотнительных систем. По его данным в настоящее время разъемные соединения для лабораторных приборов, выполненных из стекла, можно подразделить на шесть классов.
1. Шлифовые соединения: конические, сферические, цилиндрические, плоские и комбинированные шлифы, а также сферические фланцы.
2. Соединения с полированными контактными поверхностями: прецизионные соединения с прозрачными контактными поверхностями и калиброванные прецизионные трубы.
3. Соединения с прокладками (за исключением винтовых соединений): соединения с упругими сферическими элементами, конусные соединения с уплотнительным кольцом, плоские фланцы с уплотнительным кольцом, сферические фланцы с уплотнительным вкладышем.
4, Винтовые разъемные соединения: сальниковые уплотнения, винтовые соединения с накидной гайкой, муфтовые винтовые соединения.
5. Фланцевые болтовые соединения: пластмассовые фланцы на резьбе, фланцы с сальниковыми соединениями, накидные фланцы.
6. Другие типы соединений и вспомогательные материалы: заглушки, шланги, манжеты, сильфоны, фиксаторы шлифовых соединений, смазки для шлифов и и т. п.
По свидетельству Крамера [16] в настоящее время винтовые разъемные соединения с накидными гайками выпускаются в трех различных исполнениях фирмами Квикфит, Витег и Совирель.
3,2. ОПРЕДЕЛЕНИЕ ОСНОВНЫХ ПОНЯТИЙ
Часто приходится сталкиваться с тем, что в опубликованных работах по технике перегонки одним и тем же основным понятиям даются неодинаковые определения, что нередко приводит к недоразумениям. Даже само понятие «перегонка» применяют для обозначения ее разновидностей, весьма различающихся в техническом отношении. В связи с этим целесообразно ввести принципиальное разграничение между понятиями «дистилляция» (или «простая перегонка») и «ректификация *. Таким образом, термин «перегонка» приобретает значение собирательного понятия для обозначения методов разделения смесей жидкостей путем испарения смеси й конденсации отходящих паров.
В своей работе [7а]Хампель уделил внимание сложной проблеме «чистоты» растворителей и обсудил понятие «ультрачистый». Под этим понятием оно подразумевает предельную чистоту, которая вообще достижима на сегодняшний день. Нередко содержание тех или иных примесей должно быть уменьшено вплоть до нескольких триллионных долей. Для глубокой очистки наряду с экстракцией используют и ректификацию.
Применительно к перегонке в промышленных условиях еще в 1943 г. был разработан стандарт DIN 7052 «Разделение смесей жидкостей посредством дистилляции и ректификации». Однако он уже не отвечает современному уровню техники перегонки, и к тому же в нем не учитывается специфика лабораторной перегонки. Ввиду этого и в соответствии с предложениями автора [8], бывшего Комитета по стандартизации лабораторных приборов (рабочей комиссии «Приборы для дистилляции и ректификации») и Комитета по стандартизации в химическом машиностроении были выработаны новые определения (которые используются по мере необходимости в данной книге).
* Вместо принятых в СССР терминов «дистилляция» и «ректификация» | автор здесь и далее применяет термины соответственно прямоточная перегонка ? и противоточная перегонка. —Прим. ред.
32
3.3. ЕДИНИЦЫ ИЗМЕРЕНИЯ
Новые правила выбора единиц измерения базируются на авторитетных решениях X Генеральной конференции по мерам и весам, состоявшейся в 1954 г. В ГДР переход к новым единицам измерения начался с первого постановления от 14.08.58 г. по физико-техническим единицам измерений, которое, однако, было аннулировано новым постановлением от 31.05.67 г. В ФРГ постановление о переходе к новым единицам измерений появилось в Вестнике законов 28 июня 1970 г. В книгах Падельта и Лапорта [9], Ферстера [10], а также Хедера и Гертнера [11] приводится информация о практическом применении и сферах приложения новой международной системы единиц измерений (СИ) со сравнительным анализом старых систем. Между тем в ГДР дополнительно были утверждены новые стандарты (табл. 3).
Единицей измерения силы в СИ является ньютон (Н, м-кг/с2), который заменяет старую килограмм-силу (кгс) при следующем соотношении:
1 кгс = 1 кг-9,80665 м/с2 = 9,80665 Н
Для измерения давления жидкости и газа следует применять единицу СИ — паскаль (Па, Н/м2). Дополнительно вводится более крупная единица — бар (1 бар = Ю5 Па). Пересчет проводится по соотношениям:
1 ат = 1 кгс/см2 = 0,980665 бар =735,56 мм рт. ст.
1 мм рт. ст. = 1,333-Ю~3 бар
Применявшаяся до сих пор единица тепла — калория (кал) заменяется джоулем (Дж):
1 кал = 4,1868 Дж
Термодинамическую температуру выражают в градусах Кельвина (К). Градус Цельсия (°C) является специальной единицей для выражения температуры по шкале Цельсия. Температура по этой шкале равна Т — То, где То — температура таяния льда (плавления) при нормальном давлении равная 273,15 °C. Для разности температур обозначение «град» больше не применяется.
При составлении списка обозначений основных величин, используемых в книге, приходилось следить за тем, чтобы для обозначения различных величин не применялся один и тот же символ. Поскольку оказалось невозможным обеспечить необходимое разнообразие символов с помощью одного только немецкого алфавита, то пришлось прибегнуть к буквам греческого алфавита и индексам, расширившим возможности варьирования [12].
Таблица 3.
Стандарты иа условные обозначения, физические величины и единицы измерения
TGL 0-1304 Общепринятые условные обозначения, условные обозначения
физических величин
TGL 18-762 Лист 1 — Физические величины и единицы измерения; найме-
нования, сокращения
Лист 2 — Пояснения
DIN 1301 Единицы измерения, сокращения
DIN 1313 Способ записи применяемых в науке и технике уравнений,
связывающих физические величины
2 Э. Крсль
33
3.4. СПОСОБЫ ВЫРАЖЕНИЯ
И ПЕРЕСЧЕТА КОНЦЕНТРАЦИИ
При расчетах в технике перегонки концентрацию принято выражать в мольных долях или процентах, так как при определении объема паров, скоростей потока паров и других величин это очень упрощает вычисления. Поскольку большей частью рассматривают разделение двух компонентов, то на практике расчеты многокомпонентных смесей часто сводят к расчету бинарных смесей, принимай за второй компонент совокупность остальных компонентов смеси. В этом случае среднюю массу 1 моля смеси можно определить по уравнению:
м________ОТ1 Ч~ от2 Ч~ тз__ -1- -1- отз /1.
СР т^/М^т2/М2т3/М3 N±-{- N3 N3
где tn — масса; М.—молекулярная масса; N = mlМ — число молей соответствующего компонента.
Пример расчета средней массы 1 моля смеси
= 300 г бензола
т2 = 400 г толуола
ffZg = 500 г ксилола __________
тг + т2 -|- т3 = 1200 г смеси Mj = 78,11; М2 = 92,13;
М3 = 106,16;
m-jM-^ 300/78,11 = 3,84 т2/М2 = 400/92,13 = 4,35 щ3/Л43 = 500/106,16 = 4,71
Ml + = 12,90
Л4ср = 1200/12,9 = 93,0 г
Состав смеси всегда выражают концентрацией легколетучего компонента, если это особо не оговорено. Концентрацию указывают в объемных, массовых или мольных долях или же в объемных, массовых или мольных процентах.
Под концентрацией понимают отношение количества одного компонента к суммарному количеству всех компонентов.
Объемная доля : компонент 1 = К1/(К1 + К2) (2)
компонент 2 С2 = V2/(V1 + V2) (3)
Массовая доля : компонент 1 «1 = «1/("11 + «г) (4)
компонент 2 п2 = m3/(mi + т2) (5)
Мольная доля : компонент 1 Х1 = Mi/(Mi + M2) (6)
компонент 2 Хз = N2I(N1 + N2) (7)
При умножении этих долей на 100 получают значения концентрации в процентах.
Пример определения мольной доли и мольного процента для смеси
Компонент 1 300 г бензола mj/Mj = 3,84
Компонент 2 400 г толуола т2/Л42 = 4,35
700 г смеси М + М2 + М = 8,19
Мольная доля = 3,84/8,19 = 0,47
Мольная доля х2 = 4,35/8,19= 0,53
Мольный % компонента / 0,47-100 = 47,0% (мол.)
Мольный % компонента 2 0,53-100 = 53,0% (мол.)
34
При расчете концентраций многокомпонентных смесей знаменатель в соответствующих уравнениях увеличивается на дополнительное число слагаемых (компонентов), например для трехкомпонентной смеси:
Мольная доля xt = + Na) (8)
Мольная доля х2 = + ^з) (9)
Мольная доля ха = Na/(Ni + М2 + ^з) (10)
Лессельс [13] приводит удобную номограмму для пересчета % (масс.) в в % (мол.) и наоборот для трехкомпонентных смесей.
Для бинарных смесей пересчет концентраций проводят по следующим уравнениям, отнесенным к легколетучему компоненту 1.
Пересчет % (масс.) в % (мол.):
% (мол.) = —’ 100% ' -]- т2/Л42 (И)
Пересчет % (об.) в % (масс.):
Пересчет % % (масс.) = • 100% 1 1Р1-Ь г 2Р2 (мол.) в % (масс.) (12)
Пересчет % %(МаСС-)-Л1Л+Х2'100% (масс.) в % (об.) ' % (об.) = - 100% ^h/Pi + тз/Рз (13) (14)
Пересчет % (мол.) в % (об.):
«.«./Z.+X./p, 1Ю% (15)
Ввиду того, что такие пересчеты громоздки, были разработаны различные номограммы, обеспечивающие быстрый и простой пересчет. При переходе от мольных или массовых процентов к объемным следует учитывать, что пересчет дает правильный результат лишь в том случае, если при смешении компонентов не происходит изменения объема (отсутствует контрактация). Орличек, Пёлль и Валенда [14] разработали универсальную номограмму (рис. 21), применение которой пояснено следующей схемой:
При пересчете Вместо Q подставить Для компонента с индексом 1*** следует понимать
под Z под X
% (мол.) в % (масс.) % (об.) в % (мол.) % (об.) в % (масс.) Л41/Л42* Умол, 1/УмОЛ, 2 ** Р2/Р1 % (масс.)/100 % (об.)/100 % (об.)/100 % (мол.)/100, т. е. мольную долю % (мол.)/100, т. е. мольную долю % (масс.)/100, т. е. массовую долю
* М — молекулярная масса; ** ^мол — мольный объем;*** индексом 1 отмечают величины, относящиеся к компоненту с большей молекулярной массой.
Пользование номограммой поясняется построениями, приведенными на рис. 21, для следующего примера: скольким % (масс.) соответствуют 88% (мол.), если- компоненты имеют молекулярные массы 150 и 60?
= 150 __ 2 5 X =88% (мол.) или 0,88 мольных долей
Л42 = 60 Z = 94,8% (мол.) или 0,948 мольных долей
2*
35
Рис. 21.
Номограмма Орличека, Пелля и Валенды для пересчета концентраций.
Рис. 22.
Номограмма Бэра для пересчета мольных процентов в массовые и наоборот.
Разумеется, с помощью этой номограммы можно с таким же успехом провести обратный пересчет из % (масс.) в % (мол.), из % (мол.) в % (об.) и из % (масс.) в % (об.).
Для газообразных смесей значения концентрации, выраженные в мольных и объемных процентах, совпадают; объемы относят к атмосферному давлению, принимая, как обычно, что объем 1 моль равен 22,4 л при 0°С и 760 мм рт. ст. или 24,4 л при 15° С и 735,5 мм рт. ст.
Для пересчета из % (мол.) в % (масс.) и наоборот особенно удобна ая обеспечивает быстрое и надежное
50 круглая номограмма Бэра [15], получение результата, хотя достигаемая при этом точность не так велика, как при расчете с применением формул.
Пользование номограммой иллюстрируется примером, приведенным на рис. 22. Требуется определить, скольким мольным процентам соответствуют 30% (масс.) бензола в смеси бензол—толуол. Известно, что для бензола = 78,11, а для толуола М2 = 02,13. Тогда = 78,11/92,13 — 0,848 ~ 0,85.
Через точку 30% (масс.) на нижней полуокружности (см. рис. 22) и точку 0,85 на горизонтальной оси M-JМг проводим прямую, которая пересекает верхнюю полуокружность в точке 33,6% (мол.).
Концентрации изотопов в смеси принято выражать в атомных процентах. Например, природная вода имеет следующий состав:
99,9844% (ат.) Н
0,0156% (ат.) D
100,0000% (ат.) водорода
99,757% (ат.) 16О 0,039% (ат.) 17О 0,204% (ат.) 18О
100,000% (ат.) кислорода
4
Физические основы процесса разделения
4.1. ПРИНЦИПЫ дистилляции и ректификации
Как указывалось выше, термин «перегонка» можно заменить на «разделение по каплям» (см. гл. 2). Этот термин имеет, таким образом, собирательное значение и относится к процессам разделения смесей взаиморастворимых жидкостей путем испарения жидкости и конденсации паров, обогащенных легколетучим компонентом. Поэтому термин «перегонка» не содержит никаких пояснений относительно характера процесса. Понятия «дистилляция» (или простая перегонка) и «ректификация» четко определяют также и способ работы (рис. 23).
Со строго физической точки зрения перегонка не является обязательно процессом разделения. Например, перегонкой можно назвать также процесс, в ходе которого чистое жидкое вещество испаряют, пары этого вещества конденсируют и конденсат собирают. Ректификация с бесконечным флегмовым числом представляет собой предельный режим перегонки, при котором конденсат (дистиллят) не отбирают.
Массообмен можно описать следующим отношением:
т — К.а, Лх I ср
где т — масса переносимого вещества; К — коэффициент массопередачи, а/ — активная поверхность контакта фаз; Ал'ср — движущая сила процесса массооб-мена.
Обмен веществом происходит путем его диффузии через межфазную поверхность. На скорость массоотдачи в пределах одной фазы влияют коэффициент диффузии D, расстояние Ь, на которое передается вещество, и концентрация х. От площади межфазной поверхности at, приходящейся на единицу высоты ректификационной колонны, зависит количество вещества, переходящего из одной фазы в другую.
Движущая сила массообмена между двумя фазами определяется разностью концентраций фаз (у—х) *. При термодинамическом равновесии наступает состояние насыщения, движущая сила становится равной нулю, и процесс массообмена, протекавший до установления равновесия, прекращается.
* Т. е. разностью у* —у между концентрацией пара у*, находящегося в термодинамическом равновесии с жидкостью состава х, и средней действительной концентрацией пара у, соприкасающегося с этой жидкостью. — Прим, ред-38
Рис. 23.
Схема, поясняющая принцип перегонки: а — дистилляция (простая перегонка); б — ректификация.
’ -жшкость
При дистилляции (или простой перегонке) молекулы, отрывающиеся с поверхности испарения, движутся в одном и том же направлении до момента достижения поверхности конденсации. Отличительная же особенность ректификации состоит в том, что поток жидко-. сти (как правило сконденсированных паров) направляется навстречу поднимающемуся потоку паров. Если дистилляция состоит всего лишь из процессов испарения и конденсации, то при ректификации благодаря тесному контакту двух фаз в колонне имеет место массо- и теплообмен. Рассмотрим в общих чертах процесс, протекающий на тарелке колонны (рис. 24). При установившемся режиме составы пара и жидкости на одной и той же тарелке изменяются в направлении достижения термодинамического равновесия между ними под влиянием градиентов температур и концентраций. Вследствие переноса вещества в вертикальном направлении (парами вверх, а жидкостью вниз) это равновесие нарушается, что благоприятствует дальнейшему обогащению паров легколетучими компонентами [1]. Другими словами, поток жидкости (флегма) на своем пути из зоны более низких температур (вверх колонны) в зону более высоких температур (кипятильник) поглощает из потока паров высококипящие компоненты и выделяет легколетучие компоненты. Температурному градиенту в колонне соответствует перепад концентраций в парах Г и в жидкости. При этом в кипятильнике пар менее насыщен легколетучим компонентом, чем в головной части колонны, а ^жидкость (флегма) в верху колонны содержит больше легколетучего компонента, чем на входе в кипятильник.
Физические процессы массо- и теплообмена протекают на границе раздела фаз, поэтому очень важно создать максимально •возможную поверхность контакта. Такая поверхность контакта имеет место в полой орошаемой колонне, она может быть образована насыпной насадкой, тарелками, сетками и различными (вращающимися устройствами (см. разд. 4.3 и 7.3.).
На процесс разделения влияют многие факторы и в первую Зчередь физико-химические свойства смеси, геометрические пара-етры и колонны и ее насадки, а также условия работы колонны.
Как правило, компоненты жидкой смеси, температуры кипения которых различаются более, чем на 50 °C, мсжно до опре-
( 39
деленной степени разделить, применяя простое упаривание или дистилляцию. Поэтому дистилляцию применяют преимущественно в тех случаях, когда жидкая смесь содержит в незначительных количествах компоненты с очень высокой температурой кипения или нелетучие примеси. В качестве примера можно указать на отделение растворенных нелетучих примесей при простой перегонке воды или на очистку растворителей от высококипящих примесей. Исключением является так называемая перегонка по Энглеру [2], которую применяют для определения температурных пределов кипения смесей весьма близкокипящих компонентов с точками кипения примерно до 200 °C. В данном случае при дистилляции создаются легко воспроизводимые условия, а именно адиабатический режим без орошения. Смеси близкокипящих компонентов нельзя разделить простой перегонкой, поскольку хотя бы даже вследствие брызгоуноса высококипящий компонент может механически увлекаться парами в дистиллят.
По способу работы различают периодическую и непрерывную перегонку. Периодическая перегонка (разгонка) — это простая дистилляция или ректификация, в процессе которой содержимое куба частично или полностью отгоняется. Непрерывная перегонка —это простая дистилляция или ректификация, в процессе которой исходную смесь непрерывно вводят в перегонный аппарат, а различные выделенные из смеси продукты непрерывно выводят из аппарата.
Ректификация обеспечивает разделение компонентов и в том случае, если их температуры кипения различаются всего лишь на 0,5 °C. Применяя ректификационные колонны с числом теоретических тарелок порядка 400—500, как это требуется при разделении изотопов, удается разделять смеси компонентов с разностью температур кипения всего лишь 0,05 °C. С помощью
Гэлодка колонны
Темп, низкая Конц.бысокая
Жидкостьх^
И £ И МММ
& * А А тш
Тарелка
— пар, обогащенный легколетучим компонентом
— пар при входе на тарелку
— жидкость, обогащенная труднолетучим компонентом
- жидкость при входе на тарелку '
Куб
Темп, высокая Конц, низкая
Рис. 24.
Схема процесса разделения на тарелке ректификационной колонны.
40
Таблица 4.
Рабочая схема решения поставленной задачи разделения
Факторы, подлежащие рассмотрению Определяемые условия или рассчитываемые параметры Ссылка на соответствующий раздел
Разность температур ки- Способ перегонки: дистилляция или 4.1
пения ректификация 4.7.5.1
Кривые давления паров Оптимальное давление процесса 4.4 4.6.2 5.3 5.4
Кривая равновесия Число теоретических ступеней разделения и условия ректификации 4.6—4.7.4
Требуемая степень раз- Способ перегонки и режим процесса 4.7.5
деления 4.8
* 4.9 5 6
Требуемая пропускная Тип применяемой аппаратуры, разме- 4.11
способность ры прибора и тепловые характеристики 4.12
процесса 5.1
5.2 7
Эффективность колонны Метод испытания эффективности колонны 4.10
Автоматизация Тип контрольно-измерительной и регулирующей аппаратуры 8
[ селективных методов, а в трудных случаях дополняя ректифи-[ кацию другими методами разделения, например экстракцией, | ионообменным методом и особенно газовой хроматографией, В ,, удается разделять смеси, прежде считавшиеся неразделимыми. I/ В последующих главах более подробно рассматриваются сложные Вл" процессы ректификации, которые были теоретически детально исследованы Куном, а также Куном и Риффелем [3].
Е До проведения перегонки, дистилляции или ректификации, г прежде чем начать расчеты, необходимо решить ряд вопросов. I В табл. 4 приведена рабочая схема, которая должна помочь £ при решении поставленной задачи разделения. Более подробные i , инструкции по проведению процесса перегонки даны в разд. 4.14. £ у Сведения, касающиеся сборки установок, приведены в разд. 9.3, | а рекомендации по пуску аппаратов — в разд. 9.5.
II 4.2. ОСОБЕННОСТИ ДВИЖЕНИЯ ЖИДКОЙ
М. ФАЗЫ В КОЛОННАХ С ОРОШАЕМОЙ НАСАДКОЙ
4.2.1. РАСПРЕДЕЛЕНИЕ ЖИДКОСТИ ПО НАСАДКЕ
Для разделения веществ путем ректификации применяют в основ-йЯ'.ном следующие колонны: тарельчатые (см. разд. 7.3.3), пленоч-S ные (см. разд. 5.4.3), с регулярной насадкой (см. разд. 7.3.4), ЙЯ5 с насыпной насадкой (см. разд. 7.3.2.).
41
Рис. 25.
Схематическое изображение характера работы колонн различных типов (по данным Штаге):
а — с колпачковыми тарелками; б — с ситчатыми тарелками; в — с провальными тарелками; г — с насадкой «Спрейпак»; д — насадочная; е — пленочная.
Работа колонн этих четырех типов основана на общем принципе противоточного движения контактирующих фаз. Существенные различия в характере движения контактирующих материальных потоков определяются геометрическими особенностями применяемых типов колонн.
Колонны последних трех типов можно объединить общим понятием колонны с орошаемой насадкой. Их отличительным признаком является образование пленки жидкости в процессе встречного движения сплошных потоков . контактирующих фаз при полном отсутствии их взаимного проникновения. Напротив, тарельчатая колонна характеризуется тем, что при ее работе жидкая фаза пронизывается, более или менее раздробленными пузырьками газа, которые вновь объединяются на вышележащей тарелке (рис. 25). В качестве эталонных жидкостей для исследования массообмена в противоточных колоннах пригодны фреоны, а также растворы неорганических хлористых соединений иода [За]. При исследовании характера движения жидкой фазы применяли также радиоактивные изотопы, например при изучении процесса ректификации бутадиена был использован изотоп 82Вг [36].
Важной особенностью колонны с регулярной насадкой является то, что ее насадка имеет регулярную геометрическую форму. «Пакетная насадка» Зульцера представляет собой орошаемую насадку из проволочной сетки, снабженной гофрами, вызывающими движение контактирующих потоков по удлиненной зигзагообразной траектории. В распылительной же колонне 42
''а'"
L?
«Спрейпак» жидкая фаза под воздействием паров, обладающих р значительной кинетической энергией, приводится в распыленное Ь состояние, а газовая фаза проходит через насадку в виде сплош-Е кого потока. Уже из сопоставления этих устройств можно полу-чить представление о сложности гидродинамического режима Is в насадочной колонне. Если в колоннах с регулярной насадкой материальные потоки движутся по определенным траекториям, I то при хаотическом расположении насыпной насадки происходит | неравномерное и неупорядоченное движение обеих контакти-Е рующих фаз.
fc Для колонн с насыпной насадкой характерна йеравномер-Г ность распределения как газовой фазы, так и жидкости. Нерав-g номерное распределение жидкости вызывается следующими при-К чинами [4].
В- 1. Жидкость перетекает по насадочным телам предпочти-к‘ тельно в направлении к стенкам колонны, а не наоборот. В ре-Ц" зультате частичной конденсации паров, вызванной тепловыми № потерями, процесс накопления жидкости у стенок колонны еще к более усиливается. Этот вид неравномерности орошения назы-К’ вают пристеночным эффектом (см. разд. 4.8.1).
К • 2. Вследствие неудовлетворительной смачиваемости самих на-
IV садочных тел случайно образовавшаяся струйка жидкости не В разрушается (каналообразование, см. разд. 4.8.1).
₽> 3. Насадка может состоять из отдельных слоев с предпочти-
|Г тельными направлениями для стока жидкости. И в этом случае № имеет место каналообразование. Расслоение насадки может быть К вызвано особенностями выбранного способа загрузки насадочных тел в колонну или причинами, связанными с геометрической №• Формой насадочных тел.
Ж 4. Колонна может быть установлена не точно вертикально. Н 5. Не всегда обеспечивается равномерная подача флегмы Иг на насадку.
. Результат действия всех этих факторов можно условно предик ставить следующим образом:
Неудовлетворительное распределение = Каналообразование ф-
№Ё..' + Пристеночный эффект
Иг На пристеночный эффект оказывают влияние высота колон-Икны Нк, ее диаметр dK, а также диаметр насадочных тел da. При Игртношении djd,, 20 поток жидкости, стекающей по стенкам ИЕколонны, составляет от 10 до 20% общего количества жидкости МО зависимости от отношения высоты колонны к ее диаметру к^к/^к).
|Ик Влияние неудовлетворительного распределения жидкости тем ИВшачительнее, чем большим числом теоретических ступеней раз-Миеления обладает колонна. При коэффициенте неравномерности ИяЬрошения 10% число теоретических ступеней разделения, полу-ИЕяаемых в колонне, эквивалентной, например, 100 ступеням,
43
снижается до 30, а в колонне, эквивалентной 10 теоретическим ступеням разделения, — всего лишь до 9 ступеней. Ввиду того что в промышленности также наблюдается устойчивая тенденция к повышению эффективности колонн, вопрос распределения жидкой фазы приобретает особо важное значение.
Трудности, связанные с установкой колонны в строго вертикальном положении или с первоначальным распределением жидкости, могут быть так или иначе преодолены. Поэтому основное внимание при исследовании насадочных колонн в настоящее время уделяется тому, чтобы получить ясное представление о характере распределения жидкой фазы в ходе процесса разделения. Эффективность этого процесса снижается с увеличением как диаметра, так и высоты колонны.
Неравномерность в распределении жидкости по колонне всегда вызывает снижение числа теоретических ступеней разделения вследствие того, что соотношение расходов пара и жидкости в различных точках насадки отклоняется от нормы. Глубокий теоретический анализ влияния неравномерности распределения жидкости на эффективность колонны был выполнен Хьюбером и Хильтенбруннером [5]. Они приняли за основу модель, представляющую собой колонну с четырехугольным поперечным сечением, разделенную воображаемой продольной перегородкой на два отдельных отсека, работающих при неодинаковых нагрузках. В качестве критерия (коэффициента) неравномерности орошения I было принято относительное отклонение локальной плотности орошения от ее среднего значения, рассчитанного для поперечного сечения колонны
/ = (£ — £)/£
где L — локальная плотность орошения (в определенной точке); L — средняя (по поперечному сечению колонны) плотность орошения.
Воображаемая перегородка многократно пересекается движущимися вихрями, что связано с переносом вещества в радиальном направлении, и параллельные потоки частично смешиваются между собой в количестве, характеризуемом степенью перемешивания tnq (рис. 26).
Коэффициент использования насадочной колонны п/п0 является функцией коэффициента неравномерности орошения I и степени перемешивания tnq, отнесенной к одной теоретической ступени разделения:
п/п0 « 1/[1 + (/8/3,75/п^)]
где mq — степень перемешивания, отиесеииая к одной теоретической ступени и показывающая, какую долю от всего поперечного сечения колонны составляет площадка, на которой происходит смешение струй при движении потока в пределах ступени разделения, п0, п — число теоретических ступеней, получаемых в колонне соответственно при равномерном и при неравномерном орошении.
44
Степень перемешивания mq, отнесенную к одной ступени разделения, можно оценить на основе теоретических выкладок Хьюбера по формуле
mq = 27 (dH/dK)2
Например, для dK/da = 10 получаем mq — 0,27, а для dJdH = = 30 имеем mq = 0,025. В первом случае степень перемешивания tnq настолько велика, что даже значительное повышение коэффициента неравномерности орошения мало снижает коэффициент использования насадочной колонны. Во втором случае такая зависимость не имеет места. Этот пример иллюстрирует эмпирическое правило, которое требует, чтобы отношение dKlda находилось в интервале от 10 до 30. Поэтому представительное значение числа теоретических ступеней разделения для колонны можно получить лишь при отношении djda = 10 и при I 0.
Рис. 26.
Схема модели колонны без перемешивания в поперечном направлении по Хьюберу —Хильтеибруииеру [5].
Колонна разделена продольной перегородкой на две секции 1 н 2. Внутри каждой секции расходы газа G и жидкости L постоянны, хотя они могут быть и не одинаковыми в разных секциях. На данной схеме расходы газа в секциях 1 и 2 одинаковы, а расходы жидкости — неодинаковы.
уИ — концентрация легколетучего компонента (ЛК) в газовой фазе на входе в колонну; х0 — концентрация ЛК в жидкой фазе на входе в колонну; Х/ю, Уго — концентрация ЛК в газовой фазе на выходе из секций 1 и 2 соответственно; х1и, х2и — концентрация ЛК в жидкой фазе иа выходе из секций 1 и 2; I — критерий неравномерности распределения потоков в колонне.
Любое неравномерное распределение можно приближенно моделировать, представив колонну в виде достаточно большого числа продольных секций. Изменение концентраций показано для бесконечного числа теоретических ступеней.
Рис. 27.
Схема, иллюстрирующая явление смачивания твердого тела жидкостью (<р — краевой угол смачивания):
а - полное смачивание; б - - частичное смачивание; в — полное отсутствие смачивания.
45
Кафаров, Дорохов и Шестопалов [6] подробно исследовали взаимосвязь между нагрузками колонны по обеим фазам и различными гидродинамическими параметрами, например динамической или статической удерживающей способностью колонны (см. разд._4.10.5), продольным перемешиванием перепадом давления (разд. 4.11). Они установили л количественную связь между динамической удерживающей способностьюхи перепадом давления, а также зависимость статической удерживающей способности от нагрузки, изменявшейся в широком интервале. С использованием понятий эффективного и «мертвого» объема была выведена теоретическая модель нестационарного движения жидкости в насыпной насадке; модель была использована для предварительного расчета параметров движения жидкости. Исследована также зависимость коэффициента продольного перемешивания от нагрузок по газу и жидкости, а также от физико-химических свойств жидкости. Ионас [7] проанализировал основные факторы, приводящие к продольному перемешиванию в насадочных колоннах. В своих экспериментах Тимофеев и Аэров ([65] к гл. 7) основное внимание уделили вопросам влияния диаметра колонны на эффективность разделения.
Неодинаковое поверхностное натяжение компонентов смеси может даже заметно отразиться на ходе процесса разделения в насадочной колонне [8]. Если в процессе ректификации бинарной смеси по мере отбора дистиллята поверхностное натяжение о флегмы увеличивается (такие смеси называют положительными), то массообмен может быть значительно более интенсивным, чем при разделении смесей, при ректификации которых поверхностное натяжение флегмы постепенно уменьшается (такие смеси называют отрицательными). Если смесь отрицательна, то поток жидкости, омывающий насадку, дробится на мелкие струйки и капли. Разумеется, этот эффект проявляется лишь в том случае, когда соблюдается условие До > 3 дин/см. Смеси компонентов, для которых коэффициенты поверхностного натяжения различаются менее чем на 3 дин/см, называются нейтральными.
В технических руководствах отношение смоченной поверхности насадки aw к ее полной поверхности а называют долей смоченной поверхности
фзд, = dwl а
Для расчета доли смоченной поверхности предложено много формул, которые, однако, приводят к весьма неодинаковым результатам [9]*.
* В специальной литературе (см., например, Рамм В. М., Абсорбция газов. М., Химия, 1976) проводят различие между смоченной поверхностью aw и активной поверхностью de. Это обусловлено тем, что участки поверхности насадки, покрытые неподвижной пленкой жидкости, не участвуют в массообмене, т. е. не являются активными. Соответственно вводится понятие доли активной поверхности -- ае!а. — Прим. ред.
46
Весьма показательно, что в подобных зависимостях совершенно игнорируется влияние таких факторов, как поверхностная энергия жидких смесей, стремление жидкостей уменьшить свою поверхность (особенно при пленочном течении отрицательных жидких смесей), а также влияние краевого угла смачивания, образующегося при контакте жидкости с поверхностью твердого тела (рис. 27). Возможно, что пренебрежение указанными факторами и является причиной значительных расхождений между вычисленными и экспериментальными величинами. Комбинируя уравнения Дюпре и Янга,, приходим к следующему простому соотношению для определения работы, затрачиваемой на образование единицы площади поверхности раздела фаз (или поверхностной энергии):
^s-i = <T(l+cosq>)
Из этого уравнения следует вывод, что возрастание поверхностного натяжения и уменьшение краевого угла смачивания приводят к увеличению поверхностной энергии IK.._j.
Так как поверхностное натяжение целиком определяется природой разделяемой смеси, то при подборе материала насадки следует стремиться к тому, чтобы зависящий от природы материала краевой угол смачивания был как можно меньше. В табл. 5
Таблица 5.
Поверхностная энергия IV's-i для систем твердое тело —
жидкость (вода прн 20 °C)
Класс веществ Материал Средняя высота выступов шероховатости, мкм Кр аевой угол смачивания (р, ° ^s-r эрг/см2
Металлы Вольфрам 62 109,0
Платина 71 96,5
Медь 0,1—0,5 84 80,4
Алюминий 85 79,2
Никель 86 77,9
Сплавы Сталь St.37 79 86,7
Сталь X-10-CnNiTi-18,9 0,1—0,5 83 81,7
Фосфористая бронза 84 80,4
Полимеры Полиамид 72 96,3
Полимер сложного эфира 72 96,3
Пол иметилм ета кр и л ат 72 96,3
Поливинилхлорид 73 94,1
Поликарбонат Менее 0,1 77 89,2
Полистирол 80 85,2
Полипропилен 90 72,8
Полиэтилен 90 72,8
Тефлон 106 52,5
Керамика Фарфор 5-15 50 119,0
Стекло 0,1 20 140,8
47
приведены рассчитанные значения поверхностной энергии для различных систем твердое тело—жидкость [11].
Очевидно, чем больше поверхностная энергия, тем более высокую стабильность пленки жидкости следует ожидать при смачивании твердого тела, но тем труднее, однако, добиться полного смачивания жидкой фазой элемента насадки [11]. Предварительным затоплением насадки (см. разд. 4.10.8) и выбором оптимальной конфигурации рабочей поверхности насадки можно значительно улучшить ее смачиваемость [9]. Титов и Зельвен-ский [10] предложили три метода расчета активной поверхности ае в колоннах с насыпной насадкой. Получены графические зависимости доли активной поверхности, высоты единицы переноса и коэффициентов массопередачи от плотности орошения.
Квасняк [11а ] исследовал эффекты конденсации и испарения при ректификации в насадочных колоннах, предположив, что в любом поперечном сечении колонны между паром и жидкостью всегда имеется разность температур. Поэтому несмоченные участки поверхности насадки можно рассматривать как поверхность теплообмена. Элементы сравниваемых насадок имели идентичную конфигурацию, но одни элементы представляли собой сплошные медные пластинки, а другие — пластмассовые пластинки, облицованные медью, благодаря чему обеспечивались различные коэффициенты теплопроводности. Пластинки были размещены в насадке так, что их нижняя сторона в процессе ректификации не орошалась. Насадки очень сильно различались по разделяющей способности, что можно объяснить эффектами конденсации и испарения, возникающими на сплошных медных пластинках. Влияние подобных эффектов следует всегда учитывать. Основываясь на этих результатах, Квасняк разработал новую регулярную насадку, состоящую из зигзагообразно изогнутых и различно ориентированных металлических листов. Такая конструкция обеспечивает дополнительную турбулизацию жидкой и газовой фаз и лучшую смачиваемость рабочей поверхности.
4.2.2. ПЛЕНОЧНОЕ ТЕЧЕНИЕ ЖИДКОСТИ
Йилмаз и Брауэр [116] при теоретическом анализе гидродинамических закономерностей движения жидкости в слоях насыпной насадки использовали математическую модель, соответствующую неустановившемуся пленочному безволновому течению вдоль гладкой пластины. Теоретические выкладки были подтверждены экспериментом. Исследование условий волнообразования на поверхности ускоренно движущейся пленки дали материал для построения универсальной диаграммы, представляющей различные режимы движения жидкости в насадках. Из диаграммы следует, в частности, что пленочное течение в слоях насадки является практически всегда ламинарным и безволновым. Эти данные послужили фундаментом при выводе теоретически обос
48
нованных зависимостей для расчета динамической удерживающей способности жидкости в слоях насадки из колец Рашига (см. разд. 4.10.5).
Кюнне [Не] подробно исследовал гидравлику противоточных колонн с насадкой из орошаемых пластин. Опыты были проведены при комнатной температуре и атмосферном давлении с применением системы воздух—вода. Кюнне приводит методику измерений и предлагает метод расчета гидравлического сопротивления и верхней предельной нагрузки по газовой фазе. Один из методов экспериментального определения площади межфазной поверхности в системе газ—жидкость для пленочных колонн предложен Антоновым с сотр. [11 з].
На основе теоретической модели, которая описывает движение пленок жидкости, механически перемешиваемых в колоннах с вращающейся насадкой, Диц с сотр. [11г] вывел уравнения для расчета толщины пленки. Уравнения базируются на ряде упрощающих посылок и справедливы в том случае, когда толщина пленки жидкости не превышает величины зазора между ротором и корпусом аппарата:
6 = /p + J/flK|-i/8p Р = Х2НГ1/(4Р«''г)
& = Г/(ХР£)
В = ^/(gn)
где 8 — средняя толщина пленки; % = 0,304; rt — внутренний радиус колонны; р — плотность жидкости; п — число оборотов ротора в 1 мин; Г — массовая линейная плотность орошения; jt — динамическая вязкость; г2 — расстояние от наружной кромки скребка до оси аппарата.
Годау [11д ] исследовал некоторые закономерности движения жидкостей в пленочных испарителях. Биллет [lie] анализирует непрерывный процесс пленочной дистилляции на примере пленочного испарителя «Липотерм», оперируя критериями подобия и приводя математические зависимости. Вычисления с помощью полученных уравнений показывают, что для простой перегонки с дефлегмацией существуют определенные соотношения нагрузок, при которых достигается максимальное обогащение дистиллята.
4.2.3. ДИНАМИЧЕСКИЕ ХАРАКТЕРИСТИКИ КОЛОНН
Динамические характеристики ректификационных колонн пытаются рассчи-; тывать, применяя различные математические модели. По Кёллеру и Шоберу [264] динамика колонн становится объектом изучения в тех случаях, когда нашей целью [, является: 1) исследование выходных параметров колонн во времени после простого или комбинированного возмущающего воздействия на процесс ректификации; ’ 2) моделирование процессов ввода и вывода колонн из рабочего режима, а также ^отклонений от него (предусмотренных или случайных); 3) поверочный расчет [нестационарных режимов промышленных установок; 4) расчет стационарных режимов как предельных случаев переходного процесса ректификации; 5) модели-?рование процессов управления установками; 6) улучшение динамических харак-
' теристик колонн с учетом существенных факторов, проявляющихся в неустано-
49
вившемся режиме (при изменении нагрузок по газу и жидкости); 7) теоретическое объяснение пульсационных явлений в ректификационных установках и проведение альтернативных расчетов в попытке устранить подобные явления.
Адольфи [11 ж] разработал графический метод расчета динамических характеристик колонн для ректификации бинарных смесей; в основе метода лежит диаграмма Мак-Кэба и Тиле. Вагнером с сотр. [162 а, б] исследована динамическая характеристика насадочных ректификационных колонн. Приводится информация о динамике колонн при разделении бинарных идеальных и неидеальных смесей, а также тройных систем. Исследована динамическая характеристика последовательно соединенных ректификационных колонн. Для расчета динамики колонн широко применяются ЭВМ (см. разд. 4.15).
4.3. РАЗДЕЛЕНИЕ СМЕСЕЙ ВЗАИМНО
РАСТВОРИМЫХ КОМПОНЕНТОВ
Взаимная растворимость жидкостей тем выше, чем ближе их химическое сродство (например, принадлежность к одному гомологическому ряду). Данные о закономерностях, наблюдаемых при смешении органических соединений, приведены в работе Штау-дингера [12]. В большинстве случаев взаимная растворимость возрастает с повышением температуры; выше критической температуры смешения Ткр имеет место неограниченная растворимость. На рис. 28 приведена диаграмма растворимости для смеси вода—фенол.
Перегонке подвергают, как правило, гомогенные растворы; при расслоении смеси ее сначала разделяют декантацией. Разу-
Рис. 28.
Диаграмма состояния системы вода—фенол.
50
Рис. 29.
Диаграммы равновесия пар—жидкость для различных бинарных смесей: . а — бензол—вода, вода — хлорбензол, вода—ксилол; б — вода—фурфурол; в — вода—н-бутанол, вода —изобутанол; г — emop-бутанол—вода; а — уксусная кислота—н-октан, этанол—вода, сероуглерод—ацетон; е — * н-гексан—бензол, н-гептан —бензол, метанол—вода; ж — бензол—толуол, j этан—бутан, циклогексан—н-октан; з — ацетальдегид—фурфурол, аце-
у той—уксусная кислота, ;. азотная кислота—вода,
метилацетилкетон — пропионовая кислота; и — соляная кислота—вода, циклогексанол—фенол.
имеется, следует учитывать, что полностью взаимнонерастворимых ^веществ не существует. Растворенные вещества нельзя разделить
(механическими способами, и эффективным методом разделения г в подобных случаях является перегонка.
Перегонку в токе носителя, главным образом перегонку
= с водяным паром, и азеотропную ректификацию применяют также
для разделения смесей совершенно нерастворимых или частично растворимых компонентов. К перегонке в токе пара-носителя прибегают для снижения температуры процесса. При азеотропной же ректификации специальной добавкой определенного (вещества вызывают образование азеотропа между этим веществом
•R одним из компонентов разделяемой смеси, отделяя его таким ^Образом от остальных компонентов; получаемый при этом азеотропный дистиллят должен легко поддаваться разделению на ^составные части (см. разд. 6.2.1). Следовательно, в таких случаях необходимо знать диаграмму растворимости для азеотропной
51
Таблица 6.
Особенности кипения бинарных смесей
Тип смеси (см. рис. 29) Степень взаимной Свойства кипящего раствора Сравнение сил притяжения между одинаковыми (^1/1 н <*2/2) и неодинаковыми (aj/2) молекулами
растворимости компонентов
а Полностью нерас- Имеет минимальную тем- О|/2 «1/!
творимы пературу кипения “1/2 °2/2
б Частично раство- То же а1/2 а1/1
в г римы с областью расслаивания «1/2 «2/2
д Полностью растворимы » а1/2 а1/1 а1/2 а2/2
е То же Н ей деа л ьн ый р а створ, азеотропа не образует а1/2 < а1/1 «1/2 < «2/2
ж » Идеальный раствор а1/2 ~ а1/1
«1/2 ~ а2/2
3 Полностью рас- Неидеальный раствор, а1/2 > а1/1
творимы азеотропа не образует а1/2 > а2/2
и То же Имеет максимальную температуру кипения а1/2 > «1/1 «1/2 > а2/2
к » Образуются новые химические соединения -н с-1 Q Q с-1 с-1 сГ о"
смеси, чтобы выбрать соответствующую температуру, при которой данная смесь расслаивается. Из рис. 28 можно, например, видеть, что при температуре выше 68,8 °C гомогенный раствор существует при любом соотношении компонентов, а ниже этой температуры наличие или отсутствие расслоения определяется составом смеси.
Взаимная растворимость двух компонентов может изменяться в пределах от почти полной нерастворимости до полной смешиваемости; для характера же кипения смесей имеется гораздо больше возможных вариантов. В идеальных смесях молекулярные силы притяжения, действующие между одинаковыми и неодинаковыми молекулами, равны. Если силы притяжения между неодинаковыми молекулами меньше, чем между одинаковыми, то следует ожидать образования азеотропной смеси с минимальной температурой кипения (положительный азеотроп); в противоположном случае образуется азеотроп с максимальной температурой кипения (отрицательный азеотроп). Между этими двумя крайними смесями расположены неидеальные смеси, для которых кривая равновесия на соответствующей диаграмме
52
сверху или снизу асимптотически приближается к диагонали диаграммы. Штаге [13] наглядно представил эти взаимосвязи (табл. 6), а рис. 29 иллюстрирует соответствующие типы кривых равновесия. Более подробно эти вопросы рассматриваются в разд. 4.6 и 6.2.
Гильдебранд и Ротариу [14], указывая на различия в энтальпии, энтропии и активности, подразделяют растворы на идеальные, правильные, атермальные*, ассоциированные и сольватированные. Как можно судить по классификации бинарных растворов, предложенной Маузером и Кортюмом [15], теория бинарных жидких смесей, несмотря на многочисленные фундаментальные исследования, до сих пор еще находится в неудовлетворительном состоянии. В монографиях Манхена [16] и Шуберта [17] подробно излагаются основы термодинамики растворов.
4.4. ЗАВИСИМОСТЬ МЕЖДУ ДАВЛЕНИЕМ
ПАРА И ТЕМПЕРАТУРОЙ рt
При решении задачи разделения необходимо прежде всего установить связь между давлением р и температурой t для перегоняемых смесей, которую изображают в виде кривых давления паров. Если на миллиметровой бумаге построить график зависимости давления насыщенных паров от температуры, то с его помощью можно определить, при каком давлении лучше проводить дистилляцию или ректификацию (см. рис. 39). При этом для температуры лучше использовать логарифмическую шкалу. Выбор давления разгонки зависит от того, какая из следующих операций должна быть проведена: а) аналитическая разгонка; б) препаративная ректификация; в) перегонка с целью накопления продукта; г) сравнительная ректификация с целью моделирования промышленной ректификации в лабораторных условиях. При этом необходимо учитывать, принимая во внимание гидравлическое сопротивление колонны, что ректификацию следует проводить под давлением, исключающим опасность термического разложения вещества и обеспечивающим такую температуру в конденсаторе, при которой имеющаяся в распоряжении охлаждающая среда будет пригодна для конденсации паров.
При аналитической разгонке следует добиваться максимальной эффективности разделения; поэтому выбирают такое рабочее давление, при котором кривые давления паров для разделяемых компонентов находятся как можно дальше одна от другой, в результате чего обеспечивается более высокая относительная летучесть компонентов (см. разд. 4.6.2). То же относится и к препара-
* Гильдебранд называет атермальными такие растворы, в которых теплота смешения компонентов равна нулю, причем компоненты имеют различные мольные объемы. — Прим. ред.
53
тивной ректификации. При перегонке же с целью накопления продукта следует иметь в виду, что с уменьшением давления значительно возрастает объем паров, и тем самым ограничивается пропускная способность колонны. При сравнительной ректификации рабочее давление процесса выбирают в зависимости от того, какой вакуум может быть гарантирован в промышленной установке.
Весьма подробные данные по давлению насыщенных паров приведены Драйсбахом [18], который собрал также значения физических констант для 38 гомологических рядов на диаграмме Кокса (разд. 4.4.2). В таблицах Драйсбаха [19] указаны температуры кипения примерно для 500 органических веществ при 1, 10, 100 и 760 мм рт. ст. Сталл [20] собрал данные по температурам кипения для 1200 органических веществ при 1, 5, 10, 20, 40, 60, 100, 200, 400 и 760 мм рт. ст. Кроме того, он привел аналогичные данные примерно для 300 неорганических веществ и около 100 значений температуры кипения для интервала давлений от 1 до 60 ат. Штаге опубликовал уточненные данные по давлениям насыщенных паров соединений для соединений различных классов, а именно, для насыщенных спиртов жирного ряда Cj—С18 с прямой углеродной цепью [21 ], для метиловых эфиров насыщенных жирных кислот Cj—С18 с прямой углеродной цепью [22], фенолов [23] и летучих компонентов спиртовой барды и спирта-сырца [24]. Большое число справочных таблиц подготовил /Кордан [25]. Риддик и Туппс (младший) [26] приводят данные по давлению паров и другие физические величины для 254 растворителей. Заге и Ласей [27] представили данные по давлению паров и равновесию для легких углеводородов, H2S и СО2. Глазер и Рюланд [28] впервые построили кривые давления паров в интервале от 1 до 60 ат для 33 веществ. Йекель [29] приводит список литературы, охватывающей данные по давлению паров в области среднего и высокого вакуума. Дополнительные новые материалы можно найти в «Успехах технологии» и реферативных сборниках ([16] к гл. 1).
Несмотря на обилие накопленных данных, в лабораторной практике нередко еще возникает необходимость в самостоятельном определении давления насыщенных паров. В таких случаях целесообразно проводить собственные исследования; однако можно также воспользоваться различными методами расчета, описанными в разд. 4.4.2.
4.4.1. ЭКСПЕРИМЕНТАЛЬНОЕ ОПРЕДЕЛЕНИЕ ДАВЛЕНИЯ НАСЫЩЕННЫХ ПАРОВ
Существуют два принципиально различающихся метода определения давления насыщенных паров чистого вещества: а) динамический метод — определение температуры кипения при различных давлениях; б) статический метод — определение давления паров при различных температурах. Методика проведения измерения подробно описана Киницем в сборнике Губен—Вейля [30]. Милаццо [31] приводит сведения о методах и приборах, применяемых для измерения 54
малых давлений. Он отмечает, что при измерении давления паров в интервале от 10"5 до 10 мм рт. ст. хорошие результаты дает манометр Мак-Леода.
Применение статического метода в лабораторных условиях, особенно при повышенных температурах, связано с экспериментальными трудностями и чаще всего используют динамический метод определения давления паров, тем более, что контролировать давление не сложно, благодаря наличию точно работающих регуляторов. Разумеется, при подобных измерениях важно заранее убедиться в достаточной чистоте исследуемого вещества.
Необходимым условием точного определения температуры кипения является хорошее перемешивание кипящей жидкости с образующимися паровыми пузырьками. Классическим прибором для определения температуры кипения является эбуллиометр Светославского [32]. На рис. 30 изображен дифференциальный эбуллиометр в полумикроисполнении. Его можно использовать не только для определения точки кипения, но также для контроля чистоты веществ и для изучения явления азеотропии в многокомпонентных системах.
Вещество в объеме 10—12 мл загружают в оба колена 1 и 2. На стенки колена 1 наплавлен стеклянный порошок, а снаружи это колено обмотано греющей спиралью. Образующиеся пары, захватывая за собой жидкость, поднимаются в тубус 7, в котором измеряется температура кипения вещества. Далее пары проходят через тубус 6, в котором измеряется температура конденсации, в хо-
Дифференциальный эбуллиометр Светославского в полумикроисполнеиии с рабочим объемом 10 —12 мл:
/ — колено с расширением; 2 — колено прямое; 3 — трубка; 4 — счетчик капель; 5 — холодильник; 6, 7 — тубусы.
Рис. 31.
Эбуллиометр Светославского (усовершенствованный Ольшевским) для ге-тероазеотропиых смесей.
Рис. 32.
Эбуллиоскоп Вебера с теплоизолирующим кожухом:
1 — штуцер для загрузки жидкости и присоединения к вакуумному насосу; 2 — конденсатор; 3 — кипятильная трубка; 4 — теплоизоляция из стекловолокна; 5 — спираль компенсационного электрообогрева; 6 — карман; 7 - - конец разбрызгивающей трубки; 8 — рубашка; 9 — разбрызгивающая трубка; 10 — центральное углубление; 11 — электрическая свеча; 12 — колпак.
55
Рис. 33.
Установка Вебера для определения давления насыщенных паров:
1 — укороченный манометр; 2 — форвакуумный (буферный) сосуд емкостью 3 л; 3 — адсорбционные ловушки; 4 — ртутный диффузионный насос; 5 — масляный насос; 6 — вспомогательный манометр; 7 — манометр Мозера; 8 — эбуллиоскоп; 9 — воздушная камера емкостью 10 л; 10 — ртутный манометр; 11 — масляный манометр.
лодильник5, где пары полностью конденсируются. Жидкость возвращается в обогреваемое колено 1, проходя через счетчик капель 4 и трубку 3. Счетчик капель служит для контроля скорости испарения жидкости.
Для определения азеотропного состава сильно гигроскопических веществ, например пиридиновых оснований, фенолов, применяют трехступенчатый эбуллиометр. В этом случае измеряют одну температуру кипения и две температуры конденсации. При работе с чистыми веществами или азеотропными все три температуры должны быть одинаковыми. Усовершенствованный прибор для определения температуры кипения гетероазеотропных смесей * разработан Ольшевским (рис. 31).
Аналогично работает эбуллиоскоп Вебера, изображенный на рис. 32. С помощью этого прибора можно измерять давление насыщенных паров в интервале от 10 до 760 мм рт. ст. и выполнять другие операции, такие как калибровка термометров, эбуллиоскопические измерения, изучение равновесия пар—жидкость, получение характеристик различных фракций дистиллятов, например в нефтяной и коксохимической промышленности [33].
Прибор имеет вертикальную кипятильную трубку 3 с внутренним диаметром 34 мм и длиной 500 мм. Регулируемое нагревание жидкости обеспечивается снизу электрической свечой И, размещенной в углублении 10. Наружные стенки углубления для усиления парообразования покрыты наплавленным стеклянным порошком. С помощью колпака 12, доходящего почти до дна, все образующиеся пузырьки пара собираются, смешиваются с жидкостью и направляются в разбрызгивающую трубку 9 диаметром 4 мм, где происходит дополнительное перемешивание в двух шарообразных расширениях. Затем смесь через конец 7 трубки 9 в виде брызг попадает на карман 6 термометра. Разбрызгивающая трубка вместе с шариками окружена вакуумированной рубашкой 8. Отделившаяся от пара жидкость стекает с кармана термометра и через узкую кольцевую щель между рубашкой 8 и кипятильной трубкой 3 возвращается в цикл. Кончик термометра погружен в небольшое количество ртути и защищен карманом 6 с припаянным
* Определение понятий гомогенный азеотроп, а также гетерогенный азеотроп см. в разд. 6.2. — Прим. ред.
56
козырьком, предохраняющим его от попадания капель стекающего сверху_кои-денсата. Столбик ртути окружен конденсирующимися парами.
Наблюдение с помощью лупы за подсвеченной сзади шкалой термометра и подсчет десятичных делений шкалы через пленку конденсата и не представляет трудностей, если верхнюю часть эбуллиоскопа предварительно протравить в течение 2 мин 1%-ной фтористоводородной кислотой и затем прокипятить в мыльной воде. Кипятильная трубка 3 до самого конденсатора 2 окружена изолирующим слоем стекловолокна 4, в котором оставлена узкая смотровая щель. Под теплоизоляцией 4 на трубку 3 намотана спираль компенсационного электрообогрева 5, выполненная из тонкой проволоки. Мощность обогрева можно рассчитывать, условно представляя спираль в виде охватывающей прибор бесконечно длинной цилиндрической оболочки с равномерно распределенными источниками тепла. Электрообогрев регулируют с помощью амперметров и калибровочной кривой таким образом, чтобы без включения системы подогрева кубовой жидкости приближенно устанавливалась ожидаемая температура. В этом случае даже пары труднолетучих веществ доходят до конденсатора, расположенного на 250 мм выше кармана термометра. Адиабатический режим в разбрызгивающей трубке обеспечивается четырехкратной защитной системой, включающей вакуумированную рубашку, слой нагретой до кипения жидкости, стекающей в кольцевой щели, спираль компенсационного электрообогрева и слой теплоизоляции. Через штуцер 1 обычно загружают жидкость, а при работе под вакуумом к нему присоединяют вакуумную линию.
Общая схема установки, включающей кольцевой трубопровод для присоединения насосов, эбуллиоскопа и манометра, приведена на рис. 33. Стабилизацию
Рис. 34.
Схема прибора Херрингтона—Мартина для определения температуры кипения (например, пиридиновых оснований):
1 — кипятильная колба для исследуемого вещества; 2 — кипятильная колба для чистой воды; 3 — вакуумный масляный насос; 4 — газосборник емкостью 30 л; 5 — регулятор давления; 6 — газосборник емкостью 30 л в водяной бане (400 л); 7 — высоковакуумный масляный насос; 8 — сосуд с оксидом фосфора; 9 — парортутный эжекторный иасос; 10 — манометр . Мак-Леода; 11 — баллон с азотом; 12 — сосуд с аммиаком и раствором карбоната аммоиня над металлической медью; 13 — промывная склянка ' с разбавленной серной кислотой; 14 — 19 — сосуды соответственно с 10%-* иым раствором едкого натра, концентрированной кислотой, безводным хлористым кальцием, с силикагелем, с пятиоксндом фосфора, со стекло-' ватой; 20 — охлаждаемая ловушка.
57
Рис. 35.
Вид по А
Прибор Хеиииига—Штока для измерения давления паров легколетучих веществ:
/ — колбаХдля жидкости с термометром; 2 — шлиф с крючком для термометра; 3 — кран; 4 — патрубок для присоединения к вакуумной линии;
5 — измерительное колено; 6 — барометр; 7 — чашка со ртутью.
Рис. 36.
Изотеиископ, усовершенствованный Шубертом:
1 — шарик с исследуемой жидкостью; 2 — вспомогательный манометр;
3 — охлаждаемый шарик; 4 — патрубок для присоединения к буферной емкости; 5 — термостатирующнй кожух.
давления при 760 мм рт. ст. или при любом вакууме можно проводить по методам, описанным в разд. 8.3. Аналогичная установка описана Эблином [34]. Для определения температуры кипения с точностью ±0,003 °C рекомендуется прибор Херрингтона и Мартина [35], который применяли для измерения давления паров пиридина и его гомологов в интервале от 50 до 157° С (рис. 34). Высокая точность измерения давления достигается тем, что параллельно при тех же условиях, что и в перегонной колбе, испаряют особо чистую воду, и искомое давление вычисляют по температуре паров кипящей воды.
Очень изящный метод, однако обеспечивающий точность измерения всего лишь ±0,5 мм рт. ст., описан Нательсоном и Цукерманом [36]. Он основан на том явлении, что капля жидкости, висящая на конце вертикального капилляра (трубки термометра), в результате постепенного вакуумирования окружающего пространства отрывается от капиллира. В момент отрыва капли считывают показание ртутного манометра, соответствующее достигнутому давлению.
Метод Элиша для работ в микромасштабе с загрузкой порядка нескольких миллиграммов вещества был усовершенствован таким образом, что стало возможным проводить измерения также и при остаточных давлениях ниже 100 мм рт. ст. [37]. На обогреваемой микроподложке конструкции Кофлера можно также определять точки кипения при незначительных количествах вещества [38].
Давление паров при низких температурах можно также определять, применяя радиоактивные изотопы. Несмеянов [39] подробно описывает методику, специально разработанную применительно к металлам и сплавам. Для трудно-летучих веществ, например иода, нафталина и фенола, пригоден эффузионный метод [40].
В дополнение к изложенному ниже рассмотрено еще несколько статических методов определения давления насыщенных паров, в соответствии с которыми 58
температуру поддерживают постоянной с помощью термостата и измеряют установившееся давление по манометру. При измерении давления паров легколетучих веществ соответствующего интервалу температур от —20 до +20° С можно воспользоваться методикой, описанной Хеннингом и Штоком [41]. Устройство прибора показано на рис. 35.
В колбу 1 через шлиф 2 загружают исследуемое вещество, на крючке шлифа 2 подвешивают термометр с ценой деления 1/10° С и колбу помещают в ванну с охлаждающей смесью. Патрубок 4 соединяют с вакуумным насосом; кран 3 служит для отключения прибора от вакуумной линии. Измерительное колено 5 погружают в чашку 7 со ртутью барометра 6.
Систему вакуумируют и закрывают кран 3, который следует заранее проверить на герметичность. Затем колбу 1 слегка нагревают, чтобы поднять давление паров, после чего нагревание прекращают и кран 3 открывают. Эту операцию повторяют до полного удаления воздуха из прибора. После того, как в колбе 1 установится постоянная температура, можно начинать измерение. С помощью зеркальной шкалы (а еще точнее с помощью катетометра) определяют разность уровней для менисков ртути в измерительном колене 5 и барометре 6, которая и представляет собой давление паров исследуемой жидкости (в мм рт. ст.). При каждой температуре необходимо выполнить несколько операций по измерению давления, а результат усреднить. Целесообразно предварительно проверить работоспособность этого прибора путем измерения давления паров какого-либо чистого вещества, для которого точно известны соответствующие данные.
Описанный Смитом и Мензисом [42] изотенископ благодаря применению вспомогательного манометра и буферной емкости позволяет определять давление насыщенных паров для высококипящих веществ, причем получаются относительно хорошие результаты. Рис. 36 иллюстрирует устройство прибора, усовершенствованного Шубертом. При работе этого прибора очень важно, чтобы во вспомогательный (U-образный) манометр 2 перегналось нужное количество исследуемого вещества. Кроме того, необходимо с помощью крана так отрегулировать вакуум в буферной емкости (на рисунке не показана), присоединенной к патрубку 4, чтобы показания вспомогательного манометра 2 установились на нулевой отметке. Тогда главный манометр (присоединенный к буферной емкости) покажет давление паров исследуемой жидкости, установившееся в левом колене вспомогательного манометра 2. Изотенископ размещен в термостатирующем кожухе 5, что обеспечивает равенство температур, при которых находятся манометр 2 и шарик 1 с исследуемой жидкостью.
Измерение давления насыщенных паров проводят следующим образом [43]. Шарик 1 трубки изотенископа на 3/4 объема заполняют исследуемым веществом. С помощью толстостенной вакуумной резиновой трубки прибор подсоединяют в точке 4 к буферной емкости, содержащей воздух. Прибор термостатируют, и всю систему медленно вакуумируют (с помощью вакуумного насоса, присоединенного к буферной емкости). Когда давление достаточно понизится, вещество в шарике 1 закипает, его пары конденсируются в верхнем шарике 3. Конденсат стекает в нижнюю часть вспомогательного манометра 2 и тем-самым полностью исключает контакт кипящего в шарике 1 вещества с воздухом, содержащимся в.буферной емкости. Когда убеждаются, что воздух из левой части изотенископа удален полностью, в термостатирующем кожухе 5 устанавливают постоянную температуру и регулируют вакуум, добиваясь того, чтобы мениски жидкости в U-образной трубке установились на одном и том же уровне. Сразу же после этого замеряют температуру термостатирующего кожуха и давление, показываемое ртутным (главным) манометром. Чтобы быть уверенным, что в изолированной части изотенископа (т. е. в шарике 1) больше нет воздуха, отсасывание воздуха и измерение давления повторяют несколько раз. После получения нескольких повторяющихся результатов начинают медленно повышать температуру. При этом одновременно изменяют давление, следя за тем, чтобы мениски жидкости в U-образной трубке постоянно находились почти на одном и том же уровне. Во избежание обратного проникновения воздуха в шарик 1 следует всякий раз избегать избыточного давления в буферной емкости.
Мениски жидкости в U-образной трубке окончательно устанавливают на одном и том'же уровне при постоянном давлении в буферной емкости путем не-
59
Рис. 37.
Прибор Кортюма с сотр. для измерения давления паров статическим методом:
1 — паровая камера; 2 — дроссель; 3 — сборник; 4 — платиновые контакты; 5 — поплавки; 6 — приемник жидкости; 7 — стандартный шлиф NS20; 8 — краны; 9 — токоподводящие провода для платиновых контактов; 10 — патрубки для присоединения главного манометра; 11 — конические шлифы; 12 — стандартный шлиф NS12; 13 — приемник конденсата;
14 — патрубок для присоединения промежуточного манометра; 15 — железный сердечник; 16 — магнитная катушка; 17 — пластина из пористой керамики.
значительных изменений температуры в термостатирующем кожухе.
Кортюм с сотр. [44] для получения данных по общему давлению паров в системе вода—диоксан при 35° С применил прибор (рис. 37), обеспечивающий более высокую точность измерения (до ±0,2 мм рт. ст.).' Шлюндер [45] описывает простой статический метод измерения давления паров над водными растворами солей LiCl, MgCl2 и Mg (NO3)2 при температуре 20—90° С.
Масс-спектрометрический метод измерения низких давлений паров позво-ляет определить парциальные давления определенного вещества в присутствии ' загрязняющих примесей.
Рек [46] применил три различных статических метода для измерения очень низких давлений (0,056—17,9 мм рт. ст.) паров анилина. В качестве измерительных средств наиболее пригодными оказались ртутный манометр и манометр, заполненный анилином. Для определения давления насыщенных паров при значениях до 60 ат и при температурах до 500° С Глазер и Рюланд применили автоклав из стали V4A.
4.4.2. РАСЧЕТ И СПОСОБЫ ПРЕДСТАВЛЕНИЯ
ДАННЫХ ПО ДАВЛЕНИЮ НАСЫЩЕННЫХ ПАРОВ
Если нельзя непосредственно измерить давление паров какого-либо вещества или рассчитать его на основе уже имеющихся литературных данных, то это давление можно найти, исходя из атомных и структурных составляющих. Как чисто расчетные, так и графические методы определения являются всегда приближенными.
Петер [47 ] исследовал связь между давлением паров и моле-, кулярной структурой вещества, применив уравнение Ван-дер-Ваальса:
1g (1/П) = а (1/0 — 1) (16)
где П = р/ркр — приведенное давление; 0 = 'Г/Ткр — приведенная температура; Ркр и fKp — критические давление и температура; а — константа.
При расчете констант а по правилу аддитивности, исходя из соответствующих атомных и структурных составляющих, получаются значения, хорошо согласующиеся с эксперименталь-60
В'-
I ними данными. Примерно в 50% исследованных случаев разность I между расчетными и экспериментальными значениями «составила I менее 1 %. Эти расхождения никогда не превышали отклоне-г ний а, обусловленных влиянием температуры.
| Путем обработки результатов 73 опытов и сравнительных |г- вычислений были получены следующие значения атомных и ! структурных составляющих для определения а:
С —2,154 СООН 2,247
н 1,139 СО2 (сложный эфир) 0,507
сн2 0,124 Двойная связь 2,295
О 0,266 Тройная связь 4,605
ОН (спирт) 2,327 Разветвление в цепи С—С 0,056
с6н5 1,708
N —0,765
С1 1,356
к Исходя из структурной формулы соединения, критической | температуры и критического давления, по уравнению (16) можно к рассчитать давление насыщенных паров р для любой темпера-| туры Т. Аддитивность констант а сравнима с аддитивностью к мольной рефракции и парахора. Коллар и Наги [47а ] описывают ? способ, основанный на методе Киннея, позволяющий рассчиты-£ вать кривые давления паров по молекулярным структурам для интервала температуры кипения от 0 до 400 °C. При этом поляр-ность молекулы, конечно, не должна быть слишком большой.
| Аналогичный метод для оценки температур кипения органи-Р ческих жидкостей разработал Пирсон [48], исходя из размеров Г и формы молекул и учитывая их ассоциацию введением в расчет-। ную формулу поправочного коэффициента. Уравнение Эглоффа Г для расчета температуры кипения также дает возможность обой-| тись без экспериментальных данных [49]. Если для какого-либо I вещества известны температуры кипения при двух различных К- давлениях и имеется полная кривая давления паров какого-либо г’ эталонного вещества, то с помощью правила Дюринга [50] можно | рассчитать полную кривую давления паров для данного вещества. | Правило Дюринга выражается следующим уравнением:
к. Окип, 1—Анп>г)/(01 ®г) = ? (17)
В. где /Кип>1 и /к11п,2 — температуры кипения рассматриваемого вещества А при ₽ давлениях pt и р2; 0х и 02 — температуры кипения эталонного вещества Б при Давлениях рг и р2; <7 — константа.
| Из этого правила следует, что для двух веществ А и Б отноше-Е. ние разностей температур кипения при двух различных давлениях Е остается постоянным.
К Применительно к большому числу веществ Рехенберг [51 ] Е выбрал в качестве эталона воду, для которой имеются точные К данные по давлению паров.
61
Пример расчета температуры кипения уксусной кислоты; в качестве эталона выбрана вода.
1. Известные величины:
= 760 мм рт.ст. р2 = Ю мм рт. ст.
Уксусная кислота /Кип, 1 = 118,1 °C /кип, 2 = 63, 0 °C Вода 0j= 100,0 °C 62 = 51,5 °C
По этим исходным величинам в первую очередь вычисляем константу q, значение которой необходимо для дальнейших расчетов:
<7 = (118,1 — 63,0)/(100,0 —51,5) = 1,14
2. Расчет температуры кипения уксусной кислоты при 200 мм рт. ст.: pi = 760 мм рт. ст. р2 = 200 мм рт. ст.
Уксусная кислота /КИп,1= 118,1 °C /кип.г -? Вода 0Х - 100,0 °C 02 = 66,4 °C
/кип, 2 = /кип, 1 - q (91 - 02), /2 = 118,1 - 1,14 (100,0 - 66,4) = 80 °C
Полученное расчетное значение хорошо согласуется с измеренной величиной. По такой схеме могут быть рассчитаны и другие данные, однако необходимо иметь в виду, что для других эталонных веществ опытные значения температур кипения в области низких давлений часто бывают неточными. Разумеется, йри этом и разности температур, рассчитанные по правилу Дюринга, будут отклоняться от истинных значений. Следует стремиться к тому, чтобы температуры кипения эталонного вещества были установлены с погрешностью не более ±0,05 °C. В качестве эталона рекомендуется выбирать вещества, имеющие аналогичную структуру с исследуемым веществом, например соединения из одного гомологического ряда. Янцен и Эрдманн [52] при вычислении кривой давления паров для насыщенных жирных кислот с длинной прямой углеродной цепью использовали в качестве эталона миристиновую кислоту. Как показала практика, для полярных жидкостей в качестве эталона пригодна вода, а для неполярных или слабо полярных жидкостей — и-гексан. Еще один способ подбора эталонного вещества описан Киреевым [53].
Весьма часто зависимость между давлением насыщенных паров чистого вещества и температурой описывают уравнением Антуана [54]:
lg р=- a — b/(i + с) (18)
где а, Ь, и с — константы, зависящие от природы данного вещества; константа р является групповой константой и для алифатических углеводородов ее можно принять равной 230.
Использование этого уравнения возможно при условии, что константы а и b для соответствующего вещества известны из опыта. Публикуемые данные по давлению паров очень часто представляют в виде констант а я Ь. Примером являются таблицы Драйсбаха [19]. Вихтерле и Линек [58] опубликовали значения констант уравнения Антуана, которые можно использовать в памяти ЭВМ, управляющей производственными процессами. От-62
Дельные константы рассчитаны итерационным способом, причем Г указаны погрешности их определения.
Детальные исследования Томсона [551 показали, что по i уравнению Антуана получаются значения, наиболее близкие | к экспериментальным данным. Константы а и Ь уравнения Ан-I туана можно вычислить, если известны температуры кипения К вещества при двух давлениях р и рх.
I Константу Ь в уравнении Антуана можно вычислить по фор-I муле:
I ь = (1g р- + 230) (/„ +230)/(/ -/„) (19)
f Подставляя найденное значение b и с = 230 в уравнение Ь Антуана (18), находим константу а:
а = Ig р + b/(t + 230) (20)
к Значения для р№ и можно найти в табл. 7 для соединений | каждого гомологического ряда на диаграмме Кокса.
| Если известны точка кипения при атмосферном давлении и | критическая температура вещества, то с помощью универсальной К, формулы Риделя [56 ] для расчета давления паров можно получить Ь всю кривую давлений. Если давления измерены для двух любых К температур, то можно вычислить критическую температуру. Г Формула Риделя отличается особой точностью и позволяет уста-новить, являются ли вещества ассоциированными или нет. Раз-
В работанный Рекхардом [57] метод особенно пригоден для опре-Г деления температур кипения при вакуумной перегонке таких Ц многокомпонентных смесей, как смолы, дегтярные масла. Расчет jg облегчается применением двух номограмм.
К Обзор методов расчета давления паров дан в монографии И [59]. Отмер и Пу [60] приводят обзор методов расчета, основан-И ных на использовании свойств эталонных веществ. Универсаль-Е' ное уравнение для расчета давления насыщенных паров угле-водородов предлагают Зиа и Тодос [60а]. Среднее отклонение
L вычисленных по этому уравнению значений давления паров № от экспериментальных составляет 0,38%.
Ц Графические методы определения давления паров по сравнению К с расчетными методами обычно проще и требуют меньшей затраты р времени. По правилу Дюринга кривую давления паров получают К' следующим образом. Температуры кипения данного вещества А В* и эталона Б, соответствующие одному и тому же давлению, пред-К ставляют в прямоугольной системе координат в виде точки, К абсцисса которой равна температуре кипения вещества Б, а Ор-H. Дината — температуре кипения А. Точки, нанесенные для раз-Н личных давлений, лежат все без исключения на одной и той же HL прямой. На рис. 38 показана диаграмма Дюринга, характери-В' зующая давление паров уксусной кислоты; она построена с ис-ИЕ.пользованием воды в качестве эталонного вещества. Давление И Насыщенных паров уксусной кислоты для какой-либо определенна 63
tкип воды(5)t°C
Рис. 38.
Прямая, характеризующая давление паров уксусной кислоты, построенная по правилу Дюринга.
Эталонное вещество — вода.
Рис. 39.
Кривые давления паров воды (/), бензола (2) и суммарная кривая для Паров воды и бензола (3).
ной температуры находят следующим образом: эту температуру откладывают на оси ординат диаграммы Дюринга, затем отсчитывают на оси абсцисс соответствующую температуру воды и по кривой давления водяных паров (рис. 39) определяют соответствующее этой температуре давление. На диаграмме, представленной на рис. 38, можно, конечно, предусмотреть шкалу для давлений водяного пара, но в этом случае трудно отсчитывать промежуточные значения давления (ввиду неравномерности шкалы).
[5S На рис. 39 показаны обычные кривые давления паров; совершенно ясно, что для получения надежных кривых требуется сравнительно много данных. Поэтому стали искать способы выражения этих зависимостей в виде прямых линий. В прямоугольной системе координат зависимость логарифма давления паров от обратной абсолютной температуры кипения 1/Т носит линейный характер в соответствии с уравнением Клаузиуса—Клапейрона:
lgp = _Z^L ’ +с (21)
К 1
где: ЛНу — энтальпия испарения; R — универсальная газовая постоянная; с — константа интегрирования [61].
64
На рис. 40 указанная зависимость построена для нескольких Веществ с применением специальной бумаги *; по оси ординат которой отложены значения 1g р, а по оси абсцисс — 1/71; причем наноси абсцисс также отложены значения температуры по шкале Цельсия в интервале от —23 до -ф 127 °C [62]. Очевидно, по наклону этих прямых можно определить энтальпию испарения. В соответствии с другой формой уравнения Клаузиуса—Клапейрона (21)
lg₽2 = lgPi(A#V. 1/д#У.г) + с (22)
можно построить графическую зависимость логарифма давления паров вещества 2 от логарифма давления паров вещества 1; при этом также получается прямая линия. Тангенс угла наклона этой прямой равен отношению энтальпий испарения веществ. Чтобы угол наклона прямой был по возможности ближе к 45 °, в качестве эталонного вещества для углеводородов следует принять «-гексан, а для ассоциированных веществ, например кислот и спиртов — воду, поскольку кислоты и спирты подобно воде, обладают большой энтальпией испарения. На основе урав-
* Бумага фирмы «Шефере Файнпапир», Плауэн, ФРГ; заказ № 602 для интервала температур от —24 до 726 °C; заказ № 603 для интервала температур от —23 до 127 °C. — Прим. ред.
: Зависимость давления паров некоторых веществ от температуры в системе ( координат igр — 1/7:
к 1 — пропионовая кислота; 2 — вода; 3 — этанол; 4 — «-гексан; G — диэтнловый эфир.
Р«с. 41.
Диаграмма Кокса для гомологического ряда алкилбензола.
3 Э. Крель
65
Нения (22) Отмер с сотр. [63] разработал номограмму и таблицы, с помощью которых можно определять давление паров, энтальпию испарения и другие физические величины.
Если для какого-либо вещества известны температура кипения и энтальпия испарения, то можно пользоваться приведенной у Виттенбергера [64] диаграммой Бергхольма и Фишера [65], в которой кроме специальной сетки в координатах 1/Т — 1g р имеется также верхняя шкала в килокалориях. Для расчета давления насыщенных паров Хоффман и Флорин [66] приводят метод, состоящий в том, что логарифм давления паров откладывают на так называемой «оси веществ», ведущей к полюсным лучам. Этот метод аналогичен способу с применением известной диаграммы Кокса [67]. На этих диаграммах, построенных для соединений отдельных гомологических рядов («семейств» на диаграмме Кокса) все прямые, характеризующие давление паров, соединяются в точке (Полюсе) с координатами tx, которые для веществ каждого гомологического ряда имеют определенные значения. Таким образом, достаточно знать одну температуру кипения при каком-либо давлении, чтобы путем соединения соответствующей точки на диаграмме Кокса с полюсом можно было получить прямую, выражающую зависимость давления паров от температуры. В табл. 7 приведены систематизированные Драйсбахом координаты полюсов /«, и рж для 21 гомологического ряда на диаграмме Кокса. На рис. 41 показана диаграмма Кокса для алкилбензолов.
Кокс строил график зависимости 1g р от 1// и в качестве направляющей линии проводил прямую, образующую угол с ординатой. В соответствии со значениями р, отложенными на оси ординат, на ось абсцисс были нанесены экспериментальные значения температур кипения для воды. Дэвис [68], а также Калингарт и Дэвис [69] усовершенствовали диаграмму Кокса, предложив откладывать на оси ординат 1g р, а на оси абсцисс 1/(/ + 230). В таком виде диаграмму Кокса можно рассматривать как графическую иллюстрацию уравнения Антуана (18) при групповой константе с = 230. Однако эта константа применима лишь к алифатическим углеводородам [70]. Для удобства пользования диаграммой Кокса очень важно, чтобы она была построена в оптимальном масштабе. Чтобы можно было откладывать значения температур до 200 °C с точностью до 1 °C, следует применять диаграмму Кокса размерами примерно 1,5 м х 0,95 м. Драйс-бах [19] приводит точные инструкции для построения диаграммы Кокса, позволяющей определять значения давления и температуры с достаточной для лабораторной практики точностью.
На диаграмме Кокса по оси ординат отложена логарифмическая шкала давлений от 0,1 до 107 мм рт. ст., разделенная на восемь интервалов, суммарная длина которых составляет 1,422 м. Участок оси абсцисс, на котором откладывают значения температуры в интервале от —100 до 10 000 °C, имеет длину 0,984 м.
66
Таблица 7.
Гомологические ряды и соответствующие им координаты полюсов t и рк. иа диаграмме Кокса
X §
Первый член и характеристика гомологического ряда Гомологический ряд <0». °C Ро»-ю», мм рт. ст. Множитель (/„+230) Xlg(p„/7
Бензол, насыщенные боковые цепи Галогенпроизводный бензола, насыщенные боковые цепи 1670 2,320 6 621
Бензол, ненасыщенные боковые цепи Галогенпроизводный бензола, ненасыщенные боко-,, вые цепи СпН2П_8 СцН2П_(8+т)Х щ. 1063 0,916 3 985
Нафталин Галогенпроизводный нафталина СпН2п-12 QiH2n-(i2+m)Xm 921,5 0,530 3 274
Ароматический амин Галогенпроизводный ароматического амина CnH2n-5 CnH2rt_(5+mjN Хщ 1750 3,800 7 324
Фенол Галогенпроизводный алкилфенола СпН2п-бО СпН2П-(6+ГП)ОХт 2337 11,170 10 697
Нитробензол Галогенпроизводный нитробензола CnH2rt_7NO2 CrtH2rt_(7+m)NO2X/n 2022 3,701 8 304
Ароматический альдегид Галогенпроизводный ароматического альдегида CnH2rt_8O CnH2n-(8+m)^X/n 600 0,150 1 905
Фенилэтиловый спирт Галогенпроизводный фенилэтилового спирта ОгНгп-вО СцН2п-(в+т)^Х^2 3300 20,000 15 550
Ароматический кетон Галогенпроизводный ароматического кетона QiH2n-ieO СпН2п-(1б+т)ОХт 2186 5,186 9 263
Ароматическо-алифатический кетон Галогенпроизводный ароматическо-алифатического кетона с„н2П_80 СпН2п(8-т)ОХт 2186 5,186 9 263
Ароматический эфир Смешанный ароматическо-алифатический эфир Циклогексиловый эфир, насыщенные боковые цепи СпН2П-14О СпН2п-вО СпН2дО 1850 3,400 7 597
3*
67
Продолжение табл. 7
Первый член и характеристика гомологического ряда Гомологический ряд + °C 0СО-Ю6. мм рт. ст. Множитель Ц„+230)х Xlg(p„/760)
Циклогексиловый эфир, ненасыщенные боковые цепи СпН2п_2О 1850 3,400 7 597
Циклопентан c„H2rt 1450 1,550 5 560
Циклогексан CnH2n 2076 2,076 8 104.
Алифатический углеводород (насыщенный) СпН2п+2 1240 1,300 4 753
Галогенпроизводный алифатический углеводород (насыщенный) 1450 1,860 5 693
Ненасыщенный алифатический углеводород 1725 1,6980 6 548
Первичный алкиламин (тем. кип. до 190 °C при 760 мм рт. ст.) ^•пН2п+з^ 1125 1,441 4 424
Первичный алкиламин (тем. кип. 190—350 °C при 760 мм рт. ст.) QiH2n+3N 1270 1,709 5 029
Алифатический эфир QiH2n+2O 1579 2,525 6 370
Алифатический кетон Сп^2ПО 3293 6,929 13 950
Нитропарафин QiH2n+1NO2 1350 2,000 5 404
Алифатический нитрил CnH2n+iN 2025 3,461 8 250
Алифатический сложный эфир СПН2ПО2 1639 3,800 6 913
Алифатический спирт QiH2n_20 12150 128,200 64 718
Алифатическая кислота (темп. кип. до 141 °C) Алифатическая кислота (темп. кип. выше 145 °C) Галогенпроизводная алифатическая кислота cnH2rto2 QiH2nO2 ^п^2П-т^2^-т 364 1458 0,060 9,060 1 125 6 881
Расположение делений на температурной шкале можно рассчитать по следующим формулам:
t =230(0/(37,17 — /) (23)
или
/ = 37,17^/(230 + /) (24)
где: t — температура, °C; I — длина в дюймах (1 дюйм = 24,5 мм).
Нулевая точка в средней части шкалы удалена от левого края диаграммы, соответствующего —100 °C на расстояние 0,438 м; длина I обозначает расстояние на шкале, соответствующее положительному или отрицательному отклонению откладываемого значения температуры от нуля. Рекомендуется применять температурные интервалы, приведенные в табл. 8.
Полученные для интервалов I деления подразделяют на еще более мелкие.
68
Таблица 8 А
Температурная шкала для диаграммы Кокса
Температурный интервал I, °C Цена деления, °C
—100—200 10
200—600 50
600—2000 100
2000—4000 500
4000—10000 1000
Таблица 8Б
Температурный интервал 11, °C Цена деления
первая разбивка вторая разбивка
—100—0 10 10X1
0—200 10 5X2
200—600 50 5X10
600—1000 100 2X50
Более 1000 100 Более мелкие деления не повышают точность отсчета.
В заключение рассмотрим еще один метод, специально предназначенный для расчета давления насыщенных паров веществ гомологического ряда. Согласно этому методу по оси ординат откладывают значения температуры кипения в линейном масштабе, а по оси абсцисс — число углеродных атомов в молекулах компонентов в логарифмическом масштабе; затем на диаграмме строят изобары для различных давлений, как это показано на рис. 42 применительно к парафинам С10—С20 с прямой углеродной цепью для остаточных давлений 5, 10 и 30 мм рт. ст. Таким способом можно успешно интерполировать неизмеренные значения и экстраполировать данные для соединений с числом атомов углерода в молекуле менее 10 и более 20. Кроме того, преимущество этого метода состоит в том, что в ходе
сю ci2 ci6 Cte сго Рас. 42.
Изобары предельных углеводородов С10—С20,
69
ректификации при определенном давлении в любое время можно ориентировочно оценить, из какого числа атомов углерода состоит молекула вещества в дистилляте.
Имеются еще более точные методы для расчета давления паров. В работах Хоффмана и Флорина [66], Фальдикса и Штаге [70], а также Штаге и Джуилфса [71 ] и [59] содержатся подробные обзоры этих методов. Очень важно для работы выбирать метод, отвечающий поставленным целям наилучшим образом, но всегда следует иметь в виду, что все эти методы являются приближенными и лишь путем тщательно проведенных экспериментальных измерений получают максимально точные данные.
4.5. УРАВНЕНИЕ СОСТОЯНИЯ
И ФАЗОВАЯ ДИАГРАММА р—х
Жидкость состоит из молекул, движущихся с различными скоростями, из которых самые быстрые наиболее легко пересекают и покидают поверхность жидкости, переходя в виде паров в пространство, находящееся над жидкостью (испарение). Для перевода частиц жидкости в парообразное состояние необходимо затратить определенное количество тепла, которое при этом отнимается от жидкости, в результате чего она охлаждается. При внешнем подводе тепла достигается более интенсивное испарение, и жидкость закипает.
Таким образом, пар является газом вблизи температуры кипения или конденсации. При перегонке образуются насыщенные пары, которые находятся в контакте с жидкой фазой, причем в состоянии равновесия число молекул, переходящих из жидкой фазы в газообразную, равно числу молекул, возвращающихся из газообразной фазы в жидкую. Ненасыщенный пар образуется лишь в том случае, если первоначально насыщенный пар не имеет больше контакта с жидкостью и нагревается выше температуры кипения или расширяется (при переходе в область более низкого давления). В этих условиях пар не имеет возможности принимать из жидкости дополнительное число молекул в соответствии со своей более высокой температурой.
Общее уравнение состояния (26) для газов вблизи точки сжижения при высоких давлениях и при ассоциации молекул можно рассматривать лишь как приближенное и предельное соотношение, т. е. оно справедливо только для идеальных газов, а не для насыщенных паров, с которыми обычно имеют дело при перегонке.
Вышеуказанное обстоятельство учтено в уравнении Ван-дер-Ваальса:
(р+«Мол)(^МОл-0 = ^ (25)
где а и b — константы, константа b учитывает собственный объем молекул газа, я/Умол —силы притяжения, действующие между молекулами; р — давление газа на ограничивающую его поверхность; Рмол — мольный объем газа.
70
Однако и уравнение состояния Ван-дер-Ваальса ни для какого вещества не является точным, так как межмолекулярные силы не подчиняются такому простому закону, который Ван-дер-Ваальс положил в основу своего уравнения.
Планк в качестве уравнения состояния предложил уравнение пятого порядка, которое лучше всего описывает реальное поведение газов и паров. Однако проводить расчеты по уравнениям третьего и пятого порядка очень неудобно, и поэтому в практике перегонки применяют общее уравнение состояния, в основу которого положены газовые законы Гей-Люссака и Бойля-Мариотта
pV = p0V07/273,15
pV = RT (26)
При этом делают допущение, что поведение паров подчиняется общему уравнению газового состояния с достаточной точностью. Газ тем больше приближается к идеальному состоянию, чем меньше его плотность (чем меньше давление) л чем выше температура.
Если в выражение р0 УО/273,15 = R подставить параметры для нормальных условий, а именно р0 = 1 атм (что соответствует 760 мм рт. ст. или 1,033 кгс/см2 или 1,013 бар) и Vo = 22,414 л (объем 1 моль при 0 °C), то получим мольную газовую постоянную R = 1,0-22,414/273,15 = 0,082 л-атм/К.
Для любого числа молей N газа или пара справедливо следующее уравнение состояния:
pV = N-0,082T (27)
При лабораторной перегонке имеют дело обычно с малыми количествами вещества, поэтому расчеты проводят в молях, а не в килограмм-молях, как это принято для промышленных процессов.
Если нужно рассчитать объем паров испаренного вещества при определенном давлении, то пользуются формулой
V = /V-0,0827 -760/р (28)
где V измеряется в л; р — в мм рт. ст.; N — число молей.
Для 1 моль при атмосферном давлении 760 мм рт. ст. (или 1 атм) формула (28) упрощается:
V = 7-0,082 (29)
Плотность паров рп при нормальных условиях рассчитывают, исходя из массы 1 моль, по формуле
рп = .47/22,4 (г/л) (30)
При пересчете на другое давление и другую температуру ( получаем ' _ М р 273,15
/ Рп~ 22,4 ‘ 760 ' 273,15 -|- / (3
: где р измеряется в мм рт. ст.; t — в °C.
71
Если числовые константы объединить в один множитель, то из (31) получим
рп = 0,01605/Ир/Т (32)
Масса газа вычисляется по простой формуле
G = Ирп (33)
Как было показано выше, именно межмолекулярные силы влияют на взаимную растворимость компонентов. Этим обусловлены и другие свойства бинарных смесей, представленные по Торманну [72] на рис. 43.
Если компоненты практически взаимно нерастворимы, то в смеси оба компонента и образующиеся пары ведут себя так, как будто жидкие фазы компонентов не соприкасаются между собой (см. рис. 43, тип 1). Эти условия имеют место при перегонке с паром—носителем (см. разд. 6.1).
По закону Дальтона общее давление газовой смеси складывается из давлений, которые имели бы отдельные компоненты при той же температуре и в том же объеме:
Р — Р1 + Ра (34)
Отсюда содержание легколетучего в парах смеси взаимнонерастворимых компонентов при той же температуре можно вычислить по формуле:
Pi = (Pi/P) Ю0% (мол.) (35)
Чтобы определить парциальные давления (см. рис. 43, ряд I), используемые при расчете состава паров для бинарных смесей нерастворимых компонентов, строят обе кривые давления паров и получают с их помощью суммарную кривую обоих парциальных давлений подобно тому, как это показано для смеси вода—бензол на рис. 39. При 760 мм рт. ст. температура кипения этой смеси (в том числе и температура дистиллята) составляет 69,0 °C. Содержание легколетучего компонента в парах вычисляют по формуле (35):
у* = (534/760) 100 = 70,2% (мол.) (36)
По номограмме на рис. 22 проводят пересчет и получают 91,0% (масс.).
Если рассмотреть горизонтальные ряды I и II на рис. 43, для которых на диаграммах по оси абсцисс указана концентрация в мольных процентах в расчете на легколетучий компонент (см. также рис. 29), то можно выявить следующие соотношения.
Ряд I. Парциальные давления при постоянной температуре для легколетучего компонента А и труднолетучего компонента В (диаграмма р — х).
При постоянной температуре давление паров смеси зависит только от ее состава. Если силы притяжения между одинаковыми молекулами больше, чем между неодинаковыми, то наблюдается весьма слабая тенденция к взаимному смешению компонентов. Процесс растворения протекает с поглощением тепла, что приводит к уменьшению энтальпии испарения. Но при этом увеличивается летучесть смеси, и давление паров будет выше, чем это соответствует идеальной смеси (типы 1—3). Отклонения от идеального поведения можно выразить через 72
Рис. 43.
Зависимость свойств бинарных смесей от содержания легколетучего компонента в жидкой фазе по данным Торманна [72]:
ряд I — парциальные давления легколетучего А и труднолетучего В компонентов при постоянной температуре; ряд II — общее давление прн постоянной температуре; ряд III — содержание легколетучего в парах при постоянном давлении (кривая равновесия); ряд IV — температура при постоянном давлении (кривая кипення);ряд V — температура прн постоянном давлении (пунктирная кривая—линия конденсации).
Тип I — отсутствие или незначительная взаимная растворимость компонентов; тип 2 — частичная растворимость; тнп 3 — полная растворимость с наличием азеотропной точки (максимальное давление паров и минимальная температура кипения); тип 4 — полная растворимость с приближением к идеальному раствору; тнп 5 — полная растворимость с наличием азеотропной точки (минимальное давление паров и максимальная температура кипения).
в широком интервале концентраций заметно ниже температур кипения обоих чистых компонентов (минимальная температура кипения). Для смесей типа 5, наоборот, в некотором интервале концентраций температура кипения лежит выше температур кипения обоих чистых компонентов (максимальная температура кипения). Точка соприкосновения изобарных кривых кипения и конденсации соответствует азеотропной точке смеси.
Кривая равновесия у—х показывает связь между концентрацией жидкости х и соответствующей концентрацией пара у, находящегося в состоянии равновесия с жидкостью. Следовательно, кривая равновесия является основой для расчета числа теоретических ступеней разделения по графическому методу Мак-Кэба и Тиле [77], который успешно и широко применяется благодаря своей простоте. На рис. 43 в ряду III представлены кривые равновесия для смесей различных типов. Для смесей взаимно нерастворимых компонентов кривая равновесия представляет собой прямую линию (тип 1), которая пересекает диагональ в одной точке, называемой азеотропной. В этой точке составы пара и жидкости одинаковы; обогащение паров легколетучим компонентом при более высокой концентрации жидкости х уже невозможно; напротив, в этой области концентраций пар содержит меньше легколетучего компонента, чем жидкость. При перегонке смесей взаимно нерастворимых компонентов (тип 1) или смесей только частично растворимых компонентов (тип 2) дистиллят имеет один и тот же состав в широком интервале изменения концентрации легколетучего компонента в кипящей жидкости и только в непосредственной близости от концентраций 0 и 100% появляются промежуточные составы дистиллята. Для смесей с максимумом на кривой давления паров при концентрации жидкости выше азеотропной (тип 3), а для смесей с минимумом на кривой давления паров при концентрации жидкости меньше азеотропной (тип 5) пары содержат меньше легколетучего компонента, чем исходная жидкость состава х. Для смесей типа 4 характерна форма кривой равновесия у*—х, свойственная идеальным смесям, для которых у* всегда больше х.
4.6.1. РАСЧЕТ КРИВОЙ РАВНОВЕСИЯ
Построение кривой равновесия расчетным путем возможно для смесей почти нерастворимых компонентов (т. е. в высшей степени неидеальных смесей) и идеальных смесей, а также для смесей частично растворимых компонентов. Для смесей же с максимумом или с минимумом на кривой давления паров экспериментальное определение является более предпочтительным, поскольку в данном случае для расчета кривой равновесия требуются другие, зачастую малодоступные данные по физическим свойствам, причем объем вычислений возрастает.
В последние годы опубликовано очень много статей, посвященных расчету кривых фазового равновесия. Подробный разбор 76
всех этих работ занял бы слишком много места; к тому же эта тема исчерпывающе рассмотрена Шубертом [17], а также Хала с сотр. [78]. В монографиях этих авторов приведен и проанализирован обширный материал, опубликованный ранее.
Намечается тенденция рассчитывать параметры равновесия в многокомпонентных смесях, исходя из экспериментальных данных для бинарных смесей и применяя цифровые вычислительные машины. Это вызвано тем, что данных по равновесию для бинарных смесей накоплено уже достаточно много [105, 106], в то время как исследование многокомпонентных смесей весьма трудоемко. На международном симпозиуме «Перегонка—1969» в Брайтоне [61 семь докладов были посвящены расчету кривых фазового равновесия на основе физико-химических свойств чистых компонентов. На симпозиуме обсуждались данные по равновесию для электролитов и неэлектролитов, вопросы термодинамической проверки данных по равновесию для неэлектролитов и методы расчета коэффициентов активности. Во многих случаях оправдано применение уравнения Вильсона [80]. Как сообщают Донт и Вайхманн [81 ], для расчета данных по равновесию бинарных растворов на основе зависимостей р—х (или t—х) можно использовать методы последовательных приближений. Круг, Хабер-ланд и Биттрих [82 ] дали оценку основным положениям по теории растворов, выдвинутым Маргулесом, Редлихом, Кистером, Ван-Нессом, Ван-Лааром, Скэтчардом, Хеймером, Вильсоном, Хейлем и Реноном, Праусницем (NRTL—Non—Random—Two— Liquid). Шуберт [17 ] полагает, что теоретический подход Ренона— Праусница [83] весьма перспективен. Он лишен недостатков метода Вильсона, основанного на использовании трехпараметрического уравнения. Применение метода Вильсона ограничивается нерасслаивающимися жидкими системами. Соотношение Ренона—Праусница связывает физическую модель Вильсона с теорией Скотта.
Для расчета кривой равновесия идеальных смесей, например смесей химически родственных веществ, особенно относящихся к одному гомологическому ряду, можно использовать различные методы. Чтобы дать представление о ходе расчета в целом, рассмотрим некоторые из них.
Для расчета равновесного состава паров на основе данных по давлению паров чистых компонентов следует применять закон Рауля. Этот закон устанавливает, что парциальное давление компонента равно произведению мольной доли этого компонента и давления паров компонента при температуре, равной температуре жидкости:
(40) (41) (42) (43)
77
Pi = Pixi (Х1 в мол. долях)
Pi = Pixi/100 (xj в мол. %)
Pi РзО ~ *1) р2=-р2(100 —Х1)/100
Таблица 9.
Давление паров бензола и толуола при различных температурах
i, °C Давление паров, мм рт. ст.
pj (бензол) р2 (толуол)
80 748 294 2,54
90 1013 408 2,46
100 1335 555 2,41
ПО 1739 747 2,33
Общее давление р складывается из парциальных давлений рг и р2: Р = Pi + Рч = Р?Х1 + pg (1 — х,) (Xj в мол. долях) (44)
Р= Pi+Р2 = IP?Xi + Р§(ЮО —х,)]/100 (Xj в мол. %) (45)
Так как равновесный состав паров равен отношению парциального давления компонента 1 к общему давлению, то находим Pi - P?xi____________P?*i
У1 Р1 + Р2 Р1Хг4-р^(1 — х2) р 1 >
Поясним сказанное на примере. Допустим, нужно, исходя из данных по давлению паров чистых компонентов, расчетным путем построить кривую равновесия у*—х при 760 мм рт. ст. для смеси бензол—толуол, подчиняющейся закону Рауля. Вначале в возможно более крупном масштабе строим график зависимости давления паров чистых компонентов от температуры,, затем по графику определяем давления паров обоих компонентов для различных температур (табл. 9).
Преобразуя формулу (44), получим выражение для концентрации жидкости
Х1 = -£Р^-100%(МОЛ-) (47)
Подставляя в (47) значения давления паров при 90 °C, получим
Х1 = шТз — 408 '100 = 58118% (мол-) (48)
Подставив в (46), найдем соответствующую равновесную концентрацию у*
1013-58,18/760 = 77,55% (мол.) (49)
Аналогичным образом можно вычислить и остальные точки кривой равновесия у*—х. Чем больше имеется данных, тем точнее будет построена кривая равновесия. Разумеется, такой расчет возможен не только для атмосферного давления, но и для любого другого давления, например 20 мм рт. ст. В этом случае р = 20, а температуры варьируют в интервале между температурой кипения компонента 1 при 20 мм рт. ст. и температурой кипения 78
компонента 2 при 20 мм рт. ст. Результаты таких расчетов особенно удобно представлять в виде таблицы (табл. 10).
Кривую равновесия можно найти также и графическим способом. Этот способ гораздо сложнее и отнимает больше времени, чем чисто расчетный метод, поэтому ограничимся только упоминанием о нем. Более подробное его описание приводят Торманн [72] и Розенгарт [84].
Для идеальных бинарных смесей кривую равновесия можно также рассчитать по уравнению, полученному на основе объединения законов Рауля и Дальтона:
У* = Pi xPi = х
1—У* Рг (1—*)Р2 1— X
(50)
где х — мольная доля.
Уравнение (50) можно представить в более удобной для использования форме:
у* = ах/(ах— хЦ-1) (в мол. долях) (51)
или
У* = ПхУа” ~Г(В % М0Л,) (52)
1/л -ф- ОС — 1
В эти уравнения входит величина а — рЧ/которую называют относительной летучестью компонентов смеси (см. разд. 4.6.2). Для идеальных смесей можно принять величину а постоянной в интервале между температурами кипения чистых компонентов (см. также табл. 10). Для смеси бензол—толуол в табл. 9 приведены значения а, рассчитанные по заданным р? и pl они лежат между 2,54 и 2,33, среднее арифметическое значение аср = 2,43.
Таблица 10.
Пример расчета кривой равновесия для жирных кислот С10—Сп нормального строения при общем давлении р = 20 мм рт. ст.
t, °C 0 Pl-мм рт. ст. 0 мм рт. ст. Xt по (47), % (мол.) Ух по (46), % (мол.) Относительная летучесть 0/ 0 « = pjp2
161,5 20 11,0 100 100 1,819
163 21 11,9 83,9 93,4 1,765
164 22 12,5 78,9 86,8 1,760
165 23 13,2 69,4 79,8 1,742
166 24,1 13,9 59,7 71,9 1,733
167 25,2 14,5 51,3 64,7 1,736
168 26,4 15,2 42,8 56,5 1,735
169 27,5 16,0 34,7 47,15 1,717
170 28,7 16,9 26,3 37,7 1,696
171 30,0 17,4 20,6 30,9 1,723
172 31,4 18 14,95 23,45 1,743
173 32,6 18,8 8,70 14,3 1,733
174,5 35,0 20 0 0 1,750
79
Таблица 11.
Пример определения равновесной концентрации у* для системы бензол—толуол при а = 2,43
X. % (мол.) Аналитический способ Графический способ Опытные данные Киршбаума [103]
у* вычисляют по (51) или (52), % (мол.) Д % (мол.) (по сравнению с опытными данными) у* находят по кривой. равновесия для идеальной смеси при а = 2,4 Д % (мол.) (по сравнению с экспериментальными данными)
10 21,2 —0,4 20,0 +0,8 20,8
20 37,8 —0,6 37,2 ±0,0 37,2
30 51,1 —0,4 51,0 —0,3 50,7
40 61,8 +0,1 61,0 +0,9 61,9
50 70,8 +0,5 70,8 +0,5 71,3
60 78,7 +0,4 78,4 +0,7 79,1
70 85,0 +0,7 84,8 +0,9 85,7
80 90,7 +0,5 91,0 +0,2 91,2
90 95,6 +0,3 95,6 +0,3 95,9
По двум значениям а, найденным при температурах кипения чистых компонентов, можно вычислить среднее геометрическое значение, которое практически совпадает со средним арифметическим
«ср = К (53)
или
1g «ср = (1g «1 + 1g аз)/2 (54)
Это значение а подставляют в уравнение (51) или (52) и для заданных концентраций х вычисляют состав равновесных паров у*. В табл. 11 приведены рассчитанные таким способом равновесные значения у* для системы бензол—толуол.
Для расчета равновесия в идеальных бинарных смесях можно также использовать номограмму Орличека и Пёлля [85] (см. рис. 21). Левая шкала соответствует концентрации жидкой фазы х (в мол. долях), а правая шкала показывает равновесную концентрацию паровой фазы у. Например, для смеси бензол—толуол при х = 50% (мол.) и а = 2,4 на правой шкале получим (штрих-пунктирные линии) равновесное значение у* = 70,8% (мол.); это значение совпадает с расчетным.
Номограмма на рис. 21 построена в мольных долях. При пользовании номб-граммой нужно иметь в виду, что не обведенные кружком значения на шкалах X и Z только тогда принимаются в расчет, когда величины а и Q превышают 1. Если же величины а или Q (средняя шкала) меньше 1, то на шкалах X и Z во внимание принимают только значения, обведенные кружком.
Данные по равновесию можно также очень быстро рассчитать с помощью круговой диаграммы Фурвийка [86], основанной на формуле (49).
80
Мы ограничимся изложением способов расчета бинарных смесей, поскольку многокомпонентную смесь при вычислениях можно рассматривать как совокупность бинарных смесей. Методы расчета тройных смесей частично или полностью взаимно растворимых компонентов подробно проанализированы Торманном [72] и Мацом [73]. Холло, а также Холло и Ленгьел [87] обсуждают вопросы равновесия между паром и жидкостью в трех-и четырехкомпонентных системах. Эти авторы исследовали три неидеальные четырехкомпонентные системы с применением новой аппаратуры. При этом они использовали новый метод расчета, позволивший уменьшить объем экспериментальных исследований. Равновесные данные для четырехкомпонентных систем можно представить с помощью двойных ортогональных проекций.
Специально для условий нефтяной промышленности Ум-штеттер и Флашка [88] разработали метод построения диаграмм равновесия по кривым давления паров для компонентов, которые в свою очередь могут представлять собой смеси различных веществ. Точность расчетов заметно повышается благодаря учету энтальпий испарения и растворения. Метод расчета пригоден также и для азеотропных смесей.
Для расчета паро-жидкостного равновесия в многокомпонентных смесях Вильямс и Хенли [89] разработали программу на алгоритмическом языке Фортран. Для учета реального поведения смесей предназначен ряд подпрограмм, которые позволяют вычислять фугитивности по вириальному уравнению, по соотношению Редлиха и Квонга или по способу Чао и Зидера. Коэффициенты активности можно учесть, используя рекомендации Вильсона, Ван-Лаара или Гильдебранда.
4.6.2. ЛЕТУЧЕСТЬ, ОТНОСИТЕЛЬНАЯ ЛЕТУЧЕСТЬ И КОЭФФИЦИЕНТ АКТИВНОСТИ
Летучесть F выражается в виде отношения парциального давления паров вещества к его мольной доле в жидкой смеси при определенной температуре, например для бинарной системы:
Fi = Pi/xi, F2 = р2/х2 (55)
Относительная летучесть компонентов бинарной смеси равна: «=-&- = -^т1- = (Fi > F2) (56)
F2 р2/х2 р2Х! 1 ’
Для идеальных смесей летучесть не зависит от соотношения компонентов и равна давлению паров чистого вещества. По закону Рауля для легколетучего компонента 1 идеальной смеси справедливо соотношение
Pi = P?*i (57)
а для малолетучего компонента 2
Р2 = Р^г или Pi == Ра 0 — xi) (58)
81
ОС
Рис. 45.
Зависимость относительной летучести компонентов смеси от разности температур кипения А*кип Для различных температур кипения £кИП смеси при атмосферном давлении.
Таким образом, парциальное давление компонента равно произведению давления паров чистого компонента и содержания этого же компонента в жидкости при той же температуре, выраженного в мольных долях.
По формуле (35) равновесная концентрация пара равна
У* = Pi/Р и yl = рг/р
Подставив уравнения (57) и (58) в эти два уравнения, получим:
У* = PiXjp и у* = р«х2/р (59)
При делении уравнений (59) друг на друга будем иметь
У*!у1 = Р‘.Х/(/’%'2) или У^/У-Р^ = Pl/P°2 = а (60)
Выражение (60) идентично (56), поэтому относительную летучесть а для идеальной смеси значительно проще выражать через соотношение давлений паров чистых компонентов, причем о о
Р1 > Pl.
Относительную летучесть можно также определить по точкам кипения компонентов. Применив правило Трутона при К. = = hHvIT = 20,5 кал/(моль-°С) и уравнение Клапейрона, Роуз [91] вывел соотношение между температурами кипения и относительной летучестью двух жидкостей, которое можно применять для ориентировочных расчетов:
lga = 8,2(T2-T1)/(7’2+Л) (61)
Мельпольдер и Хедингтон [90] вывели следующие более точные уравнения, которые дают погрешность от 0 до 6,2%, причем в подавляющем большинстве случаев погрешность не превышает 1%.
82
Для давления 760 мм рт. ст.:
lg а = (Z2 — /j) (3,99 + 0,0011939Т)/Т (62)
Для давлений выше и ниже 760 мм рт. ст. (от 10 до 1500 мм рт. ст.):
lg а = [(/2 - /j) (7,30 - 1,15 lg р + 77(179 1g р)]/Т (63)
где /2 — 6 — разность температур кипения компонентов, °C; Т — температура кипения смеси, К; р — рабочее давление перегонки, мм. рт. ст.
Уравнение (62) графически изображено на рис. 45 для интервала температур кипения смесей от —100 до -|-300 °C.
Роуз и Билес [901 применили ректификационную колонну для экспериментальной проверки данных по относительной летучести при конечных флегмовых числах в интервале v = 9,2—15,8. Для смеси к-гептан—метилциклогексан получено а == 1,074, что хорошо согласуется с опытными данными по фазовому равновесию. Значение а определяли с использованием опытных данных по формуле:
а = (у + xe/Xb)/[v + (1 - х£)/( 1 - х£)] (64)
Этот метод обеспечивает расчет значений а идеальных и неидеальных смесей при различных давлениях на основе экспериментальных данных, полученных при ректификации на насадочных колоннах. Метод применим также и к многокомпонентным смесям.
Разность температур кипения двух компонентов однозначно характеризует трудность разделения только идеальных смесей. Если смеси не идеальны, то решению задачи разделения может способствовать анализ зависимости относительной летучести а от давления.
Если а = 1 (1g а = 0), то кривые давления паров пересекаются, как например для смеси м-крезол — и-крезол, и в условиях, соответствующих точке пересечения, разделение двух компонентов ректификацией невозможно. При этом разделение может быть достигнуто только селективными методами перегонки (см. разд. 6.2). Чем больше величина а, тем более выпуклой становится идеальная кривая равновесия и тем легче осуществить разделение.
; Чаще всего, особенно, когда компоненты смеси принадлежат к одному гомологическому ряду, величина а увеличивается с по-V нижением температуры, поэтому разделение смеси легче прово-; дить под вакуумом. К такому выводу пришли, например, Хопкинс и Брент [92] на основании многочисленных опытов по ректификации. Колонны при пониженном давлении имеют точно
83
Рис. 46.
Зависимость коэффициентов активности компонентов системы метанол—вода от концентрации метанола [109].
Сплошные кривые построены по экспериментальным данным, пунктирные — расчетом по методу Маргулеса—Ван-Лаара.
такие же рабочие характеристики, как и при 760 мм рт. ст.*, и лишь увеличение относительной летучести в вакууме повышает степень разделения смеси. Так, Цыпкина [93] сообщает, что при разделении ректификацией смесей антрацен—карбазол и пирен—фторантен число теоретических ступеней разделения снизилось больше, чем на 50% при переходе к остаточному давлению 60—100 мм рт. ст. Есть смеси, для
которых величина а остается постоянной в широком интервале рабочих давлений, например система хлороформ — четыреххлори-стый углерод или «-гептан — метилциклогексан. Смеси подобного рода благодаря стабильным значениям а являются наиболее подходящими эталонными смесями (см. разд. 4.10.3). Однако встречаются
также смеси, относительная летучесть которых с повышением температуры также растет, например смесь 2,4-диметил пентан—2,2,3-триметилпентан. Такие смеси не имеет смысла разделять под вакуумом, скорее напротив, лучшее их разделение достигается ректификацией под давлением. Из теоретических соображений иногда целесообразнее проводить перегонку в изотермических условиях, т. е. поддерживать температуру куба постоянной за
счет понижения давления.
Для определения относительной летучести идеальной смеси можно использовать уравнение (60) после введения в него коэффициента активности у:
а = PiYi/(P2?2)
(65)
* Изменение давления иногда сопровождается изменением физико-химических свойств разделяемой смеси, а также гидродинамики потоков жидкости и пара. Например, при ректификации в кольцевом зазоре между вращающимся внутренним цилиндром и неподвижным внешним цилиндром применение вакуума приводит к ослаблению интенсивности или полному исчезновению вихрей Тейлора в паровой фазе, благоприятствующих массопереносу. Затухание вихрей Тейлора происходит вследствие повышения кинематической вязкости паров. В итоге эффективность колонны заметно снижается (см. Шафрановский А. В., Ручинский В. Р. Теор. основы хим. технол. 1971, т. V, № 1; Олевский В. М., Ручинский В. Р. Роторно-пленочные тепло- и массообменные аппараты. М., Химия, 1977. — Прим. ред.
84
Так как по формуле (35) у* ~ p-Jp (по закону Дальтона), то при р! = ру* получаем
Т? = PP?/(Pi^i) У2 = РУгЛФг) (66)
Отсюда для азеотропного состава, когда у* = х1г получим
71 = Р/Pi и ?а = Р/Р’
(67)
Если построить график зависимости у; от х1( то можно получить кривые, которые являются непосредственным наглядным и количественным показателем степени отклонения свойств реального раствора.. от закона Ра ул я_ (рис. 46).
Для идеальных смесей коэффициент активности равен 1. Отклонение значений у£ от 1 являются показателем неидеальности смеси. Если давление паров обоих компонентов больше, чем это соответствует закону Рауля (рх = Pi%i), то уг и у2 превышают 1 (1g V/ > 0); при этом говорят о положительном отклонении от закона Рауля. Если эти отклонения достаточно велики, то в смеси образуется азеотроп с максимальным давлением паров или с минимальной температурой кипения *. Наоборот, при больших' отрицательных отклонениях (1g у,- < 0) образуется азеотроп с минимальным давлением паров или с максимальной температурой кипения **. Очень большие отклонения от закона Рауля приводят к расслоению раствора на две отдельные жидкие фазы и тем самым к образованию так называемого гетероазеотропа, который ведет себя аналогично гомоазеотропу (см. разд. 6.2) [94].
Данные по коэффициентам активности и их зависимости от состава раствора нужны в первую очередь для того, чтобы уста-
новить, к какому типу относится та или иная жидкая смесь. С помощью этих данных можно также проверить надежность экспериментальных данных по равновесию. Определение коэффициентов активности приобретает большое значение также для расчета процессов азеотропной и экстрактивной ректификации, при которой имеют дело с тройными смесями. Методы расчета коэффициентов активности для подобных смесей подробно рассматриваются Кортюмом и Бухгольц-Майзенхаймером [74], а также Шубертом [17].
Коэффициенты активности уг компонентов в растворах опре-
деляют по данным экспериментального исследования парциальных
давлений в изотермических условиях (см. рис. 37). Если экспе-
риментально найденные значения р£ подставить в формулу (66),
то можно получить зависимость у£ от х. при постоянной температуре (см. разд. 4.4.1). Для систем, образующих азеотроп, коэф-
фициенты активности целесообразно определять по методу Карл-) сона и Кольбурна [95]; этот метод подробно рассматривается Орличеком и Пеллем [85]. Коэффициенты активности можно
* В советской научно-технической литературе такой азеотроп принято называть «положительным». — Прим. ред.
** Или «отрицательный» азеотроп. — Прим. ред.
85
точно рассчитать по кривой общего давления с помощью графического метода, предложенного Орличеком [96].
Данные по давлению паров и фазовому равновесию, найденные экспериментальным путем, поддаются термодинамической проверке с помощью коэффициентов активности. По уравнению (66) или (67) на основе опытных значений х и р или х и у* вычисляют коэффициенты активности и строят графическую зависимость 1g у, от х. Полученные кривые сравнивают с кривыми [17, 18], рассчитанными по уравнению Ван-Лаара [97] или Маргулеса [98]. Рис. 46 иллюстрирует этот способ на примере опытных данных Гельбина [109].
При определении коэффициентов активности, отвечающих требованиям термодинамической состоятельности, следует учитывать возможность ассоциации молекул в паровой фазе [98а].
4.6.3. ЭКСПЕРИМЕНТАЛЬНОЕ ОПРЕДЕЛЕНИЕ
КРИВЫХ РАВНОВЕСИЯ
Данные по равновесию пар—жидкость в бинарных и многокомпонентных смесях лежат в основе расчета необходимого числа теоретических ступеней разделения и других параметров процесса ректификации. Ландольт и Бернштайн [99] систематизировали данные по фазовому равновесию, опубликованные до 1933—1934 гг. включительно. Дополнительные данные по равновесию приведены в книгах Перри [100], Ю Чин Чу [101 ], а также Чу, Ванга, Леви и Пауля [102].
Штаге с сотр. опубликовал многочисленные сборники данных по фазовому равновесию; аналогичные сведения для легких углеводородов, H2S и СО2 можно найти в книге Заге и Ласея [27]. Якобс [104] приводит кривые равновесия для 50 смесей. Коган и Фридман [105] подготовили обширное собрание таблиц. Монография этих авторов, изданная в 1966 г., содержит данные но равновесию для 1765 бинарных смесей, 359 тройных смесей и 32 многокомпонентных смесей; в книге также дается введение в технику экспериментального исследования фазового равновесия в системе пар—жидкость и в методику термодинамической проверки данных по равновесию. Вопросы равновесия между паром и жидкостью подробно освещает также Хала с сотр. [78]. В первом разделе книги авторы рассматривают термодинамику растворов неэлектролитов, а во втором —технику лабораторного исследования фазовых равновесий, третий раздел содержит литературные ссылки на 1232 исследованные системы. Наиболее обширный сборник данных по фазовому равновесию при атмосферном давлении опубликован Хала с сотр. [106] в 1969 г. Долгосрочный проект PPDS* поможет накопить многочисленные данные, используемые при расчетах процессов дистилляции и ректификации [106а].
В практике лабораторной ректификации постоянно встречаются смеси, фазовое равновесие которых еще не исследовано; в таких случаях возникает вопрос о выборе наиболее целесообразной аппаратуры для получения искомых данных.
Исследователи фазового равновесия проводят различие между изотермическими и циркуляционными методами при постоянном давлении. Принцип циркуляционного метода состоит в том, что бинарную смесь известного состава испаряют и после установления фазового равновесия (т. е. состояния, при котором число молекул, покидающих жидкость, равно числу молекул, возвращающихся в жидкость) при определенном давлении измеряют концентрации кубовой жидкости и сконденсированных паров. Мюллер и Штаге ([39] к гл. 1) дают превосходное введение в технику экспериментальных измерений на примере дистилляции
* Информационная служба по физическим свойствам Британского института инженеров-химиков. — Прим. ред.
86
жирных кислот. Можно так же, как это предусмотрено в изотермическом методе, поддерживать температуру постоянной и измерять устанавливающееся при этом давление паров. Статические методы имеют то преимущество, что они позволяют работать с очень малыми количествами вещества. Кортюм и Файер [107] сообщают об очень простом статическом методе измерения давления паров при постоянной температуре в двойных и тройных жидких системах. Вебер [108] описывает универсальную дистилляционную установку для исследования фазового равновесия, состоящую из термостатов. Гельбин [109] дает сравнительную характеристику различных методов нахождения кривой равновесия.
Особенно важно точно знать ход кривой равновесия в ее начале и в конце, поэтому в интервалах концентраций хв от 0 до 10% (мол.) и от 90 до 100% (мол.) проводят как можно больше измерений. Обычно равновесную концентрацию у* определяют для следующих концентраций кубовой жидкости: 1, 3, 5, 10, 90, 95, 97, 99% (мол.). В интервале концентраций от 10 до 90% измерения проводят через каждые 5%. Разумеется, при этом учитывают характер смеси, так что при идеальном ходе кривой можно вполне обойтись без промежуточных точек в средней области.
При исследовании равновесия по циркуляционному методу в колбу 1 (рис. 47) загружают от 250 до 500 мл смеси и нагревают ее до кипения. Пары конденсируются в холодильнике 3, и при закрытом кране 4 конденсат стекает обратно в колбу /. По термометру 2 наблюдают за изменением температуры. Прибор должен быть тщательно теплоизолирован. После установления равновесия из патрубка 5 при открытом кране 4 отбирают пробу конденсата объемом 0,1—0,2 мл. Одновременно берут пробу жидкости из колбы /; отбор проводят при открытом кране 6 через патрубок 7. Измерения необходимо повторять несколько раз до повторения одной и той же концентрации проб три раза подряд.
При работе в условиях вакуума применяют пробоотборник, пригодный также и при работе под атмосферным давлением, который позволяет полностью исключить контакт пробы со смазкой крана. Концентрацию пробы определяют по показателю преломления с помощью калибровочной кривой п^ — % (масс.), которую строят заранее, составляя смеси из чистых исходных веществ. Для определения концентрации можно использовать и другие физические константы, например плотность или температуру замерзания. Серию опытов обычно начинают с наименьшей концентрации хв. После завершения опыта при одной какой-либо концентрации хв легколетучий компонент добавляют в куб в таком количестве, чтобы приблизительно получилось очередное значение концентрации;
биваться точного значения намеченной концентрации, достаточно, если она лежит вблизи желаемой точки измерения.
Чтобы получать точные данные при исследовании фазового равновесия, необходимо исключать такие источники погрешностей, как перегрев кипящей жидкости, возникновение градиента концентраций в кипящей жидкости, частичная конденсация паров над колбой, унос брызг жидкости парами, полное испарение брызг жидкости, нарушение стационарного режима вследствие чрезмерно большого объема проб и загрязнение проб при отборе. Делались попытки путем усовершенствования аппаратуры по возможности
при этом не следует
Рис. 47.
Усовершенствованный прибор Отмеря для исследования фазового равновесия;
1 — колба; 2 — термометр; 3 •— холодильник; 4, 6 — краны; 5 —- патрубок для отбора пробы пара в виде конденсата состава у* 7 — патрубок для отбора пробы кубовой жид-
кости состава х&.
87
Рис. 48.
Схема циркуляционного прибора Рёка и Зига с насосом Коттрелля:
1 — перегонная колба; 2 — нагревательная спираль; 3 — трубка Коттрелля; 4 — насос Коттрелля; 5 — участок измерения температуры; 6 — отражатель брызг; 7 — паровая камера; 8 —- холодильник; 9 — приемник дистиллята; 10 — трубка с расширением для отбора проб; 11 — стандартный шлиф NS19. Цифрами без кружков указаны размеры в мм.
Рис. 49.
Обогреваемый узел циркуляционного прибора с вакуумной рубашкой:
12 — вакуумированная рубашка; 13 — термостатирующая рубашка; 14 — стандартный шлиф NS14,5; 15 — стандартный шлиф NS9.
исключить эти источники погрешностей и обеспечить оптимальные рабочие условия. Идеальным было бы такое решение, которое обеспечивало бы измерение концентрации жидкости в колбе и конденсата пара без отбора пробы. В последнее время для этой цели стали использовать проточный рефрактометр (см. разд. 8.5). Благодаря применению такого рефрактометра Штаге с сотр. [НО] добился уменьшения времени выхода процесса на стационарный режим в циркуляционной аппаратуре до 10 мин и менее по сравнению с несколькими часами для обычного прибора Отмеря [111]. Следует отметить, что всегда выгоднее работать с возможно большим количеством Жидкости в колбе, благодаря чему периодический или непрерывный отбор проб жидкости для анализа не препятствует установлению фазового равновесия.
Если один из компонентов имеет очень высокое давление паров (например, в системе метанол — метиловый эфир муравьиной кислоты) или коэффициенты преломления компонентов очень близки, то для измерения концентрации с успехом можно применить газовый хроматографический анализ. Этот метод особенно удобен при исследовании равновесия в многокомпонентных смесях, так
88
как позволяет ограничиться очень малыми количествами жидкости для проведения анализа {112].
ЧтобыХизбежать брызгоуноса и перегрева жидкости, Джиллеспи [113] воспользовался насосом Коттрелля. Применение насоса Коттрелля в приборах для исследованиЯ-фазового равновесия весьма целесообразно, так как благодаря ему удается получать очень хорошие результаты,^выдерживающие термодинамическую проверку. Насос Коттрелля использован, например, в приборах Река и Зига [114] (рис. 48 и 49), в которых приемник дистиллята по емкости в 20 раз меньше колбы. Нагревательная спираль 2 (см. рис. 48), погруженная непосредственно в кубовую жидкость, обеспечивает равномерное кипение. Поднимающиеся пузырьки пара интенсивно перемешивают содержимое^колбы 1 и при прохождении через трубку Коттрелля увлекают за собой значительное количество жидкости. Образовавшаяся паро-жидкостная смесь выбрасывается на участок измерения температуры 5, защищенный отражателем брызг 6. В этом месте жидкая н паровая фазы разделяются. Жидкость стекает вниз, проходит через расширение [10] для отбора пробы и возвращается обратно в колбу 1. Пары поступают в паровую (расширительную) камеру 7 и полностью конденсируются в холодильнике 8. Конденсат стекает через приемник дистиллята 9 и затем возвращается в колбу 1.
Паровая камера 7 и насос Коттрелля 4 в усовершенствованном приборе, показанном на рис. 49, с целью исключения частичной конденсации паров, помещены в вакуумированную рубашку 12. Наружная стенка вакуумированной рубашки поддерживается при температуре кипения смеси с помощью циркуляционного термостата.
При такой конструкции прибора удается избежать искажения равновесной концентрации паров, вызванной их частичной конденсацией или полным испарением образовавшихся капель. Гидростатический напор насоса Коттрелля и уровень жидкости в колбе при правильном проведении опыта не оказывают никакого влияния на измерение давления и температуры. Исследуемое вещество ин на каком участке прибора не соприкасается с кранами или шлифами, поэтому загрязнение пробы смазкой исключено.
Прибор с насосом Коттрелля оказался пригодным также и для исследования систем с высокой относительной летучестью (А/Кипф> 100 °C) [115]. Усовершенствованный циркуляционный прибор «Лабодест» также оборудован насосом Коттрелля и снабжен электромагнитными клапанами с тефлоновыми наконечниками для отбора проб [112]. Аналогично выполнен прибор в микроисполнении, рабочий объем колбы составляет всего 100 мл жидкости (рис. 50). Указанные выше
Рис. 50.
Схема циркуляционного прибора «Лабодест» с насосом Коттрелля:
1 — стабилизатор вакуума; 2 — пульт управления; 3 — лабораторный плоский самописец; 4 — газовый хроматограф; 5 — электронный интегратор.
Изготовитель: фирма Фишера «Лабораторная и исследовательская техника», Бонн—курорт Годесберг.
89
Рис. 51.
Схема прибора Шуберта
для исследования равновесия (модель G15).
источники погрешностей в этих приборах сведены к минимуму. То же можно сказать и о приборе Шуберта [116], предназначенном для изучения равновесия, новейшая модель G15 которого показана на рис. 51.
В этом приборе соединительный узел между кипятильной колбой и паровым пространством выполнен в виде трубки Коттрелля. Конструкция трубки, соединяющей паровое пространство с холодильником, в основном такая же, как и в модели G13. Тепло, необходимое для испарения жидкости, поступает от обогревателя, обогреватель состоит из стеклянной дуги с внутренней нагревательной спиралью. На внешнюю поверхность дуги наплавлен стеклянный порошок для исключения перегрева жидкости при кипении. Эта дуга установлена на достаточном расстоянии от дна колбы так, что под нею может свободно вращаться магнитная мешалка в тефлоновой оболочке, обеспечивающая интенсивную турбулизацию жидкости. Форма дуги способствует образованию пузырьков пара преимущественно в центре кипящей жидкости, поскольку в месте наибольшего изгиба дуги наблюдается наибольший приток тепла.
Для пробоотбора служит электромагнитная маятниковая подвеска, вдоль которой стекает струя конденсата или охлажденной жидкости. Применение подобной маятниковой подвески вместо ранее использовавшейся качающейся воронки имеет то преимущество, что это позволяет исключить два стеклянных шлифа на каждом месте пробоотбора. Другой характерной особенностью при
90
бора G15 является совмещение капилляров (3 мм) для стока конденсата и отсе-парированной жидкости перед входом в среднюю часть прибора. Достигаемое при этом смешение обоих потоков жидкости способствует устранению значительных колебаний концентрации жидкости на входном участке зоны кипения, которые могут заметно нарушать равновесие. Все остальные узлы прибора не отличаются от узлов предшествующих моделей этого прибора.
Вихтерле и Хала [117] описывают микроприбор для изотермического исследования фазового равновесия в многокомпонентных системах при объеме проб всего лишь 2 мл. Нойманн и Вальх [118] разработали прибор для изучения фазового равновесия при низких температурах, который был применен для исследования системы СО2—СН4. Штаге и Фишер [118а] сообщают о дальнейшем усовершенствовании циркуляционного прибора «Лабодест», специально предназначенного для изучения смесей компонентов с ограниченной взаимной растворимостью.
Рис. 52.
Прибор Кумаркришны Рао с сотр. для исследования равновесия при давлениях до 15 кгс/см2:
1 — вентнль для отбора пробы жидкости; 2 — карман для термопары; 3 — продувочные вентили;. 4 — манометр; 5 — конденсатор; 6 — вентиль для отбора пробы пара; 7 — предохранительный клапан; 8 — куб;
9 — перфорированная медная трубка; 10 — дренажный вентнль.
Рис. 53.
Схема прибора Лидерсена—Хаммера для исследования фазового равновесия при остаточных давлениях до 1 мм рт. ст.:
1 — колбонагреватель; 2 — колба для исследуемой смеси (диаметр 6 см);
3 — колбы для отбора проб; 4, 5 — алюминиевые цилиндры с электрической спиралью; 6 — соединительная трубка; 7 — подвесной шарнир;
8 — воздушный холодильник; 9 — водяной холодильник; '10 — буферная емкость; 11 — вентнль; 12 — вакуумный насос; 13 — регулятор давления; 14, 15 — сосуды Дьюара, заполненные твердым диоксидом углерода;
16 — манометр Мак-Леода; 17 — приемник дистиллята; 18 — соединительный капилляр; 19 — вращающийся стол; 20 — ось вращения прибора.
91
Рис. 55.
Графический метод проверки термодинамической достоверности данных по равновесию в системе хлороформ—этанол по методу Редлнха—Кистера (t = 45 °C).
Рис. 54.
Схема прибора Юигханса—Вебера для исследования равновесия с применением инертного газа:
1 — сферический сосуд для исследуемой смеси; 2 — термометры с ценой деления шкалы 1/10 °C; 3 — изоляция нз стекловаты; 4 — пластина из пористой керамики.
Для исследования систем бензол —н-гексан и бензол—циклогексан при давлениях до 18 ат Кумаркришна с сотр. [119] использовал прибор, схема которого показана на рис. 52. Можно также применять обычную аппаратуру из стекла, заключенную в автоклав. Иост с сотр. [120] встроил в автоклав емкостью 20 л циркуляционный прибор из стекла с адиабатической рубашкой.
Для исследования равновесия при остаточном давлении вплоть до 1 мм рт. ст. Лидерсен и Хаммер [121] разработали прибор, который приводят во вращение, что обеспечивает периодическое обновление пленки жидкости в процессе испарения, а также интенсивное перемешивание кипящей жидкости и конденсата (рис. 53). Прибор был использован, например, для исследования фазового равновесия в системе н-дибутилфталат — н-дибутиловый эфир себациновой кислоты при остаточном давлении 1 мм рт. ст.
Метод изучения равновесия, основанный на разбавлении насыщенных паров потоком инертного газа, описан Юнгхансом и Вебером [122]. Этот метод был применен при исследовании бинарной смеси этилбензол—стирол, причем полимеризацию исключали путем добавки ингибитора. Метод, основанный на введении инертного газа в паровое пространство, имеет то важное преимущество, что он дает возможность проводить исследование в изобарных условиях; при этом изменяющиеся парциальные давления р± и р2 паров дополняются парциальным давлением инертного газа р3 до исходного наружного давления. Одновременно благодаря термостатированию обеспечивается изотермичность процесса (рис. 54).
Большое преимущество дает применение радиоактивных изотопов в исследованиях фазового равновесия [123]. Описано применение изотопа 36S [124].
Следует иметь в виду, что несмотря на тщательность измерений экспериментальные данные отдельных авторов более или менее различаются. В последнее время проводят много исследований для выявления источников погрешностей для уточнения и подтверждения экспериментальных данных.
Экспериментальные данные по равновесию можно проверить термодинамически по способам Кортюма с сотр. [44], Карлссона и Кольборна [95], Ридлиха и Кистера [125] и других исследователей [17, 74].
92
В Редлих и Кистер [125] показали, что условием термодинамической досто-
В верности данных по равновесию является соблюдение равенства
I j 1g (Ti/Ts) dXi = 0 (67a)
К
К На основе данных по равновесию вычисляют ?1/?2> ПРИ этом используют соотно-Н шение
1g (V1/V2) = 1g (//Л/Wi) — 1g (PxM) (676)
E где 4i — коэффициент активности соответствующего компонента; р, — концен-Е трация паровой фазы; х{ — концентрация компонентов в жидкой фазе; р° — дав-Е ление паров чистых Компонентов.
Е' На графике зависимости 1g Т1/Т2 от xt заштрихованные площади над и под Et осью абсцисс (рис. 55) должны быть равны в том случае, если изотермические данные надежны с термодинамической точки зрения. На рис. 55 в качестве М; примера показана зависимость подобного рода для системы хлороформ — этанол HI при 45 °C. Несовпадение площадей составляет всего 1,6%. Херингтон [127] рас-пространил этот метод на тройные системы и изобарные условия.
Вопросы термодинамической проверки равновесных данных, полученных в изобарных условиях, были рассмотрены Биттрихом [128]. Он дал критическую К оценку важнейшим известным методам. Херингтон [6] разработал новый графи-^Е ческий метод («тест на симметричность площадей»), который позволяет проверить Щ экспериментальные данные для ограниченного интервала концентраций бинар-ных и тройных смесей. Тао [129], исходя из уравнения Гиббса—Дюгема, выво-дит соотношение для проверки термодинамической достоверности данных по фа-зовому равновесию в многокомпонентных системах. В монографии Шуберта [17] ,^Е подробно обсуждаются прикладные методы измерения коэффициентов активно-сти и способы термодинамической проверки данных по фазовому равновесию би-нарных систем.
И 4.7. ЧИСЛО ТЕОРЕТИЧЕСКИХ СТУПЕНЕЙ К РАЗДЕЛЕНИЯ (ЧИСЛО ТЕОРЕТИЧЕСКИХ К ТАРЕЛОК)
[Иг' Как уже было отмечено в гл. 2, с разработкой большого числа И| различных ректификационных колонн возникла необходимость сравнительной оценки их разделяющей способности. Под разде-ляюще® способностью колонны подразумевают количество дистил-(^Е лята определенной концентрации, полученное в единицу времени из смеси определенного состава при заданных условиях ректи-Ик Фикации *•
^Е Один из первых методов определения разделяющей способности К, состоял в следующем. Результаты, полученные при ректификации ^Е в одних и тех же условиях, смеси, содержащей, например, 50% :^Е (масс.) толуола, оформляли в виде графиков зависимости темпера-i^E туры ректификации от количества отобранного дистиллята (кри-пИе вая разгонки). Сравнивали ход кривых, полученных для различ-
К * В советской литературе под разделяющей способностью колонны пони-У^Е мают эффективность ее работы, выраженную числом теоретических ступеней 1^Е( Разделения или единиц переноса. — Прим. ред.
93
Рис. 56.
Метод определения полюсного расстояния 5 для периодической ректификации по кривой разгонки (метод Боумена—Чичелли).
W — выход дистиллята.
Рис. 57.
Эволюция тарельчатой колонны:
а — простой перегонный аппарат, эквивалентный одной теоретической ступени разделения; б — тарельчатая колонна с тремя реальными тарелками.
ных разгонок, и делали качественный вывод, например о том, что разделяющая способность зависит от высоты колонны и ее нагрузки.
Более точные сведения о разделяющей способности получают путем измерения какой-либо физической характеристики дистиллята, например показателя преломления, и затем построения графика зависимости этого показателя от количества отогнанного дистиллята. Одна лишь температура кипения вещества не является однозначным критерием степени его чистоты. Эти методы оценки разделяющей способности вполне пригодны для сравнения различных колонн, но не дают возможности заранее рассчитать размеры колонны и параметры ректификации, обеспечивающие заданную степень разделения. Поэтому для характеристики разделяющей способности были предложены следующие показатели: выход дистиллята определенной степени чистоты [130]; количество промежуточной фракции, ограниченной двумя концентрациями дистиллята; наклон кривой разгонки на промежуточном участке [1311; число конечных ступеней [132]*.
* По определению [132] число конечных ступеней при периодической ректификации зависит от концентрационных пределов промежуточной фракции дистиллята, относительной летучести разделяемых компонентов и флегмового числа. — Прим. ред.
94
Оба последних показателя можно рекомендовать для оценки разделяющей способности при периодической ректификации бинарных смесей. Было введено понятие полюсного расстояния, определяемого по наклону кривой разгонки в точке, в которой дистиллят содержит точно 50% (мол.) легколетучего компонента (рис. 56).
С введением понятия теоретической тарелки или теоретической ступени разделения появилась возможность количественно оценивать как эффективность колонны, так и степень трудности разделения.
Теоретическая тарелка— это гипотетический участок колонны, соответствующий идеальной тарелке, на котором между паром, поднимающимся из нижележащего участка колонны (теоретической тарелки), и жидкостью, стекающей с вышележащего участка колонны (теоретической тарелки) наступает термодинамическое равновесие (см. рис. 24) *.
Поясним понятие теоретической тарелки более подробно на примере эволюции тарельчатой колонны. Самый простой аппарат для перегонки состоит из перегонной колбы для испарения жид-
кости и приставки для конденсации паров и отвода конденсата. При этом, согласно определению, такой аппарат соответствует одной теоретической тарелке, поскольку пары, поднимающиеся из колбы, находятся в термодинамическом равновесии с жидкостью, загруженной в колбу ** (рис. 57д). Для достижения более
высокой степени разделения Адам предложил устанавливать после-
довательно несколько перегонных колб и каждую последующую колбу нагревать парами, выходящими из предыдущей колбы. В результате частичной конденсации паров в соединительных трубках, охлаждаемых воздухом, образуется некоторое количество флегмы. Если последовательно расположенные перегонные колбы
разместить одну над другой, то получится уже известная тарельчатая колонна (рис. 576).
Для дополнительного пояснения понятия теоретической тарелки рядом со схематическим изображением тарельчатой колонны (рис. 58) приведена кривая равновесия, которая иллюстрирует процессы, протекающие в колонне. Допустим, что как и при исследовании фазового равновесия, колонна работает с бесконечным флегмовым числом, т. е. без отбора дистиллята. В колбу загружена смесь, содержащая — 10% (мол.) компонента 1 (легколетучий). Пары, образующиеся при испарении кубовой жидкости, после установления равновесия имеют концентрацию у = 25% (мол.). При конденсации этих паров образуется жидкость состава х2 на тарелке 2 (колбу рассматриваем как тарелку 1). Пары, поднимающиеся из перегонной колбы, за счет теплообмена с жид-
* При этом равновесными являются концентрации паров и жидкости,
' удаляющихся из рассматриваемого участка колонны. — Прим. ред.
** По ряду причин указанное равновесие полностью достигается в редких
случаях. — Прим.
ред.
95
Рис. 58.
Взаимосвязь между идеальной колонной с идеально работающими тарелками и кривой равновесия.
костью, находящейся на тарелке 2, вызывают испарение из нее легколетучих фракций. Концентрация легколетучего компонента во вновь образующихся парах у составляет уже 50,4% (мол.). Эти пары конденсируются на тарелке 3, в результате чего жидкость на тарелке также имеет концентрацию х3 — 50,4% (мол.). Поднимающиеся с тарелки 3 пары имеют концентрацию //з == 76% (мол.) и т. д. При отводе в холодильник паров у, поднимающихся с тарелки 4, в результате конденсации получают дистиллят концентрацией = 91% (мол.).
Для обогащения смеси, начиная с концентрации 10% (мол.), которую имеет жидкость в колбе, до концентрации 91 %, которую имеет дистиллят, требуется в общей сложности четыре теоретических ступени разделения. Для этого нужна колонна с тремя идеально работающими тарелками и куб (перегонная колба), который соответствует одной теоретической ступени разделения *. Вместо термина «теоретическая тарелка» в дальнейшем будет применяться выражение «теоретическая ступень разделения», поскольку оно более точно отражает сущность метода ступенчатых построений, который применяютдля оценки обогащения, достигаемого как в тарельчатой, так и в насадочной колонне.
Понятие теоретической ступени разделения было использовано при характеристике эффективности насадочных колонн и ряда методов разделения. Оно позволяет количественно оценить эффективность работы применяемой аппаратуры. Длину участка колонны, который необходим для перехода от концентрации жидкости х к равновесной концентрации пара у *, называют высотой,
* При этом следует учитывать, что куб не всегда эквивалентен целой теоретической ступени. — Прим. ред.
96
эквивалентной теоретической ступени (ВЭТС). В литературе на английском языке используют термин НЕТР (height eguivalent to a theoretical plate).
Если принять за ось координат г ось колонны, получим
(^l)z+B3TC (xl)z = (68)
или
(//1/1/2 )z+ВЭТс/(^1/i's) =а (69)
Для разделяющей колонны длиной I получим
ВЭТС = (/пт
Число теоретических ступеней разделения, достигаемое в колонне, является функцией большого числа параметров как аппаратурного, так и технологического характера. Путем одних только математических построений до сих пор еще не удалось разработать метод расчета ВЭТС. На основе многочисленных экспериментальных результатов получены только эмпирические соотношения (см. разд. 4.2 и 4.8).
На реальных тарелках практически никогда не достигается к. п. д. 100%, что возможно для идеальных тарелок; обычно к. п. д. составляет 50—90% *. Это вызвано, во-первых, тем, что перемешивание пара и жидкости в большинстве случаев не является совершенным, и, во-вторых, тем, что пар, особенно при больших скоростях, увлекает брызги жидкости на вышележащую тарелку. Кроме того, колонны, как правило, работают не с бесконечным флегмовым числом, а с конечным, так как целью любой ректификации является получение дистиллята. Как показал Аншюц [133], коэффициент полезного действия тарелок может быть учтен при графическом построении теоретических ступеней разделения по методу Мак-Кэба и Тиле.
Под флегмовым числом v понимают отношение количества жидкости R, возвращаемой после конденсации паров в верхнюю часть колонны, к количеству жидкости Е, отбираемой в виде дистиллята:
v = R/E
Если дистиллят не отбирают, го флегмовое число является бесконечным:
v = lim R/E — оо £->0
Количество паров D, выходящих из колонны, складывается таким образом из R и Е:
D = R Е (70)
* Известны случаи, когда для ситчатых тарелок с переливами при работе с сильно пенящимися жидкостями удавалось в определенной области нагрузок получить к. п. д. выше 100%. — Прим. ред.
4 Э. Крель 97
Рис. 59.
Кривая равновесия смеси бензол—толуол прн 760 мм рт. ст. (построение рабочих лнкик и ступеней разделения).
Это справедливо по отношению к каждому компоненту, что выражается уравнением
Dy—RxB--ExE (71)
При подстановке (70) в (71) получим
у = RxB!(R + Е) 4- ExeKR 4- Е) (72)
Разделив на Е числители и знаменатели в правой части уравнения (72), получим
RxB Ехе
У = £(/?,'£ -]-£/£) + £(£/£+£/£) (73)
Подставив вместо RIE условное обозначение для флегмового числа v, получим уравнение рабочей линии укрепляющей части колонны
У = Vxb№ + !) + хе№ +
(74)
Это линейное уравнение выражает зависимость состава пара от состава жидкости для любого поперечного сечения колонны (при условии адиабатичности процесса) и представляет собой рабочую линию а—с (рис. 59). Если мольные энтальпии испарения компонентов сильно различаются, то это необходимо учесть при расчете числа теоретических ступеней по методу Мак-Кэба и Тиле.
Различия в мольных энтальпиях испарения могут оказывать заметное влияние на число теоретических ступеней разделения особенно при малых флегмовых числах или при малой относительной летучести компонентов и высокой разделительной способности колонны. Графический метод Мак-Кэба и Тиле в этом случае заметно усложняется, так как при этом рабочие линии процесса ректификации не являются прямыми. Однако видоизменение метода Мак-Кэба и Тиле, предложенное Фишером [134], относительно упрощает графические построения. Биллет [135] вывел уравнения для расчета рабочих линий, соответствующих процессу ректификации бинарных смесей при различных мольных энтальпиях испарения компонентов. Тум [136] разработал метод прямого расчета числа теоретических ступеней разделения при ректификации идеальных бинарных смесей с конечным флегмовым числом, в котором учтены различия в энтальпиях испарения.
Неретник с сотр. [137] пришел к выводу, что для энтальпий испарения различных веществ можно найти такие поправочные коэффициенты N, что все вещества в полученной таким образом 98
новой системе измерения будут иметь одинаковые скорректированные энтальпии испарения. Тем самым рабочие линии для систем, исходные компоненты которых имеют весьма различающиеся энтальпии испарения, в результате простого преобразования можно было бы получать на диаграмме Мак-Кэба и Тиле в виде прямых линий. Если относительная летучесть компонентов не зависит от состава смеси, указанное преобразование достаточно выполнить всего для нескольких концентраций.
В системе координат с осью абсцисс х и осью ординат у рабочая линия укрепляющей части колонны пересекает ось ординат в точке
y = xE/(v+l) (75)
При бесконечном флегмовом числе (у = со) ордината этой точки становится равной 0, так как
lim(/ = 0 (76)
V->oo
Это означает, что рабочая линия при v = оо проходит через начало координат и совпадает с диагональю диаграммы равновесия пар— жидкость. Угол наклона рабочей линии к оси абсцисс определяется зависимостью
tga = y/(o+l) (77)
Сделаем упрощающее допущение, что мольные энтальпии компонентов, а следовательно и смеси, одинаковы. Тогда с помощью графического метода Мак-Кэба и Тиле [77] можно рассчитать число теоретических ступеней разделения, необходимое для разделения смеси при определенном флегмовом числе, которое превышает минимальное флегмовое число. Вычисления можно проводить как для периодического, так и для непрерывного режима работы.
1 Следует подчеркнуть, что все вышеизложенные рассуждения относились к тарельчатым колоннам. В колоннах этого типа массо-t обмен происходит при барботировании потока паров через слой жидкости на тарелке, при этом между паром и жидкостью дости-[ гается более или менее полное термодинамическое равновесие. [ Процесс изменения температуры и состава паров и жидкости повто-t ряется от тарелки к тарелке; таким образом, обогащение паров t происходит по ступенчатому закону. В насадочной колонне при I прохождении потока паров над пленкой жидкости, орошающей I насадку, имеет место непрерывный процесс массообмена. Однако Г ни в каком месте насадки пары не остаются настолько долго, чтобы [ между ними и пленкой жидкости могло установиться термодина-I мическое равновесие. Непрерывное обогащение достигается в ре-К зультате многих элементарных актов разделения. Поэтому для I характеристики насадочных колонн ввели термин «высота единицы I переноса» (heigt of transfer unit — HTU).
I Простые приемы вычисления числа ступеней, разработанные I для тарельчатых колонн, можно использовать также для наса-I- 4* .99
дочных колонн. Однако следует учитывать, что такие расчеты дают лишь хорошее приближение без строгого теоретического обоснования.
О пользе концепции теоретической ступени можно получить определенное представление, если сравнить процессы разделения в тарельчатой и насадочной колоннах по возможности одинаковых размеров, работающих в идентичных условиях. Если колонны имеют одинаковую разделяющую способность,то можно уверенно сказать, что обе колонны эквивалентны определенному числу теоретических ступеней разделения.
4.7.1. РАСЧЕТ ЧИСЛА ТЕОРЕТИЧЕСКИХ
СТУПЕНЕЙ РАЗДЕЛЕНИЯ ПО МЕТОДУ МАК-КЭБА И ТИЛЕ ДЛЯ ПЕРИОДИЧЕСКОЙ РЕКТИФИКАЦИИ
В качестве примера рассмотрим разделение смеси бензол-толуол. Поставлена следующая задача: смесь, содержащую 40% (мол.) бензола, необходимо обогатить до концентрации 98% путем периодической ректификации.
В координатах х—у строим кривую равновесия (см. рис. 59) и через точку хв = 40% (мол.) на оси абсцисс проводим прямую, параллельную оси у, до пересечения с равновесной кривой в точке Ь. Прямая, проведенная через точку хБ = 98% (мол.) параллельно оси ординат, пересекает диагональ в точке а. Соединив точки а и Ь, получим рабочую линию, которая пересекает при продолжении ее за точку b ось ординат в точке с, соответствующей у = 37% (мол.). Из уравнения (75) находим:
"мин = (МО-1 <78>
сМин = 98/37 — 1 = 1,65 (79)
Значение 1,65 представляет собой минимальное флегмовое число, при котором для разделения смеси бензол—толуол требуется бесконечно большое число ступеней разделения. Так как в этом случае точки хЕ, хв и у* лежат на рабочей линии, то минимальное флегмовое число может быть также выражено уравнением [103]
°мин = (хе-^Ж-хв) <8°)
в котором у * соответствует точке пересечения b рабочей линии а—с с кривой равновесия. Находим
%ин = (98-62)/(62-40)~ 1,65 (81)
При флегмовом числе меньше 1,65 обогащение до концентрации 98% (мол.) вообще невозможно. Если увеличить флегмовое число (у > 1,65), то потребуется конечное число ступеней разделения и оно будет тем меньше, чем больше флегмовое число. Примем, что флегмовое число v = 1,85; тогда длина отрезка, отсекаемого на оси ординат рабочей линией, будет равна:
«/ = 98/(1,85 4-1) = 34,4 (82)
100
Таблица 12.
Число теоретических ступеней разделения, требуемое для разделения смеси бензол—толуол при хв= 40%, хе — 98% (мол.), 1'мин ~ 1,65
Рабочее флегмовое число Отрезок, отсекаемый иа оси ординат, ХЕ у- о + 1 Число теоретических ступеней разделения, п
1,85 3,44 10,0
3,00 24,5 7,0
5,00 16,4 6,0
10,00 8,9 5,5
20,00 4,7 5,0
50,00 1,9 5,0
оо 0,0 4,5
Таблица 13.
Флегмовые числа, обеспечивающие разделение смеси бензол—толуол при хв от 80 до 5% (мол.), п — 10 и хе = 98% (мол.)
Хв, % (мол.) Флегмовое число
минимальное рабочее
80 0,58 0,65
60 0,94 1,09
40 1,65 1,85
20 3,46 3,90
10 7,10 8,80
5 13,00 25,00
Точку пересечения с, соответствующую у — 34,4, (см. рис. 59) соединяем с точкой а и получаем рабочую линию, характеризующую выбранные условия ректификации. Начиная от точки Ь,
наносим теоретические ступени разделения, вписывая их между равновесной кривой и рабочей линией; получим 10 теоретических ступеней разделения.
Следует выяснить, насколько уменьшается число теоретических ступеней разделения с увеличением флегмового числа. В табл. 12 приведены числа теоретических ступеней, необходимые для разделения смеси бензол—толуол при разных флегмовых числах. На рис. 60 показана кривая равновесия этой смеси с соответствующими рабочими линиями.
Очевидно в данном случае разделения нет смысла увеличивать флегмовое число выше 5; увеличение числа ступеней становится таким незначительным, что дальнейшее повышение флегмового числа себя не оправдывает.
Следует учитывать, что при периодической ректификации содержание легколетучего компонента в кубовой жидкости постепенно уменьшается в результате отбора дистиллята в головке колонны концентрация хв стремится к нулю. Предположим, что в результате разгонки концентрация кубовой жидкости xs уменьшилась до 10% (мол.). Еще раз, как вначале, проведем расчет. Ниже дано сравнение конечных рабочих параметров раз-
гонки с исходными данными, соответствующими началу разгонки:
'Начало разгонки...............
Сравнительный расчет ..........
ХВ
40
10
ХЕ
98
98
°мин °раб
1,65 1,85
7,10 8,80
п
10
10
101
Рабочие линии процесса ректификации смеси бензол—толуол в обогаща ющей части колонны для различных флегмовых чисел.
х^ — 98% (мол.). Линии построены по данным табл. 12.
Рис. 61.
Зависимость флегмового числа от концентрации кубовой жидкости xg.
Xg = 98% (мол.), число теоретических ступеней разделения п = 10.
Для того, чтобы установить, до каких пределов нужно повысить флегмовое число (при изменившемся составе кубовой жидкости) для получения дистиллята прежнего состава расчетное число теоретических ступеней разделения для начала и конца разгонки было намеренно выбрано одинаковым. Если оставить флегмовое число v = 1,85, то концентрация дистиллята хЕ с уменьшением концентрации жидкости в перегонной колбе снизилась бы.
Таким образом, при периодической ректификации недостаточно установить условия процесса для начального момента. Следует также решить, до какой концентрации кубовой жидкости целесообразно проводить разгонку или какое максимальное флегмовое число необходимо выбрать, чтобы длительность разгонки не была бы слишком большой. Если принять предельное флегмовое число и = 25 и хв = 80% (мол.), оставив постоянными хЕ ~ 98% (мол.) и число теоретических ступеней разделения п = 10, то для снижения концентрации легколетучего компонента в кубовой жидкости с 80 до 5% (мол.) необходимы флегмовые числа, указанные в табл. 13.
Из графика зависимости ораб от хв (рис. 61) легко увидеть, что в области концентрации низкокипящего компонента ниже 10% (мол.) уменьшение его концентрации на каждый мольный процент потребует значительного увеличения количества орошающей жидкости. На практике разгонку обычно начинают при флегмовом числе v = 2, а при повышении температуры в верху колонны 102
флегмовое число увеличивают до 5, и затем еще раз до 10. Целесообразность дальнейшего увеличения флегмового числа следует проверять в каждом отдельном случае. Необходимо только иметь в виду, что чем меньше соблюдаются требуемые условия ректификации, тем больше будет объем промежуточной (нецелевой) фракции.
Основываясь на уравнениях Релея, Вебер [1381 разработал диаграмму, с помощью которой для идеальных смесей при известном значении относительной летучести компонентов можно определить, какой состав имеет кубовая жидкость, если первоначальная загрузка куба равна Во, а количество кубового остатка после разгонки составляет В{. Фогельпол 1139] разработал метод расчета ректификации идеальных тройных смесей в тарельчатых колоннах. Доменех с сотр. [139а] создал математическую модель процесса ректификации. Биллет [140] представил уравнения, с помощью которых для периодической ректификации бинарных смесей при постоянной концентрации дистиллята можно рассчитать, применяя смешанные графоаналитические способы, такие параметры ректификации, как флегмовое число, количество наработанного продукта, состав кубовой жидкости и объем проходящих в колонне паров смеси для различных моментов разгонки. Применительно к глубокой очистке жидкостей путем периодической ректификации Вилкокс [140 а] вывел специальные уравнения для расчета процессов разделения, направленных на получение дистиллята весьма высокой степени чистоты.
При расчете процесса периодической ректификации следует учитывать, что по мере изменения состава кубовой жидкости одновременно повсюду в колонне (в парах и в жидкости) уменьшается доля легколетучего компонента (см. разд. 4.10.5).
4.7.2. РАСЧЕТ ЧИСЛА ТЕОРЕТИЧЕСКИХ СТУПЕНЕЙ РАЗДЕЛЕНИЯ ПО МЕТОДУ МАК-КЭБА И ТИЛЕ ДЛЯ НЕПРЕРЫВНОЙ РЕКТИФИКАЦИИ
Лабораторную ректификацию большей частью проводят в периоди-' ческом режиме, однако возможны такие процессы разделения, для которых непрерывный способ является предпочтительным или даже ) обязательным (см. разд. 5.2.2). В противоположность периодиче-( ской ректификации, при которой концентрации кубовой жидкости,
дистиллята и всего содержимого колонны непрерывно изменяются, при непрерывной ректификации эти параметры остаются неизменными. При непрерывной ректификации исходную жидкую смесь
постоянного состава предварительно подогревают до температуры, установившейся в колонне в месте ввода смеси; ее пары обога-
щаются в укрепляющей части колонны 1 (рис. 62). Участок колонны 3 между местом ввода исходной смеси 2 и кубом (перегонная колба 4) называют исчерпывающей частью. Обозначим коли-
.чество дистиллята (головной продукт) через Е, а количество про-
дукта, отбираемого из нижнего конца колонны (над кубом) или
103
Рис. 62.
Установка (модель IIIv) для непрерывной вакуумной ректификации по нормалям «Дестииорм»:
1 — укрепляющая часть колонны; 2 — место ввода исходной смеси; 3 — исчерпывающая часть колонны; 4 — перегонная колба.
Изготовитель: народное предприятие
«Комбинат технического стекла», Ильменау).
из самого куба (кубовый остаток или кубовый продукт) через А.
При работе установки] непрерывного действия следят за тем, чтобы постоянно сохранялся материальный баланс:
Z = Е + Л (83) а также
x_Z = хРЕ + х.А (84) где Z, Е, А — соответственно количество питающей жидкости, головного (целевого) продукта и кубового продукта (отходы).
Рассмотрим процесс разделения смеси жирных кислот нормального строения Св—С7, содержащей 81% (мол.) кислоты С6. Остаточное давление при разгонке должно составлять 20 мм рт. ст. Рассчитаем число теоретических ступеней, необходимое для разделения
этой смеси по непрерывному способу, если требуется получить головной продукт, содержащий 96% (мол.) низкокипя-щего компонента, а кубовый остаток — 0,5%.Кривая равновесия для этой смеси экспериментально не изучена, она была рассчитана по формуле (52) при а = 2,10 (рис. 63).
Через точки а (хЕ = 96%) и b (xz = 81%) проводят рабочую линию процесса ректификации для укрепляющей части колонны. Эта линия пересекает ось ординат в точке с, которой соответствует у = 59,8% (мол.). По уравнению (78) вычисляем минимальное флегмовое число:
ОмИа=(^)- 1 = 96/59-8 - 1 = 0’6
(85)
Чтобы не получить слишком большого числа теоретических ступеней разделения, примем флегмовое число v = 1, тогда отре-
104
зок, отсекаемый рабочей линией на оси ординат, будет равен у = 96/(1 + 1) =48 (86)
Рабочая линия а—с', соответствующая конечной стадии разгонки, пересекает ось ординат в точке 48% (мол.), а вертикальную линию, проведенную через xz = 81% —в точке d. Через ; точку хА = 0,5% проводят линию, параллельную оси ординат, ! до пересечения с диагональю в точке е. Соединяют точки d и е и j получают прямую, которая является рабочей линией процесса ( ректификации для исчерпывающей части колонны. Начиная от ;* точки d, вычерчивают ступени разделения для укрепляющей части колонны. Эти ступени располагаются между кривой равновесия
S и рабочей линией d—а для укрепляющей части колонны до-г статочно иметь 4 ступени разделения. Затем откладывают ступени разделения от точки d вниз и между равновесной кривой и рабочей ^линией процесса для исчерпывающей части колонны d — е получают «' всего 13 ступеней. Если кривая равновесия построена в мелком v масштабе (примерно 25 X 25 см) или подходит очень близко к диаго-
нали, то рекомендуется участок диаграммы, лежащей между 10 и 0%, отдельно вычертить в большем масштабе, как это показано
в левой части рис. 63; такой прием облегчает построение ступеней
(разделения.
1 Для заданного разделения необходимо в общей сложности (17 теоретических ступеней, из которых 13 приходятся на исчер-; пывающую часть колонны, а 4 — на укрепляющую часть. В разд. ( 4.10.9. и 7.3 показано, как можно практически получить эти (17 теоретических ступеней.
X, 7о(мОЛ.)
UC. 63.
клетная кривая равновесия смеси жирных кислот Св—С7 нормального роеиия при 20 мм рт. ст. и пример графического способа расчета числа Юретических ступеней разделения для непрерывной ректификации.
105
т. . °/о( мал.)
лонны увеличится на
Рис. 64.
Влияние температуры исходной смеси при непрерывной ректификации на положение рабочей линии процесса в обогащающей части колонны.
Описанный прием определения минимального флегмового числа для непрерывной ректификации основан на допущении, что разделяемая смесь поступает в колонну при температуре кипения. В этом случае количество жидкости в исчерпывающей части ко-
количество исходной смеси, т. е.
R' = R + Z
(87)
Если температура исходной смеси ниже температуры кипения, то происходит частичная конденсация паров; количество конденсирующихся паров определяется количеством тепла, необходимым для нагревания смеси до точки кипения. Это обстоятельство учитывают поправочным коэффициентом е, вследствие чего уравнение (87) принимает вид
R' = R^-eZ (88)
e==1+(^кип-^)/А77г (89)
где QzKIIII — энтальпия смеси при температуре кипения; Qz — энтальпия смеси при фактической температуре смеси в точке питания; &HV — мольная энтальпия испарения.
При этом получают значение абсциссы в точке g (рис. 64):
x'z = xz/e (90)
Прямая g — d пересекает рабочую линию в точке в, вследствие чего минимальное флегмовое число дополнительно уменьшается. Если исходная смесь поступает при температуре кипения, то е = 1, а точкам d и f соответствует одна и та же абсцисса xz [ 103 ]. Если часть смеси поступает в виде паров, то можно воспользоваться рекомендациями Каверса [142], которые позволяют и в этом случае проводить расчет числа теоретических ступеней с помощью диаграммы Мак-Кэба и Тиле.
Поль [143] описал метод расчета параметров исчерпывающей части колонны для смесей с очень низкой концентрацией легколетучего компонента. Этот метод основан на использовании диаграммы Мак-Кэба и Тиле. При этом способе расчета кривую равновесия можно приблизительно представить в виде прямой. Норват и Шуберт [144] разработали метод расчета, который можно ис-106
пользовать при высоких концентрациях легколетучего компонента в головном продукте [около 99,99% (мол.)] и при низких концентрациях этого компонента в кубовом остатке. Если диаграмму Мак-Кэба и Тиле построить в логарифмических координатах, то точность расчета увеличится. Аналитические методы определения числа теоретических ступеней и рабочих параметров для непрерывной ректификации идеальных смесей подробно изложены в гл. 4.
4.7.3. ОПРЕДЕЛЕНИЕ ЧИСЛА ТЕОРЕТИЧЕСКИХ СТУПЕНЕЙ РАЗДЕЛЕНИЯ ПО МЕТОДУ МАК-КЭБА И ТИЛЕ ДЛЯ СМЕСЕЙ С КРИВЫМИ РАВНОВЕСИЯ, ИМЕЮЩИМИ ТОЧКУ ПЕРЕГИБА И АЗЕОТРОПНУЮ ТОЧКУ
Если на кривой равновесия для какой-либо смеси имеется точка перегиба, например для смеси этанол—вода, то число теоретических ступеней разделения определяют методами, описанными в разд. 4.7.1 и 4.7.2. Однако при этом вводятся следующие ограничения, а именно: концентрация легколетучего компонента в головном продукте хЕ должна быть меньше концентрации в азеотропной точке, а при определении минимального флегмового числа < рабочую линию процесса ректификации для укрепляющей части : колонны следует 'проводить по касательной к кривой равновесия I (рис. 65). Если провести рабочую линию а—с1; как обычно, от г точки а через точку Ь2, то на кривой равновесия получатся три I точки пересечения blt b2 и Ь3. Даже при некотором увеличении I флегмового числа, начиная с его минимального значения, этой f рабочей линии соответствует бесконечно большое число теорети-I ческих ступеней разделения. Чтобы получить конечное число тео-I ретических ступеней, необходимо перейти от минимального флег-I мового числа, соответствующего касательной к кривой равнове-I сия (рабочая линия а—с), к несколько большему флегмовому числу, определяемому рабочей линией а—с'; после этого можно строить ступени разделения в обычном порядке. Касательная а—с пересекает кривую равновесия только в одной точке &4 и касается ее в точке Ьъ. Отсюда ясно, что минимальное флегмовое число остается постоянным в широком интервале концентраций. Рис. 66 иллюстрирует
Рис. 65.
Графическое определение минимального флегмового числа для разделения смесей, кривая равновесия которых имеет точку перегиба.
107
о 10 20 зо Чо 50 во
X- , Чо(мОЛ.)
Рис. 66.
Зависимость минимального фле; .OBrtr'j числа от при непрерывной решифи* кации смеси этанол—вода.
760 мм рт. ст.; = 81,6% (мол.).
зависимость умин от состава исходной смеси xz при ректи-фикации смеси этанол—вода, когда требуется получить дистиллят концентрацией 81,6% (мол.), или 92% (мол.). Как видно из рис. 66, в пределах значений xz от 7,5 до 65,3% (мол.) минимальное флегмовое число остается постоянным и равным 1,3.
Если кривая равновесия, начиная с нулевой концентрации, проходит ниже диагонали, а после пересечения с диагональю выше нее, то это означает, что данная смесь является азеотропной с максимумом на изобарной кривой кипения или минимумом на изотермической кривой давления паров. При этом точка кипения азеотропной смеси лежит выше точек кипения обоих чистых компонентов. В качестве примера можно привести смесь азотная кислота — вода (см. рис. 29 и). Температура кипения Кртео азотной кислоты 86,0° С, воды 100,0° С, азеотропа, содержащего 37,81% (мол.) кислоты, 122° С. Для этой системы Флатт [145] приводит метод графического расчета рабочих условий ректификации.
4.7.4. ОПРЕДЕЛЕНИЕ ЧИСЛА ТЕОРЕТИЧЕСКИХ
СТУПЕНЕЙ РАЗДЕЛЕНИЯ ДЛЯ СМЕСЕЙ СО СЛАБО
ВЫПУКЛЫМИ КРИВЫМИ РАВНОВЕСИЯ
И ПРИ НЕБОЛЬШОМ РАССТОЯНИИ МЕЖДУ КРИВОЙ
РАВНОВЕСИЯ И РАБОЧЕЙ ЛИНИЕЙ
Если кривая равновесия смеси, например л/-крезол — п-крезол, незначительно отклоняется от диагонали (рис. 67), то очень трудно точно построить ступени разделения в интервале низких значений хв и высоких значений хЕ. В этом случае для облегчения графических построений кривую равновесия следует вычертить в большом масштабе (порядка 2x2 м), а также дополнительно укрупнить масштаб в интервалах концентраций 0—10% и 90— 100% (мол.). При этом кривая равновесия практически принимает вид прямой (ср. с рис. 63).
Аналогичный прием можно использовать и в том случае, когда кривая равновесия достаточно выпукла, но область между кривой равновесия и рабочей линией слишком узка для точного по-
* Условное обозначение температуры кипения прн 760 мм рт. ст., принятое в немецкой литературе; в советской литературе применяется термин н. т. к. (нормальная температура кипения). — Прим. ред.
108
X , °!о(мОЛ.}
Рис. 67.
Кривая равновесия смеси л-крезол—л-крезол
Остаточное давление 30 мм рт. ст.
X, °/о(мОЛ.')
Рис. 68.
Кривая равновесия смеси толуол—н-октаи с асимптотическим приближением к диагонали.
760 мм рт. ст.
строения ступеней разделения (рис. 68). При этом число наносимых ступеней вследствие асимптотического приближения равновесной кривой к диагонали или рабочей линии является столь большим, что точность метода становится ограниченной.
Для ректификации с бесконечным флегмовым числом Штаге и Шульце [146] предлагают метод расчета числа теоретических ступеней, который связан с построением так называемой дифференциальной кривой. Согласно этому методу по возможности в большем масштабе (ось абсцисс примерно 1 м) строят график зависимости разности у* — хв от хв. Таким образом получают дифференциальную кривую выпуклой формы (рис. 69). С помощью кривой строят график зависимости «число теоретических ступеней разделения — концентрация» (рис. 70). Построение начинают с очень низкой концентрации, например 0,16% (мол.), которой на дифференциальной кривой (см. рис. 69) соответствует обогащение (у*—хв) = 0,28%. Это значение прибавляют к 0,16% и получают концентрацию жидкости на второй тарелке, равную 0,44%. Для этого значения на диаграмме снова определяют обогащение, равное 0,70%, которое прибавляют к 0,44% и получают концентрацию на следующей
20
О.2В
Я к 10
Рис. 69.
Дифференциальная кривая равновесия смеси бензол —к-гептан по данным Штаге-Шульце.
Xq — концентрация бензола в смеси.
109
35
О 20 ‘tO 60 80 100
П
Рис. 70.
Зависимость числа теоретических ступеней разделения от концентрации для смеси бензол—н-гептан (по данным Штаге—Шульце).
Рис., 71.
Зависимость числа теоретических ступеней разделения от показателя преломления для эталонной смеси н-гептан—метилциклогексан.
тарелке, равную 1,14%. Эти графические построения продолжают до достижения концентрации 99,8%, получая в результате кривую зависимости числа теоретических ступеней разделения от концентрации (см. рис. 70). Необходимое число теоретических ступеней разделения при бесконечном флегмовом числе находят по кривой следующим образом: из точек хв и хЕ на абсциссе проводят вертикальные линии до пересечения с кривой, и разность чисел по ординате дает искомое число теоретических ступеней разделения. Например, для обогащения смеси бензолом с 25% (мол.) в кубе до 85% в дистилляте потребуется 11,2—6,6 = 4,6 теоретических ступеней разделения при бесконечном флегмовом числе.
Вместо значения абсциссы хв можно также использовать и другие показатели, например плотность или показатель преломления, и аналогичным способом определять число теоретических ступеней разделения; при этом на график наносят соответствующие значения показателей для загрузки куба и дистиллята и находят разность соответствующих чисел на оси ординат. Рис. 71 иллюстрирует зависимость числа теоретических ступеней от коэффициента преломления для модельной смеси н-гептан — метилциклогексан 1147].
Для ректификации с конечным флегмовым числом Шефер разработал метод расчета теоретических ступеней с использованием двух номограмм и двух диаграмм. Этот метод, особенно пригодный для многократно повторяющихся расчетов числа теоретических ступеней разделения п, основан на приближенной гиперболической зависимости между п и флегмовым числом v, применявшейся еще Фишером:
(« - «мин) (V - ‘'мин) = («' - «мин) (°' - "мин) (91)
110
Фишер [149] предлагает метод, который в отличие от метода Шефера основан на использовании только одной номограммы. Другие графические и графоаналитические методы расчета изложены в обзоре Штаге и Джуилфса [71 ].
«Метод относительной летучести» Маца [1501 позволяет рассчитывать параметры периодической и непрерывной ректификации идеальных смесей также и в тех случаях, когда значения хЕ находятся в интервале 99—99,8%.
4.7.5. АНАЛИТИЧЕСКИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ
ЧИСЛА ТЕОРЕТИЧЕСКИХ СТУПЕНЕЙ РАЗДЕЛЕНИЯ
для ПЕРИОДИЧЕСКОЙ РЕКТИФИКАЦИИ
До сих пор мы рассматривали, главным образом, графические и графоаналитические методы определения числа теоретических ступеней, так как они более наглядно передают понятие ступени разделения. Наряду с этим разработаны многочисленные методы и уравнения, позволяющие чисто расчетным путем определить условия ректификации для заданной степени разделения.
Графические методы имеют то преимущество, что они базируются на кривой равновесия и тем самым учитывают также и неидеальное поведение смеси. Применяя чисто расчетный метод, обычно допускают, что по свойствам смесь приближается к идеа-альной.
4.7.5.1. Определение числа теоретических ступеней разделения по разности температур кипения
О взаимосвязи между разностью температур кипения и необходимым числом теоретических ступеней разделения можно судить по приведенным ниже данным для бинарных смесей углеводородов при атмосферном давлении, позаимствованным у Хильберата [1511.
Необходимое число тео-
Разность температур кипения, °C ретических ступеней разделения
1,5
3,0
5,0
7,0
100
55
30
20
Расчет облегчается, если исходить из степени очистки легколетучего компонента. Диаграмма Брэгга—Льюиса [152] позволяет сразу же найти минимальное число теоретических ступеней разделения в зависимости от разности температур кипения для бинарных эквимольных идеальных смесей (рис. 72).
111
Рис. 72.
Диаграмма Брэгга- Льюиса.
А^кип — разность температур кипения компонентов; пмнн — минимальное число теоретических ступеней разделения при полном возврате флегмы.
Для любой разности температур кипения необходимое число теоретических ступеней разделения можно также рассчитать по следующему уравнению:
п=(Т2 + 7’1)/Л(7’2-7’1) (92)
где 7 и Та — температура кипения легколетучего и труднолетучего ком-
понентов соответственно, К; К — константа, причем К = 2,5 соответствует максимальному числу теоретических ступеней разделения, К = 3 — оптимальному, а К = 4— минимальному числу теоретических ступеней разделения.
Однако в этом уравнении не учтены остальные условия ректификации такие, как флегмовое число, рабочее давление, общая удерживающая способность колонны.
По уравнениям Странджио и Трейбала [152а] можно ориентировочно рассчитать некоторые параметры ректификации (о и п) также и для многокомпонентной смеси при условии постоянства относительной летучести и при вводе исходной смеси либо в виде жидкости, либо в виде паров.
4.7.5.2. Расчет числа теоретических ступеней разделения с помощью фактора обогащения и формул Роуза
Формула (93) предназначена для расчета числа теоретических ступеней разделения по относительной летучести а и коэффициенту обогащения Fr*. Последний представляет собой эмпирическую величину, выведенную Куком на основе многочисленных опытов по ректификации:
Fr = an = xEixBJ (хЕхв^ (93)
где хв и — содержание легколетучего и труднолетучего компонентов в кубовой жидкости соответственно; хЕ^ и хЕг — содержание легколетучего и труднолетучего компонентов в дистилляте соответственно.
Величина Fr достигает 10000 при четком разделении, когда легколетучего компонента в дистилляте содержится 99% (мол.), а в кубе — 1 %:
Fr~ 99-99/1-1 =9801 » 10 000 (94)
* Коэффициент обогащения часто называют также коэффициентом разделения. — Прим. ред.
112
^В При обычном разделении результаты считаются удовлетво-рительными, если дистиллят содержит 90 %, а кубовая жидкость —
^В 1% легколетучего компонента; в этом случае величина Fr равна S 1000:
V Fr =90-99/10-1 = 891 « 1000 (95)
Отсюда получают для четкого разделения
п = 4,0/lg а теоретических ступеней (96)
Щ а для обычного разделения
п = 3,0/lg а теоретических ступеней (97)
Разумеется, коэффициент обогащения можно рассчитать также ^В и для других условий, например для концентрации дистиллята ^В 95% (мол.) и концентрации кубового остатка 1%:
Fr = 95-99/5-1 = 1850 (98)
а" = 1850 (99)
^В Тогда
К; п lg а = 1g 1850 (100)
и
в^Вг п = 3,3/lg <х теоретических ступеней разделения (101)
^В Результаты вычислений по уравнениям (96) и (97) приведены ^В в табл. 14, они использованы при построении диаграммы, пока-^В занной на рис. 73.
^В Роуз [154] усовершенствовал этот метод, исходя из того, что для большинства процессов разделения результаты очистки вполне удовлетворительны, если первые порции дистиллята, со-ставляющие 40% от всего его количества, содержат выше 95% (мол.) легколетучего компонента при условии, что компоненты исходной идеальной бинарной смеси находятся в равных коли-чествах. Дополнительно предполагается, что удерживающая спо-
М| Таблица 14.
^^В Число теоретических ступеней разделения, рассчитанное
^^В по эмпирическому коэффициенту обогащения
Относительная летучесть Разность температур кипения, °C Требуемое число теоретических ступеней разделения
при обычном разделении по кривой / на рнс. 73 и по формуле (97) при четком разделении по кривой 2, на рис. 73 н по формуле (96)
'3,0 30 5 8
(2,0 20 9 13
И ,5 10 17 22
|1,3 7 25 35
1,25 6 30 42
1,20 5 38 50
fl, 15 4 50 65
У,12 3 70 80
И ,07 2 100 150
113
Рис. 73.
Зависимость относительной летучести от разности температур кипения компонентов и зависимость необходимого числа теоретических ступеней разделения от а при различных коэффициентах обогащения (см. табл. 14): П1 — рассчитывается по формуле (97), Fr = 1000; п2 — по формуле (96), Fr = 10 000.
Рис. 74.
Зависимость числа теоретических ступеней разделения лмакс, «опт и лмин от относительной летучести данных по методу Роуза при обычной степени очистки (см. табл. 15).
собность колонны по жидкости настолько мала, что ею можно пренебречь, в силу чего справедливо равенство п — V. Кроме того, должно быть известно среднее значение относительной летучести, на основе которой можно получить максимальное пмакс, оптимальное пОПт и минимальное пмин числа теоретических ступеней разделения (рис. 74):
«макс = 3,6/1g а (102)
«опт =2,85/lga (103)
«мин = 2,3/1g а (104)
Таблица 15.
Числа теоретических ступеней разделения, определенные по методу Роуза
а 1g а пмнн попт пмакс
2,0 0,3010 8 9 12
1,5 0,1761 13 16 20
1,25 0,0696 24 29 37
1,10 0,0414 56 69 87
1,05 0,0212 108 134 170
1,03 0,0140 203
1,02 0,0085 335
114
использование формул (102—104),
Рис. 75.
Зависимость числа теоретических ступеней разделения от флегмового числа при обычной ректификации по данным Роуза.
1 — v = 3/2п [см. уравнение (105)1; n = 73/lg а, [см. (104)]; К=4 [см. (92)1; 2 — о = п [см. (106)]; п = = 2,85/lg а [см. (103)]; К = 3; 3 — о = 2/з [см. (107)]; п = 3,6/lg а [см. (102)]; К = 2,5.
Значения указанных выше величин приведены в табл. 15, которая поясняет а также графика на рис. 74.
При обычных условиях разделения принято исходить из
того, что низкое значение п можно компенсировать соответствующим приростом v, а более высокие значения п допускают меньшее значение v. Однако очень важно не выходить при этом из определенных пределов, так как слишком низкие значения п невозможно неограниченно компенсировать увеличением флегмы, а слишком высокие значения п не допускают неограниченного уменьшения флегмового числа v. В соответствии с этим по данным Роуза получаются следующие выражения для флегмовых чисел (рис. 75):
^макс = 3/2пмнн (Ю5)
^ОПТ — ^оПТ
^мин = 2/ЗпМаКс
(106)
(107)
4.7.5.3. Расчет минимального числа теоретических ступеней разделения по уравнению Фенске для идеальных смесей при v = оо
Относительная летучесть компонентов идеальной бинарной смеси может быть рассчитана по уравнению:
а = (у11у1)1(хх1х2) (108)
где у* и у*, — концентрация легколетучего и труднолетучего компонентов в парах соответственно; и х2 — концентрация легколетучего и труднолетучего компонентов в жидкости соответственно.
Если элементарное обогащение, достигнутое на одной теоретической ступени разделения, многократно увеличивается на п последовательно расположенных ступенях разделения, то суммарный эффект разделения можно выразить следующим образом:
ап = хе/^~хе) xb/Q хв)
Если прологарифмировать равенство (108а), то можно прийти к уравнению Фенске [155] для определения минимального числа
(108а)
115
Рис. 76.
Номограмма Мельпольдера—Хедингтона для определения минимального числа теоретических ступеней разделения идеальных смесей при v = оо.
теоретических ступеней разделения (см. разд. 5.1.4.2) при периодической ректификации идеальных смесей (концентрация указана в мольных процентах):
1М* 1 * * * V — хв)/хв(* — хв)1 ] П08(й
Линн------------1 (ЮоО)
или (см. разд. 5.1.4):
1 Г хе хв
«мин= ~'sT' In 1 — In “Т----
О I — Хс 1 — Хп с о
где 6 = а — 1.
По сравнению с методом Роуза расчет по этому уравнению можно проводить для различных значений хв и хЕ при условии
V = ОО.
Мельпольдер и Хедингтон [901 на основе уравнения (1086) разработали номограмму, по которой можно быстро и просто определить искомые величины (рис. 76).
На номограмме приведен пример расчета п при условии: хв = 15% (мол.), хЕ = 90%, а = 1,2. Находят точку пересечения вертикальной линии, соответствующей хв= 15% (мол.), с кривой хЕ = 90%. От точки пересечения проводят горизонтальную линию до линии а — 1,2. От новой точки пересечения опу-
116
число
(Ю9)
(ИО)
(1И)
скают перпендикуляр на нижнюю шкалу, указывающую теоретических ступеней разделения, и находят п = 22.
Используя обозначение
хЕ (ЮР — хв) хв (100 x£)
Рихтер [156] преобразовал уравнение Фенске к виду
п = (A71g а) — 1 или
К = (л+ l)lga
Методы, описанные в разд. 4.7.5.2. и 4.7.5.3, основаны на зависимости:
п— c/lga (112)
в которой величина с зависит от начальных условий разгонки. Однако применение этой формулы ограничено идеальными смесями.
Минимальное флегмовое число, необходимое для разгонки идеальной бинарной смеси, вычисляют по следующей формуле °мин = [(МХВ1) -a(X£2/XB,)l/(a- ’> (113>
Соответствующая этому уравнению номограмма Смоукера приведена в книге Харбанда [238].
4.7.5.4. Другие методы расчета числа теоретических ступеней разделения и их сравнительная оценка
Приведенными выше методами, очевидно, не исчерпывается все многообразие расчетных способов определения числа теоретических ступеней разделения. Пол [157] опубликовал относительно простые методы расчета числа теоретических ступеней разделения для периодической и непрерывной ректификации идеальных смесей при бесконечном и конечном флегмовом числе. Кроме того, следует сослаться на работу Штаге и Джуильфа [71 ], в которой, как и в книге Роуза с сотр. [1531, приведены другие точные и приближенные методы расчета. Цуидервег [158] предполагает метод, учитывающий общую удерживающую способность колонны (см. разд. 4.10.5) и размер промежуточной фракции в условиях периодической разгонки.
Все методы расчета числа теоретических ступеней разделения, описанные в разд. 4.7.5, сопоставлены между собой и с графическим методом Мак-Кэба и Тиле в табл. 16 на примере следующей задачи разделения: хв = 40% (мол.), хЕ = 98%, a = 2,4, А/кнп = = 30° С.
Из данных табл. 16 видно, что результаты расчетов по методам II и V, I и VII, а также VI при и = оо хорошо согласуются между собой.
117
Таблица 16.
Сравнение различных методов определения числа теоретических ступеней разделения
Метод расчета и п Примечание
I. Графический, Мак-Кэба и Тиле (данные табл. 12) 1,85 3,00 5,00 10,00 20,00 50,00 оо 10,0 7,0 6,0 5,5 5,0 5,0 4,5 При проведении периодической разгонки параметры хв и v изменяются, как показано ниже: хв, % (мол.) v 80 0,65 60 1,09 40 1,85 20 3,90 10 8,80 5 25,00
II. Диаграмма на рис. 72 5,0 8,0 (х£= 99%) (хЕ= 99,9%) хв = 50% (мол.)
III. Формула (92) 9,7 8,0 6,0 (макс.) (опт.) (мин.) хв и хЕ не учитываются
IV. Формула (96) Формула (97) 10,0 7,5 [х£ = 99% (мол.)] ]х£ = 90% (мол.)] Хв = 1% (мол.)
V. Формула (102) Формула (103) Формула (104) 8,8 7,1 6,0 9,0 7,1 5,8 Хв = 50% (мол.), первые порции дистиллята (40% от всего количества) имеют концентрацию выше 95% (мол.)
VI. Формула (108в) оо 3,7
VII. По Цуидер-вегу (11584 к гл. 4) 26 10 Соответствует концу разгонки при хв = = 5 % (мол.) по данным табл. 13
Методы II и IV дают лишь ориентировочное значение необходимого числа теоретических ступеней разделения. За основу берут максимальное значение п, а флегмовое число повышают по мере возрастания температуры в головке колонны.
4.8. ТЕОРИЯ ПРОЦЕССОВ, ПРОТЕКАЮЩИХ В НАСАДОЧНОЙ КОЛОННЕ
4.8.1. ПРОЦЕСС РАЗДЕЛЕНИЯ В НАСАДОЧНОЙ КОЛОННЕ
От реальной тарелки в тарельчатой колонне мы перешли к понятию теоретической ступени разделения. Что же происходит при ректификации в насадочных колоннах, содержащих упорядоченную
118
или неупорядоченную насадку? Вследствие противоточного движения орошающей жидкости и паров между'кубом и конденсатором и при наличии двух противоположных тенденций, а именно к установлению равновесия за счет диффузии в горизонтальном направлении и к смещению равновесия за счет извне созданного противотока, становится возможным разделение компонентов и обогащение паров легколетучим компонентом. Различия в траекториях движения паров и жидкости в насадочной и тарельчатой колоннах схематически показаны на рис. 77. Еще в разд. 4.2 мы познакомились с тем, что хаотически загруженный слой насадки очень сильно влияет на характер аэродинамического и гидродинамического режима в колонне.
Следует указать, что невозможно достаточно полно описать основные закономерности процесса разделения в насадочной колонне, если оперировать только такими величинами, как высота, эквивалентная теоретической ступени или единице переноса. Зицман [159] показал, что массообмен в насадочной колонне протекает тем интенсивнее, чем легче проникают компоненты из ядра одной фазы к границе раздела жидкость — газ и оттуда далее в ядро другой фазы. Поэтому необходимо принять во внимание два диффузионных сопротивления, а именно: 1ЕП при массопере-носе внутри паровой фазы и 1ЕЖ при массопереносе внутри жидкой фазы. Диффузионные сопротивления зависят от среднего пути переносимого вещества в соответствующей фазе, от степени перемешивания фазы в точках контакта между насадочными телами, от турбулентных завихрений и других факторов, которые уже были обсуждены в разд. 4.2. Соотношение между диффузионными сопротивлениями в газовой и жидкой фазах, экспериментально измеренные Зицманом для семи различных типов насадки, указаны в табл. 17. Из данных табл. 17 следует вывод, что вклад диффузионного сопротивления газовой фазы в общее сопротивление массопереносу при ректификации может составлять от 9 до 96%.
Оптимальную поверхность фазового контакта обеспечивают правильным подбором насадки, наиболее подходя щей’для заданного процесса разделения. Каналообразование J f можно ограничить, выбирая колонну с отноше-
J нием диаметра к поперечному размеру насадоч-Д ного тела, превышающим 10 : 1, применяя < > / аэродинамически выгодные насадочные тела, например седловидную насадку, и секционируя
„ 2 А Рис. 77.
* *, * * / у Схематическое изображение процесса массо- и теплообмена в на-
* 1/1 садочной колонне (а) и в тарельчатой колонне с двумя реальными
а б тарелками (^).
фф Траектории движения паров н жидкости; -> направление мас-__ сопереноса.
119
Таблица 17. Соотношение диффузионных сопротивлений в паровой Wn и жидкой №ж фазах прн различных типах насадки по данным Зицмана £159] Опыты проведены в колонне высотой 1 м и диаметром 30 мм. Эталонная смесь хлорбензол—этилбензол; наименьшая концентрация смеси — 20% (мол.) хлорбензола; относительная летучесть — 1,11. Температура дистиллята около 110 °C; турбулентное движение паров Наличие или отсутствие стадии предварительного затопления насадки <75 CU <75 <и <75 <75 Ф <75 W СХЗ СХЗ CU CU гад ЧДЧ гтжп с[ с(с[ ж ж
+ в ь + 10 1ГЭ СО ИЗ СО ИЗ СО 04 ь- lON 05 из 05 С"- -'t-ФСО 1ГЭСО1Л А А О О ООО 000 о о о* о о*
к h с Ю О О О U5 г-^ооиз счиэ—сч - — <о -Ф со о* о" о* —" о —• о" ь ь о* о 04 —
ВЭТС, см -ф СО О -Ф О ИЗ ИЗ 04 СО 00 СО СОСО 0 — 0 10 Т-Г10 coco 04 СО
Нагрузка, мл/(см2-ч) OO ООО OOiO О О У2 ю 0 r-. r-. rococo t*--ф 04 b- —00 05 b- —« — — — 04 — — — — 04 — — —
£ * ВСЮ > * О О Я я 04 04 000 Tf -ф 50 OO 00 10 t"- io ic co 05^05 А A о" о* о* о о" oo"o* о” о" o' 0 0
Материал насадки 1 Стекло » » » I Керамика 1 » » Стекло матированное Сталь V4A » им.
Размер насадки, мм о со ОС0 Тр rf тр тр тр 1Л1Л тр тр а XX XXX XXX x-х XX XX сх C0-C0 тр тр тр xt ’Cf Ю 1Л тр rf 5 S
Тип насадки Кольца Рашига Кольца Рашига Седла Берля Шары Проволочные кольца од-новитковые То же Проволочные кольца двухвитковые ♦ Измерено в трубе диа
насадку, благодаря чему орошающая жидкость чаще собирается и перераспределяется на нижележащих слоях насадки. Влияние каналообразования на эффективность работы колонны наглядно показано на рис. 78. Опыты проводили со смесью н-гептан—метилциклогексан. Очевидно, незначительное канало-образование в колоннах с 10—20 теоретическими ступенями разделения не очень заметно сказывается на эффективности процесса разделения; в колоннах же с 100 теоретическими ступенями'разде-ления каналообразование сильно ухудшает эффективность колонны. Для смесей с высокой относительной летучестью компонентов (например, при а = 1,07) эффект каналообразования незначителен; если же а < 1,07, то эффективность разделения заметно ухудшается [1601.
Несмотря на то, что чаще всего первоначальное’распределение жидкости является равномерным, нередко бывает, что насадка смачивается неравномерно и, кроме того, отдельные участки пленки стягиваются в струйки. Штуке [161] тщательно проанализировал вопросы смачивания насадки и исследовал, каким образом тип насадки, свойства жидкости, нагрузка колонны и рабочее давление процесса ректификации (и тем самым температура процесса) оказывают влияние на неравномерность образования пленки. В результате он пришел к следующим выводам (см. также разд. 4.2).
1. Струйчатое обтекание насадки имеет место главным образом при малых нагрузках; большее количество жидкости относительно лучше распределяется на поверхности насадки. 2) Чем больше плотность жидкости и чем меньше ее вязкость, тем с большей вероятностью даже на полностью смоченной насадке (под действием силы тяжести) формируется струйное течение. 3) Чем меньше поверхностное натяжение, тем легче при малых нагрузках смачивается твердое тело. 4) Высокая разделяющая способность, которую следует ожидать при использовании мелкой насадки, может быть достигнута только при достаточно равномерном смачивании всей насадки. При более высоких нагрузках на разделяющую способность отрицательно влияет возрастающая неравномерность пленки. 5) Основные закономерности, связанные с влиянием
Рис. 78. Влияние каналообразования на разделяющую способность колонны.
, F — коэффициент уменьшения разделяющей спо-
Каналоооразооание, °fo собности; п — число теоретических ступеней разделения; у — концентрация жидкости в головной ___ части колонны.
121
рабочего давления при ректификации, до сих пор не установлены. Некоторые исследователи обнаружили наличие оптимального давления при ректификации, другие же отмечали отсутствие зависимости разделяющей способности колонны от давления.
Интерес представляет также динамическое поведение насадочных колонн. Хайсе, Хиллер и Вагнер [162] исследовали поведение насадочной колонны во времени при ректификации тройной почти идеальной смеси этилацетат — бензол — трихлорэтилен при ступенчатом изменении соотношения расходов жидкости и паров.
Однако этими факторами не исчерпывается сложность процессов, протекающих в насадочной колонне. Крель (см. разд. 4.2) указал на важность состояния межфазной поверхности и перечислил факторы, определяющие процесс ректификации в насадочной колонне:
1) свойства разделяемой смеси; это — относительная летучесть, характер фазового равновесия, состав смеси, взаимная растворимость компонентов в жидкой смеси, плотность паров и орошающей жидкости, поверхностное натяжение на границе раздела паров и жидкости, вязкость паров и жидкости, удельная теплоемкость компонентов, энтальпия испарения компонентов, смачиваемость; 2) характеристика колонны (диаметр, высота слоя насадки, способ укладки насадочных тел); 3) характеристика насадки (высота, диаметр, толщина стенок насадочных тел, свойства поверхности, свободный объем, конструкционный материал и форма насадочных тел); 4) факторы, обусловленные режимом работы: это — способ и характер подачи и распределения орошающей жидкости, удерживающая способность колонны или отношение количества удерживаемой жидкости к количеству загруженной жидкости, рабочее давление, потеря напора (перепад давления), предельная скорость.
Из этих данных следует, что ректификация в насадочной колонне представляет собой сложный процесс и исследование влияния отдельных факторов на разделение часто сопряжено с большими экспериментальными трудностями. Возможно, это и является объяснением того, почему в литературе часто встречаются противоречивые сведения по одним и тем же вопросам.
4.8.2. ОПРЕДЕЛЕНИЕ ЧИСЛА ЕДИНИЦ ПЕРЕНОСА
Количественная характеристика процессов, протекающих в насадочной колонне, по- указанным выше причинам может быть получена лишь полуэмпирически на основе теории подобия. Чильтон и Кольборн [163] для оценки эффективности массообмена в насадочных колоннах ввели понятие числа единиц переноса (ЧЕП). Это понятие учитывает тот факт, что в насадочной колонне в противоположность тарельчатой колонне массо- и теплообмен осуществляется в виде бесконечно малых элементарных ступеней
122
Рис. 79-
Сравнительная характеристика теоретической ступени разделения и единицы переноса
— — — — единица переноса, теоретическая ступень разделения; а — рабочая линия (2) круче, чем кривая равновесия (/) (Ду уменьшается); б — рабочая линия (2) параллельна кривой равновесия (?) (Ду не изменяется).
разделения. Для теплопередачи движущей силой является температурный перепад, а для массопередачи — разность парциальных давлений или концентраций переносимого вещества. ЧЕП — это безразмерная величина, которая определяется по разности концентраций между соответствующими точками кривой равновесия и рабочей линии [73].
Рассмотрим часть диаграммы для графического определения числа теоретических ступеней разделения по методу Мак-Кэба и Тиле (рис. 79). В тарельчатой колонне между жидкостью состава ylt находящейся на тарелке, и поднимающимися парами устанавливается термодинамическое равновесие *. Концентрация паров, покидающих тарелку, равна у*. Такую же концентрацию (г/2) имеет жидкость, находящаяся на вышележащей тарелке **. В паровом пространстве между тарелками (а следовательно, между точками у± и у2) массообмен практически не происходит.
Иначе протекает процесс в насадочной колонне. Изменение
концентрации здесь в каждом слое между сечениями г/х и у2 пропорционально у* — у. Только когда кривая равновесия и рабочая линии расположены параллельно (см. рис. 796), имеет место случай, при котором ЧЕП и число теоретических ступеней разделения п совпадают, поскольку в рассматриваемой области концентраций разность у* — у остается постоянной. Такая зависимость наблюдается при ректификации идеальных смесей, компоненты которых имеют мало различающиеся температуры кипения. По-> добные смеси используют главным образом при испытании колонн.
Вообще,’ по'обогащающему эффекту единица переноса идентична теоретической ступени разделения, рассчитанной для разности концентраций у*—у, являющейся средней между значением у*—у и соответствующей разностью концентраций уь—уь в конце единицы переноса [71].
Если движущая сила у увеличивается по мере продвижения
потока паров в колонне, то для одного и того же процесса разделения единиц переноса требуется меньше, чем теоретических ступеней разделения. При уменьшении у (см. рис. 79а) имеет место другая зависимость. Если смеси идеальны или почти идеальны,
* Термодинамическое равновесие имеет место между жидкостью, стекающей с тарелки, и парами, уходящими с тарелки (конечно, при идеальной работе тарелки). — Прим. ред.
' ** Т. е. жидкость, стекающая с вышележащей тарелки. — Прим. ред.
123
Таблица 18.
Сравнение числа теоретических ступеней разделений и числа единиц переноса
а Уи % (мол.) (соответству-ет хв) У г. % (МОЛ.) (соответству-те л-£) ЧЕП по урав нению (120) м по уравнению (1086)
1,08 10 99 119,5 . 119,5
1,08 50 99 61,0 59,8
2,50 10 99 10,7 10,0
2,50 50 99 6,9 5,0
то между ЧЕП и числом теоретических ступеней разделения нет существенных различий. Наоборот, эти различия весьма значительны и должньГучитываться для неидеальных смесей и смесей с высокой относительной летучестью, как это следует из данных, приведенных в табл. 18 [164].
При условии *, что все сопротивление массопереносу сосредоточено в паровой фазе, пол чи ; согласно определению 1163] уравнение
у* л
ЧЕП=| -Д- (114)
Vi аналогично уравнению ке л
НА <"5>
*в
По аналогии с высотой, эквивалентной теоретической ступени разделения
Высота колонны РЭГС =--------------- см
п
(ПЕТР—height equivalent to a theoretical plate) (116)
получают высоту единицы переноса
ВЕП =-------гГпТт----см (HTU—height of a transfer unit) (117)
4hl 1
Высота единицы переноса является мерой сопротивления массо-перепосу. Она слабо зависит от скорости газа **.
Мац [73] отмечает, что процессы массо- и теплообмена в отличие от термодинамически обратимых процессов протекают не
* Формулы (114) и (115) справедливы и без принятия такого допущения. — Прим. ред.
** Иногда влиянге скорости газа на ВЕП (HTU) довольно ощутимо (см., например, Шафрановсхий А. В., Ручинский В. Р. Теор. основы хим. техи., 1967, т. I, № 1, с. 111 или 1970, т. IV, № 1, с. 106). — Прим. ред.
121
бесконечно медленно, а в измеримое время и с конечной скоростью. Он предлагает использовать для решения интеграла (114) выражение
(118)
где аН — характеристическая величина для поверхности массообмена; а — удельная поверхность массообмена, м2/м3; И — высота колонны, м; K/w — коэффициент скорости; К — скорость массопередачи, м/ч; ш — скорость потока паров, м/с.
Исходя из этого выражения, Мац вывел уравнения, с помощью которых легко рассчитать ЧЕП при бесконечном флегмовом числе
ЧЕП = —Ц- 1п -* ~У1 + In (119)
т — 1 1 — т — 1 г/1
; где т — тангенс угла, который образует с осью абсцисс касательную к кривой равновесия в рассматриваемой точке.
В книге Маца 173 ] даны примеры расчета ЧЕП как для v = оо, так и для конечного флегмового числа, превышающего минималь-[ ное флегмовое число.
1 Для идеальных смесей и бесконечного флегмового числа урав-I нение (114) поддается интегрированию; при этом получают
I 2 303 хР (1 — xR) 1— Хд
I ЧЕП = 1g -Ц-.---------Ц + 2,303 1g --® (120)
I а —1 xB(l-%V Х~хе
| Для конечного флегмового числа рассчитать ЧЕП по такому I же методу значительно сложнее [71]. Чильтон и Кольборн [163] I описали приближенный графический метод расчета ЧЕП, который К представляет собой дальнейшее развитие метода Мак-Кэба и | Тиле. Он пригоден для всех смесей, для которых известна кривая К равновесия. При этом в рассматриваемом интервале концентраций рабочая линия процесса ректификации для укрепляющей части Е колонны не должна подходить слишком близко к кривой равно-Г весия. Задача сводится к тому, чтобы подобрать подходящее среднее значение движущей силы, совпадающее с величиной обогащения, соответствующей единице переноса. На диаграмме равновесия в обычном порядке вычерчивают рабочую линию и Е через точку хв проводят вертикальную “линию, которая пересе-К кает рабочую линию в точке ylt а кривую равновесия — в точке у’. На рис. 79 такое построение схематически пояснено для отдель-К ного участка диаграммы равновесия; при этом на рис. 79а кривая К равновесия проходит менее круто, чем рабочая линия, а на рис. К 796 линии параллельны между собой.
К Как показано на рис. 79а, одна единица переноса дает обога-Вщение у'ь — yi, которое является средним значением для разно-Встей уь—уь и у’—уг. Одна теоретическая ступень разделения дает В обогащение t/2—уг.
к Единицы переноса можно строить в виде ступенек на диаграмме В-Мак-Кэба и Тиле, но при этом надо следить за тем, чтобы степень
125
Рис. 80.
Энтальпийная диаграмма смеси этанол—вода при 760 мм рт. ст.
обогащения уь—уъ соответствующая единице переноса, была равна среднему значению для у!—у и у*ъ—уъ-Точку уь следует наметить на рабочей линии таким образом, чтобы соблюдалось условие
у'ь - Ух = 0,5 [(^ - j/i) + (уь - у&)]
(121)
Построения ступенек, соответствующих единицам переноса, прово-
дят методом проб и ошибок, затраты времени на это не очень велики. Если кривая равновесия и рабочая линия являются прямыми (хотя и не параллельными), то точность метода можно повысить, используя среднее логарифмическое значение для у*—у и уь—уь-Однако в большинстве случаев погрешность, вносимая спрямлением одной или обеих кривых или использованием среднего арифметического вместо среднего логарифмического, не существенна. Чтобы наглядно представить взаимосвязь между числом теоретических ступеней разделения и числом единиц переноса, выше был намеренно рассмотрен наиболее простой из графических методов. Другие точные и приближенные методы описаны в работах Штате и Джуилфса [71 ]. В весьма интересной статье, содержащей математическое описание понятий п и ЧЕП, Аркенбут и Смит [166] показали, что согласно их расчетам нередко ВЭТС оказывается более удобной единицей длины колонны, чем ВЕП.
Укажем еще на два метода расчета числа теоретических ступеней, которые не зависят от конструктивных особенностей ко-
лонны и поэтому могут применяться как для тарельчатых, так и для насадочных колонн, а также для колонн с другими видами насадок. Меркель [167] разработал метод, в соответствии с которым процессы противоточного массообмена представляют в энтальпийной диаграмме Н—х—у. По ней находят изменение состава жидкости и пара, их количества, а также подводимую и отводимую теплоту (рис. 80). К сожалению, получено незначительное число энтальпийных диаграмм, и применение этого метода ограничивается небольшим числом смесей. Некоторые сведения по этому методу можно найти в литературе [73, 75, 103]. Биттер [261 ] дал сводку различных приемов вычислений для определения числа теоретических ступеней разделения при ректификации бинарных смесей; эти приемы основаны только на уравнениях рабочих линий и служат основой для графических методов решения с применением энтальпийной диаграммы.
126
4.8.3. ИНТЕНСИВНОСТЬ ПРОТИВОТОЧНОГО
МАССООБМЕНА И ВРЕМЯ, НЕОБХОДИМОЕ
ДЛЯ РАЗГОНКИ
Удерживающая способность (УС) колонны, т. е. удерживание некоторого количества жидкости в колонне, является необходимым условием для проведения ректификации (см. разд. 4.10.5). УС определяет время, требуемое для проведения процесса (см. разд. 4.10.5). Если число теоретических ступеней разделения и флегмовое число увеличиваются пропорционально 1/lg а, то время разгонки, обусловленное соотношением количеств удерживаемой и загруженной жидкости, возрастает в квадрате. Таким образом, время разгонки однозначно характеризует степень трудности разделения. Если составить отношение
Число теоретических ступеней • Расход флегмы
УС колонны (122)
для колонн 1 и 2, исходя из условий их работы, то отношение времен tn : /2, необходимых для проведения одного и того же процесса разделения в этих колоннах, будет соответствовать отношению факторов интенсивности противоточного массообмена /2 : /j при условии, что во всем остальном колонны в одинаковой мере отвечают поставленной задаче ректификации. Величина I выражается соотношением [168]:
/ = tiRjH (с'1) (123)
Фактор / по единицам измерения является обратной величиной времени; эти же единицы измерения имеет частота. Фактор интенсивности не зависит от размеров аппаратуры и может быть вычислен для колонны любого объема, любой высоты и любого поперечного сечения.
Составной частью времени разгонки является также пусковое время tn, в течение которого не следует еще отбирать дистиллят. Отбор начинают после достижения достаточного обогащения легколетучим компонентом жидкости в головке колонны. Пусковое время рассчитывают по уравнению Вебера [170):
(ra4-l)2/(2Z) (124)
Время, необходимое для введения колонны в режим, увеличивается примерно пропорционально квадрату числа теоретических ступеней разделения, т. е. высоты колонны [152, 169]. Пусковое время колонны будет более подробно рассмотрено в разделе, посвященном получению стабильных изотопов (см. разд. 5.1.4); пусковому времени придается особое значение при п > 100.
Приняв за основу противоточного устройства модель Куна [3], состоящую иЗ двух плоскопараллельных вертикальных стенок, £ебер [170] рассчитал фактор интенсивности для эталонной смеси н-гептан — метилциклогексан; при этом он исходил из максимального числа теоретических ступеней (3,54 на 1 см высоты колонны) и оптимальной скорости паров 0,1525 см/с (табл. 19).
127
Таблица 19.
Значения фактора интенсивности при различных числах теоретических ступеней разделения и скоростях потока паров
Ректификации эталонной смеси м-гептан — метилциклогексан в противоточном устройстве согласно модели Куна
Скорость потока паров, см/с Число теоретических ступеней разделения иа 1 см высоты колонны Фактор интенсивности /•10», с“‘
0,1525 3,54 260
0,305 2,83 367
0,4575 2,12 381
0,610 1,66 375
1,525 0,70 322
7,62 0,14 217
Из данных табл. 19 видно, что максимальное число теоретических ступеней разделения (3,54 на 1 см) не соответствует максимальному фактору интенсивности, равному 381 при числе теоретических ступеней 2,12 на 1 см и при скорости паров 0,4575 см/с. Далее можно видеть, что после максимального значения 381 фактор интенсивности снова уменьшается, но намного медленнее, чем следовало бы ожидать в связи с сильно уменьшившимся числом теоретических ступеней разделения. Таким образом, для получения высоких значений фактора интенсивности не имеет смысла работать при низких скоростях паров. При повышенных нагрузках движение паров становится турбулентным, что оказывает благоприятное действие на массопередачу аналогично увеличению коэффициента диффузии. С возрастанием нагрузки колонны выше оптимального значения количество орошающей жидкости увеличивается, и под действием поднимающихся паров происходит подвисание жидкости в колонне. Удерживающая способность возрастает в степени, превышающей 2, вследствие чего фактор интенсивности снижается.
Сравнивая, по данным Вебера 1170], разделяющую способность колонн различного диаметра с насадкой в виде дисков из проволочной сетки при нагрузке, составляющей 2/3 от максимального значения, получим результаты, приведенные в табл. 20.
Таблица 20.
Условия ректификации в колонне с орошаемыми сетчатыми дисками при оптимальной нагрузке, составляющей а/3 от максимального значения
Внутренний диаметр, мм Нагрузка, г/с Число теоретических ступеней разделения на 1 см высоты колоииы УС, г/см Фактор интенсивности /•10», с-1
17,0 0,139 0,460 0,239 268
25,4 0,333 0,370 0,228 537
50,8 1,530 0,144 0,328 670
128
Таблица 21.
Условия ректификации для заданной степени разделения
при постоинном числе теоретических ступеней и постоянном отношении УС к количеству загруженной жидкости
Загрузка, кг Диаметр колонны, мм Высота колонны, м Время разгонки, ч
1,0 17,0 1,о 100
1,18 25,4 1,24 49
4,40 50,8 3,20 40
Если те же данные пересчитать на постоянное число теоретических ступеней и постоянное отношение УС к количеству загруженной жидкости, то для заданных условий разделения будут получены значения времени разгонки, приведенные в табл. 21.
Последняя колонна (см. табл. 21) является наилучшей, поскольку она работает наиболее интенсивно.
Чтобы показать порядок значения фактора интенсивности, приведем еще три максимальные значения этого фактора, полученные Вебером [170] при нагрузке колонны 2/3 от максимальной:
Насадочная колонна с насадкой из стеклянных спиралей диаметром 0,3 см..................165-10~3, с-1
Насадочная колонна с матированными кольцами
Рашига 2,4X2,4 мм....................... 370-10"3, с*1
Тарельчатая колонна Брууна ...............140-10~3, с-1
Таким образом, фактор интенсивности является параметром, который легко рассчитать. Его можно использовать для сравнения различных типов колонн, поскольку он представляет собой величину, которую можно определить даже для весьма сложных процессов ректификации.
Практически к тем же выводам пришел Коллинг [171], который исследовал производительность различных насадок и сформулировал определение производительности как количества дистил-
Таблица 22.
< Условия ректификации для получения дистиллята, содержащего 90% (мол.)
i я-гептана
Таблица 23.
Условия ректификации при постоянной скорости отбора дистиллята (40 мл/ч)
Флегмовое число Нагрузка колоииы, мл/(см*.ч) Концентрация дистиллята, % (мол.)
10 70 71,5
15 100 78,5
25 160 84,5
50 300 88
' 5 Э. Крель
129
Рис. 81.
Зависимость степени чистоты дистиллята от числа теоретических ступеней разделения и флегмового числа для исходной смеси м-гептан—метилцикло-гексаи [(50% (мол.)]
*£ — содержание «-гептана в дистилляте.
Рис. 82.
Зависимость степени чистоты дистиллята от скорости отбора дистиллята при различных флегмовых числах для исходной смеси «-гептан—метил-циклогексаи [50% (мол.)].
Цифры на кривых обозначают нагрузку в мл/(см2, ч).
лята определенной степени чистоты, полученного в единицу вре-мени. На рис. 81 показана зависимость степени чистоты дистиллята от числа теоретических ступеней и флегмового числа для смеси 50% (мол.) я-гептана и 50% метилциклогексана. Если нужно получить дистиллят концентрацией 90% (мол.), то рекомендуется проводить разделение в условиях, указанных в табл. 22.
Таким образом, можно получить в два раза больше дистиллята той же чистоты, если нагрузку увеличить в 4,6 раз, а флегмовое
число удвоить.
Если же исходить из постоянного количества отбираемого дистиллята, то в соответствии с рис. 82 можно получить значения, указанные в табл. 23.
Результаты исследования фактора интенсивности указали на необходимость пересмотра прежнего мнения о том, что скорость потока паров, а следовательно, и нагрузку ко-
Рис. 83.
Зависимость ВЭТС от скорости паров для иасадки из колец Рашига (/) и седел (2) размером 25 мм.
130
лонны следует поддерживать возможно более низкими для достижения максимальной разделяющей способности. Чем выше нагрузка колонны, тем лучше результаты разделения при одинаковой скорости отбора дистиллята. Не следует забывать и о том факте, что большее количество жидкости относительно легче распределить по насадке, в результате чего достигается лучшее ее смачивание. Это действительно до определенного предела, после чего разделяющая способность снова уменьшается. Такая зависимость становится особенно очевидной, если построить графическую зависимость ВЭТС от скорости потока паров [103]. Например, для колец Ра-шига диаметром 25 мм ВЭТС остается постоянной при скоростях парового потока от 0,2 до 1,0 м/с, т. е. при пятикратном увеличении нагрузки (рис. 83). Седловидная насадка имеет максимальную ВЭТС при линейных скоростях 0,08 и 1,6 м/с. Как видно из диаграммы, работать в интервале между этими двумя скоростями паровых потоков нецелесообразно, так как при этом разделяющая способность снижается.
4.9. ОПРЕДЕЛЕНИЕ ЧИСЛА ТЕОРЕТИЧЕСКИХ СТУПЕНЕЙ РАЗДЕЛЕНИЯ ПРИ ПЕРИОДИЧЕСКОЙ И НЕПРЕРЫВНОЙ
РЕКТИФИКАЦИИ МНОГОКОМПОНЕНТНЫХ СМЕСЕЙ
Под многокомпонентной смесью подразумевают систему с числом компонентов больше двух. При этом различают смеси с точно определенными компонентами и сложные смеси, состоящие из очень большого числа компонентов, часто с весьма близкими температурами кипения. Такими сложными смесями являются, например, нефть и бензин. Как правило, для подобных смесей определяют лишь кривую разгонки. В зависимости от требуемой точности для разделения применяют либо прибор Энглера (см. разд. 7.2), либо эффективную колонну, с помощью которой легко выделить близкокипящие фракции.
Для тройных смесей условия ректификации могут быть установлены с помощью треугольной системы координат [72—74 , 78]. Корт [172] предлагает метод расчета минимальных флегмовых чисел при непрерывной ректификации трех- и четырехкомпонентных смесей, как идеальных, так и азеотропных.
Хаузену [173] удалось решить дифференциальное уравнение для идеальной трехкомпонентной смеси, а Вирн и Тийссен [174] разработали графический метод определения числа теоретических ступеней, основанный на приближенном представлении многокомпонентных смесей в виде бинарных. На эту возможность указал еще ранее Львов [175 ], который в своей теоретической работе рассматривал процесс ректификации любой многокомпонентной смеси как процесс разделения, состоящий из совокупности параллельно
б* 131
Выход дисггщппята, г
Рис. 84.
Диаграмма ректификации смеси жирных кислот С4—Св нормального строения при остаточном давлении 20 мм рт. ст.;
---температура кипения при 20 мм рт. ст.;----кислотное число (к.ч.);
50
---------показатель преломления пр.
Горизонтальные отрезки у кривых указывают на теоретическое значение физических величин.
протекающих процессов разделения бинарных смесей. Таким образом, задача сводится к тому, чтобы многокомпонентную смесь представить в виде бинарной, содержащей «ключевые» компоненты, которые труднее всего разделить (следовательно, смесь с наименьшей относительной летучестью компонентов), и в основу расчетов положить кривую равновесия для этой смеси. Для лабораторной практики применительно к разделению идеальных смесей метод «ключевых» компонентов является наиболее рациональным, к тому же приближенные расчеты можно проверить с помощью сравнительно простых пробных разгонок.
Мостафа [175а] представляет многокомпонентную смесь в виде эквивалентных бинарных смесей, обладающих различной относительной летучестью, и определяет число теоретических ступеней разделения с помощью графического метода Мак-Кэба и Тиле. Если компонентов много, то можно выбрать один «ключевой» компонент, относительная летучесть которого является средним значением для Относительных летучестей всех компонентов.
Шьен [1756] приводит точный метод расчета минимального числа теоретических ступеней разделения для ректификации многокомпонентной смеси. Приближенный метод разработан Серовым с сотр. [175в].
132
Непрерывный способ ректификации имеет то преимущество, что условия процесса остаются неизменными. Кроме того, при этом имеется возможность в качестве головного или кубового продукта получать как отдельные компоненты, так и смесь нескольких компонентов. Если необходимо получить х различных фракций, то для разделения требуется (х—1) колонн. Возьмем в качестве примера предварительно отогнанную смесь жирных кислот С4—С8, для которой на рис. 84 показана диаграмма разгонки [176]. Очевидно, для непрерывного разделения этой смеси на отдельные фракции (по числу атомов углерода) потребовалось бы четыре колонны. Опыт показывает, что лучше работать, отбирая отдельные фракции по возможности в виде головного продукта (рис. 85); при этом фракции получаются в виде прозрачного и бесцветного дистиллята. Однако можно проводить ректификацию и по схеме, изображенной на рис. 86.
Сначала рассмотрим колонну IV на рис. 85. В этой колонне разделяется бинарная смесь С7—С8. Условия ректификации для этого случая рассчитать весьма просто. В колонну III вводят тройную смесь и получают один компонент в головном продукте и два компонента в кубовом продукте. Сравнивая кривые равновесия смесей С6—С7 и С,—С8, выбирают для расчетов ту кривую равновесия, которой соответствует более низкое значение относительной летучести компонентов а.
В колонну II поступает смесь из четырех компонентов. Снова находят относительную летучесть компонентов каждой из бинарных смесей С5—С6, С6—С7 и С,—С8 и для расчетов выбирают ту кривую равновесия, которой соответствует наименьшее значение а. Разумеется, следует иметь в виду, что в процессе массообмена компоненты С7 и С8 почти не содержатся в паровой фазе. Эти компоненты можно рассматривать скорее как балластное или
Рис. 85.
Схема непрерывной ректификации смеси жирных кислот С4—С8 нормального строения с отбором компонентов С4—С7 в дистилляте и компонента С8 в кубе.
Рис. 86.
Схема непрерывной ректификации смеси жирных кислот С4—С8 нормального строения с отбором компонентов С4, С6 и С7 в дистилляте н компонентов Св и С8 в кубе.
133
«вытесняющее» вещество. Поэтому рекомендуется внести поправку в значение а с учетом ожидаемой температуры, состава кубового продукта и его летучести. Если температура кубовой жидкости сравнительно высока, то из имеющихся значений а для трех бинарных смесей, выбирают наименьшее значение а. Если температура куба сравнительно низка, то за основу достаточно взять наименьшее значение а для двух первых из названных смесей. Ввиду постоянного присутствия в работающей колонне определенного количества жидкости и очень незначительного содержания компонента СБ рекомендуется исходить при расчете из смеси С6—С7, а остальные компоненты рассматривать как балластное вещество.
Аналогичные условия имеют место в колонне I. По отношению к температуре головного продукта температура в кубе является сравнительно высокой, поэтому из значений относительной летучести для смесей С4—С6 и С5—С6 выбирают наименьшее.
Если компоненты смеси принадлежат к одному гомологическому ряду, как в рассмотренном примере, то значения а снижаются по мере увеличения числа атомов в молекуле компонентов, образующих смесь:
2,66
2,50
2,10
2,13
Таким образом, с увеличением молекулярной массы компонентов разделение становится все более трудным.
В качестве головного и кубового продуктов можно отбирать и смеси различных компонентов, как показано на рис. 86 для колонны I. В дистилляте получают фракцию С4—С6, а в кубе — фракцию С7—С8; следовательно, граница раздела смеси лежит между компонентами С6 и С7. В этом случае прежде всего выбирают кривые равновесия для крайних пар компонентов, т. е. для С4—Сй и С,—С8, и рассчитывают число теоретических ступеней разделения и другие условия ректификации, необходимые для обогащения смеси до хЕ = 95% (мол.) при непрерывном режиме работы колонны. Для двух полученных чисел ступеней вычисляют среднее значение. За основу можно взять также кривую равновесия для смеси С6—С7, поскольку количественно фракция С4—С6 преобладает, а компонент С8 почти не участвует в массообмене.
Исследования Фальдикса и Штаге [176 а] позволили найти оптимальные схемы системы, состоящей из двух ректификационных колонн. Мюллер [1766] разработал различные схемы соединений колонн с боковым отбором продукта из колонн при разделении многокомпонентных смесей.
Биллет [176в] отмечает, что отклонения от оптимальных условий ректификации могут привести к значительному увеличению производственных затрат. Он рассматривает различные схемы 134
соединения основных и вспомогательных колонн, а также вопросы рационального использования обогащающего эффекта дефлегмации при дистилляции и ректификации. Эти данные следует учитывать также при работе в лаборатории и на опытных установках.
Разработаны многочисленные методы расчета параметров процесса ректификации для идеальных многокомпонентных смесей, которые подробно изложены Торманном [177], а также Эллисом и Фрешуотером [178]. Особо следует отметить приближенную формулу Кольборна [179] и Андервуда [180], позволяющую определять минимальные флегмовые числа. Простой приближенный метод расчета минимального числа теоретических ступеней разделения при v = оо принадлежит Фенске [181], который с целью упрощения рассматривает многокомпонентную смесь как бинарную. При этом условно принимается, что в смеси преимущественно содержатся «ключевые» компоненты, температуры кипения которых образуют постепенно возрастающую последовательность, а разности температур кипения для различных соседних компонентов смеси примерно одинаковы. Если через х1 обозначить содержание низкокипящего «ключевого» компонента, содержание которого в кубовом продукте невелико, а через х2 — содержание высоко-кипящего «ключевого» компонента, содержание которого невелико в головном продукте, то уравнение Андервуда—Фенске для расчета минимального числа теоретических ступеней разделения будет иметь вид:
(ДгАзг) (х2в/х1в)
«мин =--------------- (1 20)
jg а12
где а12 — отношение давлений паров «ключевых» компонентов.
Хаберт [182] разработал метод расчета минимального числа Г теоретических ступеней разделения, согласно которому давление паров каждого компонента смеси относят к давлению паров самого 1 летучего компонента. В своем превосходном обзоре Бруийн [1831 ! рассматривает теорию ректификации многокомпонентных смесей I при минимальном флегмовом числе.
| Для точного расчета параметров колонн и условий ректифика-| ции многокомпонентных смесей необходимо применение электрон-| ных вычислительных машин. При этом проблемы моделирования I и оптимизации поддаются решению с минимальными затратами I времени. В разд. 4.15 обсуждены возможности применения цифро- вой и аналоговой вычислительной техники для решения задач Е разделения.
I 4.10. АППАРАТУРНО-ТЕХНОЛОГИЧЕСКИЕ | ПАРАМЕТРЫ ТАРЕЛЬЧАТЫХ
Ви НАСАДОЧНЫХ КОЛОНН. ИСПЫТАНИЯ колонн
До сих пор были рассмотрены лишь методы, позволяющие рас-
считать необходимое для разделения число теоретических ступеней или число единиц переноса. Испытания колонны с применением В 135
определенной эталонной смеси проводят, чтобы установить, скольким теоретическим ступеням или единицам переноса эквивалентна колонна. Такие испытания необходимы, так как отсутствуют подходящие методы, позволяющие рассчитать эффективность разделения в лабораторной колонне. При испытаниях в куб загружают эталонную смесь известного состава и доводят ее до кипения. Обычно испытание проводят при бесконечном флегмовом числе, т. е. без отбора дистиллята, так как это условие наилучшим образом поддается воспроизведению. Из головки колонны и из куба пробы отбирают по каплям, благодаря чему стационарный режим в колонне не нарушается. Анализом проб устанавливают достигнутое обогащение, соответствующее разнице в концентрациях кубовой жидкости и дистиллята. Методические вопросы испытания колонн подробно рассмотрены Андервудом [ 184 ] в одной из его ранних работ.
Определив число теоретических ступеней разделения, обычно обнаруживают, что это число меньше числа реальных тарелок. Следовательно, реальная тарелка работает не идеально, и поэтому работу тарелки оценивают по отношению найденного числа теоретических ступеней разделения к числу реальных тарелок. Это отношение называют средним относительным обогащением или средним коэффициентом полезного действия тарелки (по Киршбауму) [103 ]
Sep = П/Пр (126)
где Sep—средний к. п. д. тарелки; п — число теоретических ступеней разделения; Лр — число реальных тарелок.
Для насадочной колонны определяют ВЭТС
Высота колонны
ВЭТС =-----------------см (127)
Исследования Хюбнера и Шлюндера [103а] показывают, что средний коэффициент полезного действия тарелки (или к. п. д. Мерфри) можно с достаточной точностью рассчитать, используя модель турбулентной диффузии, при условии, что известны коэффициенты массоотдачи или турбулентной диффузии в газовой и жидкой фазах. Фогельпол и Серетто [1036] приводят сведения о значениях к. п. д. тарелки при ректификации тройных смесей.
Если высота, эквивалентная теоретической ступени разделения, совпадает с расстоянием между соседними реальными тарелками колонны, то это свидетельствует об идеальной работе реальной тарелки. В насадочной колонне высота теоретической ступени разделения соответствует ВЭТС.
Число теоретических ступеней разделения, установленное при испытании колонны, определяется кривой равновесия эталонной смеси и зависит от соблюдаемых условий ректификации и, разумеется, от размеров колонны. Если завод-изготовитель указывает для ректификационной колонны лишь число теоретических ступеней разделения без каких-либо дополнительных пояснений, то подобная информация практически бесполезна. Только при 136
Таблица 24.
Аппаратурно-технологические параметры, необходимые для оценки разделяющей способности колонн (в дополнение к числу теоретических ступеней)
Насадочные колонны Тарельчатые колонны Колонны с другими насадками См. р аздел
Диаметр колонны мм Пункты 1,3—7 те же, что и для насадочных колонн Пункты 1,3—7 те же, что и для насадочных колонн 4.10.1
Длина разделяющего участка (высота насадки), мм Эталонная смесь [состав загруженной в куб смеси, % (мол.)] Флегмовое число и расход орошающей жидкости, отнесенный к свободному поперечному сечению, мл/(см2-ч) УС колонны Рабочее давление, мм рт. ст. Нагрузка (мл/ч) или скорость потока паров (м/с) Длина разделяющего участка — рабочая высота колонны Длина разделяющего участка — рабочая высота^колонны 4.10.2 4.10.3 4.10.4 4.10.5 4.10.6 4.10.7
Форма насадочных тел, материал насадки (состояние поверхности), размеры насадочного тела (высота, диаметр, толщина стенки), активная поверхность насадки Расстояние между тарелками Размеры реальных тарелок Режим работы (например, число оборотов ротора в колоннах роторного типа) Размеры насадки 7.8
наличии дополнительных сведений относительно аппаратурнотехнологических параметров (табл. 24) возможна однозначная оценка и опытная проверка разделяющей способности колонны.
4.10.1. ДИАМЕТР КОЛОННЫ
По экспериментальным данным Мерча [185] при постоянных размерах насадочных тел ВЭТС возрастает с увеличением диаметра колонны. Однако для насадок из проволочной сетки влияние диаметра колонны (по данным Стедмана и Мак-Магона) не так ощутимо, как, например, для насадки из колец Рашига или из седел. На основе систематических исследований пристеночного эффекта (растекание жидкости к стенкам колонны), проведенных Муллином [186], пришли к выводу, что наибольшая эффективность колонны достигается при соотношении: диаметр колонны/ .диаметр насадочного тела = 10—12. Если это соотношение не
137
выдерживается, то следует предусмотреть как можно больше пере-распределителей жидкости по высоте колонны (см. разд. 4.2 и 4.8.1).
4.10.2. РАБОЧАЯ ВЫСОТА КОЛОННЫ
И СПОСОБЫ УКЛАДКИ НАСАДОЧНЫХ ТЕЛ
Под рабочей высотой колонны подразумевают высоту участка колонны (любой конструкции), на котором непосредственно протекает процесс разделения. Следовательно, рабочая высота в насадочных колоннах представляет собой высоту слоя насадки. Вследствие каналообразования и пристеночного эффекта ректифицирующий участок необходимо секционировать по всей высоте колонны, иначе в ее нижней части не будет обеспечен противоточный массообмен ([39] к гл. 1).
Киршбаум [187] по результатам испытания промышленных колонн установил, что число теоретических ступеней разделения не увеличивается пропорционально высоте слоя насадки. Казанский [188] тщательно исследовал пристеночный эффект в лабораторных колоннах. В частности, он обнаружил, что эффективность несекционированной колонны высотой 149 см, составляющая при определенных условиях 18 теоретических ступеней разделения, увеличивается до 24 ступеней после секционирования колонны на три участка. Работы Бушмакина и Лызловой [189] подтвердили эти результаты. При использовании в качестве насадочных тел константановых спиралей диаметром 1,8 мм было показано, что секционирование колонны на участки длиной по 25 см с целью сбора и перераспределения орошающей жидкости обеспечивает ее максимальную разделяющую способность. При увеличении числа секций от 1 и до оптимального значения каждое перераспределительное устройство повышает эффективность на 1,5 теоретической ступени. Автором проведены испытания насадки из фарфоровых седловидных насадочных тел размером 4X4 мм при v = оо. Результаты испытаний приведены в табл. 25.
Таблица 25.
БЭТС в зависимости от рабочей высоты колоииы и числа секций
Эталонная смесь четыреххлорнстый углерод — бензол [40%
(мол.)], исходный объем смесн 300 мл, давление атмосферное
Нагрузка, мл/г ВЭТС (см) при рабочей высоте колонны']
450 мм 900 мм
1 секция 2 секции
200 4,73 6,93 6,93
600 6,00 11,25 7,83
138
Рис. 87,
Устройство Алленби— Лере для неупорядоченной загрузки мелкой насадки:
1 — трубка; 2 — корковая пробка с отверстием; 3 — выступ.
Эти опыты выявили следующие факты, которые до сих пор недостаточно учитывались при лабораторной ректификации: 1) ВЭТС при одной и той же нагрузке зависит от высоты ректифицирующего участка; 2) если при малых нагрузках секционирование колонны не дает эффекта, то при более высоких нагрузках с введением секционирования эффективность разделения увеличивается. Суммарная поверхность насадки в слое определенной высоты зависит от способа ее укладки, влияющего также и на распределение жидкости [190]. Влияние способа укладки на перепад давления в колонне и ее разделяющую способность весьма значительно [191]. Для обеспечения беспорядочной укладки насадки Майлс с сотр. [192] применил способ, в соответствии с которым колонну наполняют минеральным маслом и бросают в нее насадку по одному элементу. Проще заполнять колонну, опускдя в нее по три-четыре насадочных тела при постоянном постукивании деревянной палочкой по стенкам колонны. Небольшие насадочные тела можно очень быстро загрузить с помощью устройства, описанного Алленби и Лёре [193] (рис. 87). Каждое насадочное тело попадает в колонну отдельно, благодаря чему обусловливается неупорядоченность расположения насадки. Насадочные тела насыпают на дно колбы в виде слоя высотой примерно 1 см. В трубку 1 (см. рис. 87) с помощью газодувки или воздухопровода, присоединенного к напорному патрубку вакуумного насоса, вдувают воздух, при этом насадочные тела приподнимаются и начинают вращаться. Выступ 3 притормаживает насадочные тела, которые через отверстие в корковой пробке 2 проскакивают по одному в соединительную трубку, ведущую в колонну.
В тарельчатых колоннах и в колоннах с иными насадками длина разделяющего участка соответствует рабочей высоте колонны. В тарельчатых колоннах эта высота равна расстоянию от нижнего края нижней тарелки до нижнего края верхней тарелки плюс один шаг между тарелками. Для колонн с другими насадками в качестве рабочей высоты указывают общую высоту специальной насадки, например рабочей высотой для колонн с вращающейся насадкой является высота ротора.
4.10.3. ЭТАЛОННАЯ СМЕСЬ И СОСТАВ ЗАГРУЖЕННОЙ ЖИДКОСТИ
В принципе, при испытании ректификационной колонны следует использовать те смеси, для разделения которых предназначена колонна. При отсутствии достаточно чистых веществ можно
139
ограничиться применением эталонных смесей, отвечающих определенным требованиям. Эталонные смеси следует выбирать в первую очередь с таким расчетом, чтобы степень трудности их разделения находилась в полном соответствии с предполагаемой разделяющей способностью колонны.
Эталонную смесь с низким значением относительной летучести компонентов а можно применять для колонн как с большим, так и с малым числом теоретических ступеней разделения; эталонную же смесь с высоким значением а можно использовать только для колонн с малым числом теоретических ступеней разделения.
Хальденвангер [194] наиболее полно сформулировал требования, предъявляемые к эталонным смесям: 1) по свойствам эталонная смесь должна приближаться к идеальному раствору, т. е. практически без отклонений подчиняться закону Рауля и иметь относительную летучесть компонентов, постоянную для всех концентраций; 2) данные по равновесию пар— жидкость должны быть известны или их можно легко рассчитать; 3) смесь должна состоять только из двух компонентов во избежание трудностей при измерениях и расчетах; 4) относительная летучесть компонентов должна иметь такое значение, чтобы в испытуемой колонне достигалось достаточное, но не слишком большое разделение; 5) температуры кипения смеси должны лежать в интервале, для которого нетрудно подобрать надежную тепловую изоляцию колонны; 6) компоненты смеси должны быть термически стойкими в условиях ректификации; 7) вещества и их смеси не должны вызывать коррозии конструкционных материалов, использованных в аппаратуре; 8) исходные вещества должны быть легко доступными; 9) вещества не должны содержать примесей; их чистота должна поддаваться проверке доступными методами; 10) смеси с любой концентрацией компонентов должны легко поддаваться анализу.
Международное сотрудничество в рамках Европейской федерации химической технологии (European Federation of Chemical Engineering) дало возможность стандартизировать эталонные смеси благодаря обширному экспериментальному материалу, накопленному членами федерации. В брошюре, опубликованной Цуидервегом [195], приведены 11 систем с данными по равновесию и со всеми необходимыми физическими свойствами, а также данные по химической стойкости компонентов.
Чтобы можно было сравнивать различные насадки и колонны по их разделяющей способности, рекомендуется использовать эталонные смеси, указанные в табл. 26. Для испытания колонн при атмосферном давлении в соответствии с международными нормами рекомендуется применять в первую очередь смеси «-гептан— метилциклогексан—бензол—1,2-дихлорэтан и четыреххлористый углерод — бензол для числа теоретических ступеней разделения п=50—60, бензол—этиленхлорид для п = 50—60 и четыреххло-140
ристый углерод—бензол для п < 30*. Брандт и Рёк [196] исследовали степень отклонения смеси «-гептан—метилциклогексан от идеальной и показали,что эту смесь можно назвать псевдоиде-альной, поскольку по свойствам она не соответствует традиционной классификации. Данную смесь можно с успехом применять в качестве эталонной для работы при остаточном давлении до 300 мм рт. ст., тем более, что мольные энтальпии испарения компонентов практически одинаковы (7,575 ккал/моль). Для колонн с числом теоретических ступеней разделения от 100 до 500 в качестве эталонного вещества пригодна тяжелая вода. Система 16Н2О—18НгО позволяет проводить испытания, когда п достигает 1000; однако при этом время, в течение которого устанавливается стационарный режим, измеряется месяцами (см. разд. 5.1.4). Для проведения точных измерений п можно также применять эталонные вещества, меченые радиоактивными изотопами 14С, 35 S и 36С1.
В интервале давлений от 20 до 400 мм рт. ст. смесь циклогексилциклопентан — «-додекан ведет себя почти как идеальная, а смесь дициклогексил — w-тридекан можно приближенно считать идеальной только в среднем интервале концентраций, т. е. от 30 до 90% дициклогексила [197].
Мерчу [185] удалось количественно описать влияние физических свойств жидкой смеси на разделяющую способность колонны. Он установил, что ВЭТС прямо пропорциональна выражению ат]/р (где а — относительная летучесть; т] — динамическая вязкость в сП; р — плотность в г/мл). Результаты своих исследований Мерч обобщил следующей эмпирической формулой:
ВЭТС = ft1A4ft*dfc«AI/3aT)/p (128)
где М — удельная нагрузка по паровой фазе, кг/(м2-ч); d — диаметр колонны, м; h — высота слоя насадки (рабочая высота колонны), м; ku и k3 — константы, которые зависят от типа и размеров насадки (табл. 27).
Формула (128) справедлива только при следующих условиях: 1) ректификация при атмосферном давлении; 2) величина М должна находиться в интервале 25—80% от предельной нагрузки; 3) отношение диаметра колонны к диаметру насадочного тела должно превышать 8—10; 4) ВЭТС рассчитывают при бесконечном или очень большом флегмовом числе.
Погрешность расчетов по формуле (128) не превышает ±10%. Автор, однако, предлагает, что для повышения надежности вычисленную величину следует увеличивать на 20%. Формула (128), таким образом, дает только грубое приближение, которое нужно проверять опытным путем.
Так как ВЭТС зависит от состава исходной смеси в кубе [92, 199], то, как показывает опыт, лучше всего поддерживать кон-
* Здесь смесь бензол—четыреххлористый углерод рекомендована одновременно для двух интервалов теоретических ступеней (я = 50—60 и п 30). Эту смесь предпочтительнее использовать при п 30. — Прим. ред.
141
X Таблица 26.
Характеристика эталонных смесей, применяемых при испытаниях колонн
Рабочее давление ректификации Бниариая смесь А + В Температура кипения, °C Коэффициент 20 преломления /гр Разность коэффициентов преломления А«Ь° Относительная летучесть п для и ?пы-тания колонн с ожидаемой эф-1 фективио- 1 стью
<А ZB А/ А В
760 мм рт. ст. Бензол—толуол * 80,1 110,6 30,5 1,50122 1,49647 0,0048 2,36—2,61 2—7
Бензол-н-гептан 80,1 98,4 18,3 1,5011 1,3876 0,1135 2,12—1,11 5—20
Метилциклогексан—толуол 100,98 110,6 9,62 1,4232 1,4968 0,0736 1,306—1,328 5—25
Бензол-1,2-дихлор-этан 80,1 83,5 3,4 1,5011 1,4448 0,0563 1,162—1,107 15—20
н-Гептан—метилциклогексан 98,4 100,85 2,45 1,3878 1,4232 0,0354 1,076—1,074 20—80
п-Ксилол-л-кси-лол ** 139,10 138,35 0,75 1,4960 1,4973 0,0013 1,0203—1,0204 50—250
5—100 мм рт. ст. (возможно до 1 мм рт. ст.) 2-Метилнафталин— 1-метилнафталин * — — — — — 0,0050 (<Ч°) 1,1400—1,1417 (5 мм рт. ст.); 1,1312—1,1328 (10 мм рт. ст.); 1,1035—1,1047 (100 мм рт. ст.) 15—45
5—100 мм рт. ст. транс- Декали н— ^ис-декалин * — — — 1,46932 1,48098 0,01166 1,483—1,460 (5 мм рт. ст.); 1,440—1,420 (10 мм рт. ст.); 1,314—1,300 (100 мм рт. ст.) 4—20
50—760 мм рт. ст. (возможно 20 мм рт. ст.)
Бензол—этилбензол
1,5246 1,4958 0,0288 1,188—1,194 (50 мм рт. ст.); 1,140—1,143 10—30
(300 мм рт. ст.); 1,130 (760 мм рт.
ст.)
8—16 ат Изобутан—н-бутан — — — — — — 1,308—1,241 (8 ат); 1,261— 1,217 (12 ат); 1,220—1,195 (16 ат) 6—25
1 —10 ат Метанол—этанол * — — — — — — 1,644—1,866 (1 ат); 1,474— 1,559 (3 ат); 1,395—1,447 (5 ат); 1,289— 1,319 (10 ат) 4—20
• Для анализа состава смесей лучше применять газовую хроматографию; ** Для анализа состава смесей лучше применять ннфра-красную спектроскопию нлн газовую хроматографию.
Таблица 27.
Константы klt k2 и k3 в формуле Мерча [185]
Тип насадки Поперечный размер насадочного тела, мм &3
Кольца Рашига 9,5 13,58 —0,37 1,24
12,7 44,95 —0,24 1,24
Седловидные насадочные тела 12,7 25,61 —0,45 1,11
Насадка Мак-Магона 6,35 0,0114 0,5 1,0
9,5 0,199 0,25 1,0
12,7 0,356 0,20 1,0
центрацию легколетучего компонента в кубовой жидкости на уровне не ниже 40% (мол.); при этом количество загруженной в куб жидкости должно превышать общую удерживающую способность колонны по жидкости примерно в восемь раз. Эталонную смесь следует выбирать с таким расчетом, чтобы обогащение по легколетучему компоненту, достигаемое в колонне, не превышало 90% (мол.). По разности температур кипения компонентов эталонной смеси можно выделить следующие группы смесей:
Для большого числа теоретических ступеней (п > 40)...........................
Для среднего числа теоретических ступеней (п = 10—40)........................
Для малого числа теоретических ступеней
(п < 10)...............................
разность температур кипения компонентов эталонной смеси, °C
0,5—3,0
3,0—10,0
Более 10,0
Для важнейших эталонных смесей составлены таблицы [92, 146, 147, 195], позволяющие вычертить диаграмму зависимости nD—п, аналогичную показанной на рис. 71 (зависимость числа теоретических ступеней от показателя преломления смеси). По оси абсцисс отсчитывают показатели преломления дистиллята и кубового продукта и с помощью оси ординат определяют соответствующие им числа теоретических ступеней разделения; разность этих чисел равна числу теоретических ступеней испытуемой колонны (см. разд. 4.7.4).
144
4.10.4. ФЛЕГМОВОЕ ЧИСЛО И КОЛИЧЕСТВО ОРОШАЮЩЕЙ ЖИДКОСТИ
Как уже было указано, число теоретических ступеней разделения обычно определяют при бесконечном флегмовом числе, т. е. без непрерывного отбора дистиллята, поскольку это условие поддается наиболее точному воспроизведению.
На практике часто необходимо знать, как изменяется число теоретических ступеней разделения при конечном флегмовом числе или какой поправочный коэффициент следует ввести в найденное число п при ректификации с конечным флегмовым числом, если в основу расчета положены результаты испытания эффективности при v = оо.
Число теоретических ступеней при конечном флегмовом числе определяют следующим образом: работают при определенной нагрузке с конечным флегмовым числом, а непрерывно отбираемый дистиллят так же непрерывно возвращают в куб (рис. 88). Затем в определенные промежутки времени из головки колонны и куба одновременно отбирают пробы и определяют их состав ф164].
Рис. 88.
Установка для испытания эффективности колонны при конечном флегмовом числе.
Рис. 89.
Зависимость поправочного коэффициента F для насадки из спиралей 2 X Х2Х0.2 мм от флегмового числа при различных нагрузках колонны.
145
Выход дистиллята, °/о(од.)
Рис. 90.
Зависимость состава дистиллята и кубовой жидкости от выхода дистиллята при ректификации смеси хлорбензол—этилбензол.
Нагрузка колонны 44 мл/ч; v = 8; рабочее давление 760 мм рт. ст. (/) н 20 мм рт. ст. (2); х — содержание хлорбензола, начальная концентрация ХВ = 37 % <мол->-
Рис. 91.
Зависимость числа эквивалентных теоретических ступеней разделения от выхода дистиллята в интервале 0—50% (об.) для смеси хлорбензол—этилбензол.
Условия ректификации:
1 — v = 24, 20 мм рт. ст.; 2 — v = 24, 760 мм рт. ст.; 3 — v = 8, 20 мм рт. ст.; 4 — v = 8, 760 мм рт. ст.
В колонне постепенно устанавливается стационарный режим, который выражается в постоянстве составов дистиллята и кубовой жидкости и соответствует любому моменту периодической разгонки. Рекомендуется после отбора последней пробы в течение некоторого времени отбирать дистиллят обычным образом (не возвращая его в куб) и затем повторить процесс при измененном составе кубовой жидкости.
На рис. 89 представлены поправочные коэффициенты F для насадки из спиралей размером 2x2x0,2 мм, которые были экспериментально определены автором по указанной методике (колонна диаметром 30 мм с рабочей высотой 1,15 м). Эти результаты были подтверждены исследованиями Науманна и Лейбница [201 ].
Под величиной F понимают поправочный коэффициент, на который нужно умножить число теоретических ступеней разделения, определенное при v = оо, чтобы получить фактическую разделяющую способность колонны при заданном конечном флегмовом числе:
«оо-Р = «КОН (129)
Из рис. 89 следует, что при испытании колонны с 60—70 теоретическими ступенями нецелесообразно увеличивать флегмовое число свыше 70. Это согласуется с уравнением (106), полученным Роузом.
Рекомендуется проводить периодическую ректификацию с использованием эталонной смеси при определенной нагрузке и при определенном флегмовом числе с отбором небольших проб дистиллята (около 1% от загруженной смеси); одновременно с каждой второй пробой дистиллята следует проводить капельный отбор и анализ жидкости из куба. Строят график зависимости состава
146
дистиллята и кубовой жидкости от выхода дистиллята (рис. 90) [92]. Эти данные сопоставляют с результатами испытания при бесконечном флегмовом числе, причем для этой цели используют понятия «число эквивалентных теоретических ступеней разделения» и «коэффициент полезного действия колонны».
Число эквивалентных теоретических ступеней разделения — это минимальное число теоретических ступеней, которое требуется для того, чтобы при бесконечном флегмовом числе вызвать такое же обогащение (хв —-> хЕ), как и при ректификации с конечным флегмовым числом. Все условия разгонки за исключением флегмового
числа остаются при этом одинаковыми.
При построении ступеней разделения на диаграмме Мак-Кэба и Тиле по указанному методу рабочую линию не используют, а ступени вписывают между диагональю и кривой равновесия, как это делается для бесконечного флегмового числа. Этот прием можно проиллюстрировать с помощью рис. 90. Так, после отбора 30% от общего объема дистиллята (при 20 мм рт. ст. и v = 8) получается, что 17% (мол.) хлорбензола содержится в кубовой жидкости, а 60% хлорбензола — в дистилляте. Если обе эти концентрации [хв = 17% (мол.) и хЕ = 60% (мол.)] нанести на диаграмму Мак-Кэба и Тиле или подставить их в уравнение Фенске (108в), то для v = оо получим обогащение, эквивалентное 17,5 теоретическим ступеням разделения. Эти расчеты можно выполнить для ряда точек, получая в итоге кривые, показанные на рис. 91. Как видно из сравнения кривых, число эквивалентных теоретических ступеней при высоком флегмовом числе больше, причем в начале ректификации при низком флегмовом числе (и = 8) наблю-
дается значительное уменьшение числа эквивалентных теоретических ступеней разделения. Из рис. 91 также следует, что давление
не оказывает существенного влияния на число эквивалентных
теоретических ступеней.
Диаграмма, построенная по опытным данным Коллинза и Ланца [200] (рис. 92) иллюстрирует взаимосвязь между числом теоретических ступеней при v = оо и 27 и числом эквивалентных
теоретических тарелок. При ректификации эталонной смеси «-гептан — метилциклогексан колонна с 30 реальными тарелками при нагрузке 3000 мл/ч имела при v = оо 19 теоретических ступеней, при v = 27 — 17 теоретических ступеней, а при v =
Рис. 92.
Зависимость числа теоретических ступеней (/, 2) и числа эквивалентных теоретических ступеней разделения (3) от нагрузки колонны:
1 — п при v — оо; 2~то же при и=27; 3 — пэкв при v = 27; 4 — кривая зависимости удерживающей способности колонны от нагрузки.
147
Рис. 93.
Устройство для измерения количества жидкости, стекающей в куб, и динамической удерживающей способности колонны.
Устройство изготовлено из стандартных деталей «Дестинорм».
Рис. 94.
Измеритель расхода жидкости с сифоном конструкции Зигварта—Штаге: / — шлиф для соединения с колонной; 2 — сифон; 3 — камера для сбора жидкости; 4 — шлиф для соединения с колбой.
Рис. 95.
Мерник жидкости «Лабодест» с магнитным клапаном конструкции Штаге.
= 27—12,5 эквивалентных теоретических ступеней. Это означает, что обогащение, достигнутое при v = 27, можно получить, применяя 12,5 теоретических ступеней разделения при v = оо. И, наоборот, необходимо подбирать колонну, которая при нагрузке 3000 мл/ч дает обогащение, соответствующее 19 теоретическим ступеням при v = оо.
Если отнести число эквивалентных теоретических ступеней пэкв к числу теоретических ступеней ru при v = оо и при такой же нагрузке, то, согласно Оболонцеву и Фросту [202], получим коэффициент полезного действия колонны:
К. п. д. = пэкв/п^ (при одной и той же нагрузке) (130)
Максимальное значение этой величины равно единице, оно тем ближе к 1, чем выше рабочее флегмовое число. 1
Изложенный выше метод расчета числа эквивалентных теоретических ступеней разделения и к. п. д. колонны имеет то существенное преимущество, что он позволяет сравнивать колонны по их разделяющей способности при различных рабочих условиях, поскольку все найденные числа теоретических ступеней разделения можно пересчитать относительно бесконечного флегмового числа v = оо.
Количество орошающей жидкости в основном измеряют в головке колонны. При условии, что колонна работает адиабатически, т. е. без рассеяния и подвода тепла, и мольные энтальпии 148
испарения компонентов различаются незначительно, количество жидкости, измеренное непосредственно над кубом, должно совпадать с количеством флегмы, измеренным в головке. Однако на практике вследствие тепловых потерь количество жидкости, стекающей в куб, как правило, больше количества флегмы, возвращаемой в верхнюю часть колонны. Поэтому целесообразно для одновременной проверки адиабатичности процесса ректификации измерять также и количество жидкости, стекающей в куб. Для этого применяют устройство, показанное на рис. 93. При измерениях кран устанавливают в такое положение, чтобы жидкость не стекала сразу в куб, а сначала попадала в градуированную воронку. С помощью секундомера измеряют количество жидкости, поступающее в единицу времени в воронку (т. е. расход жидкости). Этот прибор имеет то преимущество, что измеряемый расход жидкости можно варьировать в зависимости от условий разгонки. Недостатком устройства является возможность растворения смазки крана.
Это исключается в устройствах, предложенных Зигвартом и Штаге. На рис. 94 показано устройство с сифоном, а на рис. 95 — более удачная конструкция с магнитным клапаном. Геммекер и Штаге [245 ] дают превосходный обзор устройств для измерения количества орошающей жидкости.
4.10.5. ОБЩАЯ, СТАТИЧЕСКАЯ И ДИНАМИЧЕСКАЯ УДЕРЖИВАЮЩАЯ СПОСОБНОСТЬ КОЛОННЫ
Под общей удерживающей способностью колонны — УС (total holdup*) подразумевают количество вещества, которое в виде жидкости и паров находится в ректификационной колонне между кубом и конденсатором. Общая УС складывается из статической УС («захват» жидкости), называемой также УС в состоянии покоя, и динамической УС колонны (рабочая задержка — operating holdup*).
Информация об общей УС колонны очень важна, поскольку, как уже было показано в разд. 4.7.1, влияние общей УС усиливается с увеличением числа теоретических ступеней разделения. Общая задержка при большом флегмовом числе ухудшает разделение, а при малом флегмовом числе, наоборот, благоприятствует разделению. При очень большой общей задержке флегмовое число почти не оказывает влияния на разделяющую способность колонны. Для насадки из колец Рашига возможен теоретический расчет динамической УС, предложенный Йилмазом и Брауэром [11 в] (см. разд. 4.2.2).
Статической УС колонны называют количество жидкости, которое остается в колонне после предварительного затопления насадки или окончания разгонки и охлаждения. При определе
* Термины total holdup, operating holdup приняты в научно-технической литературе на английском языке. — Прим. ред.
149
нии статической УС в куб загружают жидкость в пятикратном количестве по сравнению с предполагаемой УС колонны и в течение I ч проводят ректификацию с бесконечным флегмовым числом. После охлаждения колонны измеряют количество жидкости, оставшееся в кубе. Разница между первоначально загруженным количеством и оставшимся количеством и представляет собой статическую УС. В насадочных колоннах статическая УС складывается из капель жидкости, оставшихся на насадочных телах и между ними, а также на стенках колонны, приставки и конденсатора *. В тарельчатых колоннах основную часть статической задержки составляют слои жидкости, оставшиеся на отдельных тарелках.
В соответствии с более простой методикой определения статической УС через верхний конец конденсатора в установку заливают отмеренный объем разделяемой смеси и по количеству собранной внизу жидкости устанавливают, сколько жидкости задержится в колонне. Измерение следует повторять несколько раз, чтобы достигнуть полного смачивания насадки. Это позволит получить надежное среднее значение, которое, однако, соответствует более низким температурам, чем рабочая температура.
Динамической (рабочей) УС колонны является количество жидкости, которое дополнительно удерживается во время ректификации в аппарате над кубом. Динамическую УС определяют следующим образом. Во время разгонки быстро удаляют колбонагре-ватель, по возможности также отсоединяют и колбу **, а всю стекающую жидкость собирают в измерительный сосуд устройства, показанного на рис. 95; целесообразно при этом отводить жидкость через холодильник.
Для косвенного определения общей удерживающей способности (УС) как насадочных, так и тарельчатых колонн в куб с находящимся в нем эталонным веществом, например к-гептаном, добавляют небольшое количество высококипящего вещества, например стеариновой кислоты или а-бромнафталина (пд = = 1,6580). В ходе разгонки легколетучий компонент смеси накапливается в колонне и соответственно увеличивается доля высококипящего вещества в расчете на оставшееся в кубе количество эталонного вещества. Во время разгонки через определенные промежутки времени из куба отбирают пробы и определяют концентрацию добавленного высококипящего вещества. Если добавляет стеариновую кислоту, то ее содержание определяют либо титрованием, либо взвешиванием перед и после упаривания пробы. Содержание
* Следует дополнительно учитывать утечку паров за пределы конденсатора (например, в ловушку или в производственное помещение), которая за 1 ч может достигнуть ощутимых размеров. Утечка вещества из системы происходит иногда в результате недостаточно полной конденсации паров (особенно легколетучих веществ). — Прим. ред.
** При работе с ядовитыми или легковоспламеняющимися веществами этого делать не рекомендуется по положениям техники безопасности работ в лабораториях. — Прим. ред.
150
Рис.
Зависимость общей удерживающей способности колонны от нагрузки для различных насадочных тел (при 730 мм рт. ст.):
/ — перфорированные полуцилиндры; 2 — седла из проволочной сетки; 3 — кольца Рашнга. Эталонное вещество — н-гептаи.
а-броинафталина можно определить рефрактометрическим методом. Общую УС колонны вычисляют по формуле:
УС=Ув[1-(х1/х2)] (131)
где УС — общая удерживающая способность; Ев — объем загруженной смеси; Xj и х2— концентрация добавленного вещества в куб перед разгонкой и во время перегонки соответственно.
Для получения сравнимых результатов статическую УС, а также динамическую и общую УС целесообразно указывать в пересчете на одну теоретическую ступень разделения или реальную тарелку. О зависимости общей УС от нагрузки опубликовано мало сведений. Коллинз и Ланц [200] приводят результаты, полученные для ситчатой колонны Ольдершоу диаметром 28 мм с 30 реальными тарелками (см. рис. 92). В зависимости от нагрузки общая УС изменяется между 43 и 60 мл, так что на одну реальную тарелку в результате перерасчета приходится в среднем 1,4—2,0 мл, а одной теоретической ступени соответствует в среднем 2,5—3,5 мл. В насадочных колоннах, по собственным данным автора, УС в расчете на одну теоретическую ступень разделения имеет величину того же порядка, что и для тарельчатых колонн, как это видно из рис. 96, иллюстрирующего зависимость общего удерживаемого количества w-гептана при 97 °C от нагрузки при остаточном давлении 730 мм рт. ст. [203]. Ректификацию проводили в колонне диаметром 19 мм, рабочая высота колонны составляла 812 мм.
Цуидервег [158] при расчете числа теоретических ступеней разделения п для периодической ректификации учитывает влияние УС на п и размер промежуточной фракции. Им была исследована зависимость «четкости» разделения от удельной общей УС, флегмового числа и числа теоретических ступеней для смесей с относительной летучестью от 1,07 до 2,42. С помощью введенного Цуидервегом так называемого полюсного расстояния можно определить оптимальное флегмовое число. Способ соответствует расчету по методу Мак-Кэба и Тиле при конечной концентрации хв = 5% (мол.).
4.10.6. РАБОЧЕЕ ДАВЛЕНИЕ
Под рабочим давлением понимают давление, установившееся при перегонке в головке аппарата. Его обычно измеряют в газовом (или воздушном) пространстве непосредственно за зоной конденсации. Рабочее давление следовало бы измерять в паровой фазе
151
Рис. 97.
Зависимость числа теоретических ступеней разделения, приходящихся иа 1 м высоты иасадки, луд от перепада давления по данным Дэвида.
Эталонная смесь бензол—этилеихлорид, концентрация бензола в кубовой жидкости х& =И4% (мол.).
перед зоной конденсации, т. е. на том же самом участке, где измеряют температуру. Но это сложно, поэтому трубки конденсатора подбирают с достаточно большим диаметром, чтобы
между давлением в паровом пространстве (перед зоной конденсации) и давлением в газовом пространстве (после зоны конденсации) не мог
вознйкнуть перепад. Возможность создания перепада давления особенно реальна при работе в условиях высокого вакуума; в этом
случае рекомендуется проводить сравнительные измерения перед и после зоны конденсации. Давление, установившееся в кубе, равно сумме давления в газовом пространстве (перед зоной конденсации) и перепада давления (гидравлическое сопротивление потока) по высоте колонны. Так как при работе под вакуумом увеличившийся объем паров сильно влияет на процесс разделения, то часто при ректификации применяют более крупные насадочные тела, чем это следует из соотношения диаметр колонны/диаметр насадочного тела = 10, или, по крайней мере, в верхнюю часть колонны загружают более крупные насадочные тела. В головке колонны устанавливается самое низкое давление, поэтому в верхней части колонны из-за более высокой скорости потока паров легко может произойти затопление насадки.
Вопрос о том, насколько благоприятно влияет применение вакуума при перегонке на число теоретических ступеней разделения, еще не выяснен окончательно. Вебер [204] установил, что разрежение оказывает лишь слабое влияние на число теоретических ступеней; при этом пропускная способность колонны снижается вследствие большего объема паров, а относительная летучесть под вакуумом обычно увеличивается (см. разд. 4.6.2); Майлз с сотр. [192] установил, что для различных насадочных тел ВЭТС имеет минимум при остаточном давлении порядка 200 мм рт. ст. Страк и Кинней [203] выяснили, что значения ВЭТС в интервале 50—100 мм рт. ст. проходят через слабо выраженный минимум, и при 100 мм рт. ст. разделяющая способность такова же, как и при атмосферном давлении (см. также разд. 4.8 и 4.10.4).
На основе обширного экспериментального материала Дэвид [191 ] однозначно показал, что число теоретических ступеней разделения тем меньше, чем ниже рабочее давление ректификации. Эти результаты находятся в полном согласии с данными Цуидер-вега [205], а также частично соответствуют опытным данным
152
Майлза [192]. Если построить графическую зависимость числа теоретических ступеней разделения, приходящегося на 1 м высоты насадки, пуц от перепада давления в колонне, то можно наглядно проследить влияние вакуума- на разделение. На рис. 97 подобные кривые построены по данным Дэвида [191] для смеси бензол— этиленхлорид. К аналогичным выводам пришел Киршбаум с сотр. [206], показавший, что в результате влияния относительной летучести изменение числа теоретических ступеней разделения, приходящегося на 1 м насадки, при переходе к вакууму не всегда сопровождается параллельным изменением степени обогащения, достигаемой в ректификационной колонне.
При исследовании влияния давления на массообмен в насадочной колонне с помощью меченых атомов 14С, 35S и 36С1 Зельвенский с сотр. [209] обнаружил, что снижение давления вызывает уменьшение разделяющей способности колонны.
Шапиро [207] вывел уравнение для расчета состава кубовой жидкости и жидкости на n-ой тарелке в зависимости от давления при ректификации идеальных бинарных систем с бесконечным флегмовым числом.
4.10.7. НАГРУЗКА И СКОРОСТЬ ПОТОКА ПАРОВ
Под нагрузкой понимают количество вещества, прошедшее в единицу времени через колонну и получаемое в виде конденсата в головке колонны. Нагрузка складывается из количеств флегмы и отбираемого дистиллята. В промышленности принято выражать нагрузку колонны через скорость потока паров, отнесенную к свободному сечению колонны. В лабораторной практике количество паров, проходящих через поперечное сечение колонны в единицу времени, рассчитывают только в тех случаях, когда нужно провести сравнение с полупромышленными или промышленными установками. Принято также измерять нагрузку в мл/(см2-ч).
Разделяющая способность колонны зависит от нагрузки (рис. 98) и тем сильнее, чем больше флегмовое число. Поэтому при испытаниях эффективности колонны необходимо точно указывать нагрузку; по предложению Вебера для получения сравнимых результатов испытания следует, как правило, проводить при нагрузке, равной 2/3 максимальной нагрузки. Под максимальной нагрузкой понимают нагрузку, несколько меньшую той нагрузки, при которой орошающая жидкость под действием поднимающихся паров удерживается во взвешенном состоянии и не может стекать вниз. При этом колонна захлебывается и не может работать в режиме противоточного массообмена.
Важная особенность насадочных колонн заключается в том, что эффективность насадочных тел небольшого диаметра (2—4 мм) сильнее зависит от нагрузки, чем эффективность насадочных тел большего диаметра (5—10 мм) (см. рис. 98). По некоторым данным [214], как было уже установлено выше, оптимальной нагрузкой
153
Рис. 98.
Зависимость числа теоретических ступеней, приходящихся на 1 м высоты, Луд от нагрузки по данным Штаге ([39 ] к гл. 1) для различных насадок: 1,2 — спирали 2х2х 0 0,2 мм и 4X4 х 0 0,4 мм, сталь V2A; 3,4 — коль-' ца Рашнга 6,5х 0 6 мм и 4,5х 0 4,5 мм, стекло; 5 — шарики 0 3 мм, стекло; 6, 7 — шарики 0 7,5 мм, 0 4 мм, матированное стекло; 8, 12 — кольца Рашнга 8X0,8 мм и 5,5х 0 5,5 мм, фарфор; 9 — кольца Рашнга 10Х 10 мм, глнна; 10 — спираль 0 3 мм, длина 1—5 мм, стекло; И — токарная стружка 0 1— 0 3 мм, ширина —2 мм; 13 — 17 — спирали соответственно 0 2 мм, 0 3 мм, 0 4 мм, 0 6 мм, 0 8 мм, сталь V2A; 18 — 20 — спирали с разомкнутыми витками соответственно 3x3x0,4 мм, 4х4х X 0 0,45 мм, ЗхЗХ 0 0,45 мм, сталь V2A; 21 — 23 — спнралн с сомкнутыми витками соответственно 2X2 х 0 2 мм, 3X3x0,45 мм, 4Х4х X 0 0,4 мм, сталь V2A.
Эталонная смесь: бензол — 1,2-днхлорэтан для кривых 1 —12 (опыты Шульце—Штаге); н-гептан —метнлциклогексаи для кривых 13—17 (опыты Кол-линга); н-гептан —метилцнклогексан для кривых 18—23 (опыты Брауэра).
является не самая низкая, а несколько меньшая, чем нагрузка, вызывающая явление захлебывания (см. рис. 83).
В лабораторных колоннах максимальная эффективность наблюдается не при верхней предельной нагрузке, как это имеет место по данным Дэвида для полупромышленных колонн, а при значительно меньших нагрузках, хотя и превышающих минимально допустимую нагрузку.
При сравнении лабораторных колонн с полупромышленными или промышленными колоннами необходимо иметь в виду, что скорости потока паров в лабораторных колоннах в среднем ниже,
154
т. е. находятся в интервале 0,02—0,5 м/с, в то время как в промышленных установках эта скорость достигает 0,1—2,0 м/с и выше [184]. В насадочных колоннах очень важно поддерживать скорость потока паров постоянной; в тарельчатых же колоннах колебания скорости потока паров меньше сказываются на эффективности процесса разделения.
От нагрузки зависят динамическая и общая УС, перепад давления и предельная скорость потока паров, которая в свою очередь определяется формой и размерами насадочных тел или размерами и конструкцией реальной тарелки, а также свойствами разделяемых смесей. В разд. 4.11 об этом сказано подробнее. О соотношении нагрузок в насадочных колоннах исчерпывающую информацию предоставили Штаге и Бозе ([39] к гл. 1).
4.10.8. ПРОВЕДЕНИЕ ИСПЫТАНИЙ
Подготовка колонн к работе. При испытании колонн на эффективность очень важна аккуратность в работе. Всю аппаратуру следует тщательно промыть и просушить. Ни в коем случае в колонне не должно оставаться даже следов воды. По этой причине перед испытанием рекомендуется оставлять включенным на ночь обогрев кожуха колонны. Насадочные тела перед загрузкой также следует тщательно очистить. При этом рекомендуется их промыть сначала в четыреххлористом углероде и трихлорэтилене, затем в горячем бензоле и, наконец, снова в трихлорэтилене. При заполнении колонны насадкой нужно следить за тем, чтобы не касаться руками насадочных тел. При работе с насадочными колоннами большое внимание уделяется способу укладки насадочных тел. Лучше всего опускать одновременно по 3—4 насадочных тела при постоянном постукивании деревянной палочкой по корпусу колонны. С помощью приспособления, показанного на рис. 87, достигается быстрая и неупорядоченная укладка мелких насадочных тел. После завершения очередного испытания насадочные тела выгружают из колонны, промывают, просушивают, снова загружают, после этого можно приступить к новому испытанию. Таким путем проверяют влияние способа укладки насадки на разделяющую способность колонны.
Очистку тарельчатых колонн проводить труднее, так как на тарелках может оставаться некоторое количество жидкости. В этом случае колонну пропаривают чистым легколетучим растворителем, а затем просушивают теплым воздухом, предварительно надев на нее обогреваемый кожух.
Эталонную смесь загружают в количестве, соответствующем восьмикратной УС колонны, которую определяют заранее, как это описано в разд. 4.10.5. Перегонную колбу подбирают такого размера, чтобы исходная смесь в начале испытания заполняла бы колбу на 2/3 объема. Содержание легколетучего компонента в эталонной смеси должно составлять 30—40% (мол.). Пока еще не
155
5
Рис. 99.
Пробоотборник по нормалям <Дестинорм» для работы при атмосферном давлении и под вакуумом.
Рис. 100.
Головка колонны по нормалям <Дестинорм» с устройством для капельного пробоотбора без застойных зон:
1 — паровая камера; 2 — термометр; 3 — капилляр; 4 — воронка; 5 — кран для сообщения с атмосферой; 6 — кран для отбора пробы; 7 — патрубок; 8 — приемник жидкости; 9 — мерный сосуд для флегмы.
установлено однозначно, в какой мере состав кубовой жидкости влияет на степень разделения [208].
Большое внимание уделяют приготовлению эталонной смеси. Нельзя без проверки применять выпускаемые промышленностью реактивы квалификации «чистый для анализа» или «чистый». Часто для контроля чистоты недостаточно определения одного только показателя преломления. Точный анализ возможен с помощью газовой хроматографии и инфракрасной спектроскопии [195]. Дополнительная очистка эталонного вещества не требуется в том случае, если экспериментально определенные физико-химические константы совпадают с теоретическими значениями и температура кипения вещества, измеренная термометром с ценой деления 1/10 °C, имеет отклонение, не превышающее 0,1 °C с учетом влияния колебаний атмосферного давления. Большинство веществ нуждается в химической очистке от сопутствующих примесей [210—212] и в последующей четкой ректификации при высоком флегмовом числе. При использовании недостаточно очищенных веществ возможно смещение калибровочной кривой По — содержание % (масс.), а также концентрирование сопутствующих примесей в головке колонны или кубе при испытаниях. Это может привести к искажению результатов измерения разделяющей способности колонн.
При построении калибровочной прямой п2° — содержание, % (масс.) берут тщательно очищенные вещества, приготавливают из них растворы, содержащие от 5 до 95% легколетучего компо-156
нента, и определяют их показатели преломления. Найденные значения наносят на диаграмму — содержание, % (масс.). Иногда с помощью уравнения Фенске (108в) для v = оо вычисляют необходимое число теоретических ступеней для ряда обогащений и строят график зависимости числа теоретических ступеней разделения от показателя преломления (см. рис. 71). По этому графику после отбора пробы и определения п® можно сразу же найти число теоретических ступеней разделения, соответствующее эффективности колонны.
При отборе проб объемом 0,1—0,2 мл из головки колонны и из куба нужно следить за тем, чтобы проба по возможности не соприкасалась со смазкой шлифа. До сих пор еще недооценивают возможность искажения значений измеряемых величин вследствие загрязнения пробы смазкой для кранов. Загрязнения пробы можно избежать, если для отбора проб из куба при атмосферном давлении и под вакуумом пользоваться устройством, показанным на рис. 99.
Применяемая при испытании приставка или головка также должна гарантировать при минимальном «мертвом» объеме возможность отбора жидкости по каплям без ее соприкосновения со смазкой крана. Аналогичное пробоотборное устройство показано на рис. 100. До установления стационарного режима работы колонны маленькая воронка 4 повернута вниз. Для отбора пробы воронку 4 поворачивают вверх и осторожно подсасывают жидкость путем присоединения патрубка 7 к вакуумной линии при медленном открывании зажима. После отбора 0,1—0,2 мл жидкости, которая капает в приемник 8 через большое отверстие в пробке крана 6, этот кран, а также зажим на вакуумной линии закрывают, а кран 5 осторожно и ненадолго открывают; благодаря этому находящаяся в воронке 4 жидкость вытесняется обратно в колонну. Приемник 8 сообщается с' атмосферой через патрубок 7. При работе в условиях атмосферного давления в патрубок 7 подают воздух для полного удаления жидкости из трубки для отбора проб, расположенной в нижней части приемника 8. Другие возможные способы пробоотбора обсуждаются в разд. 7.5.3 (см. также [39] к гл. 1).
Условием, обеспечивающим точность результатов испытаний, является также адиабатичность режима работы аппаратуры. Поэтому во избежание тепловых потерь необходимо тщательно изолировать асбестовым шнуром и стекловатой как куб, так и остальные узлы, вплоть до конденсатора (см. разд. 7.7). Так как при температуре даже ниже 80 °C всегда приходится принимать в расчет «дикую флегму», то при испытаниях нужно обязательно компенсировать потери тепла в колонне с помощью обогревающего кожуха. При этом регулирование обогрева’осуществляют автоматически по температуре внутри колонны (см. разд. 8.2.2). Обогревающие кожухи с продольным разъемом (см. разд. 7.7.3) имеют то
157
преимущество, что их можно монтировать после сборки стеклянной аппаратуры, а во время разгонки их можно в любой момент снять (при каких-либо неполадках). Участок между кубом и обогревающим кожухом, между кожухом и головкой колонны, а также участок, примыкающий к конденсатору, изолируют сначала слоем асбестового шнура толщиной 20 мм, на который накладывают обмотку из стеклянной шерсти, а поверх нее ленту из стеклоткани. Толщина изоляции должна составлять около 60 мм. Для большей надежности целесообразно применять ленточный электрообогрев.
Особенное внимание нужно обращать на правильный выбор точки для измерения температуры паров в приставке или в головке колонны, при котором было бы исключено смачивание шарика термометра переохлажденной жидкой флегмой. Кроме того, при перегонке под вакуумом должно быть гарантировано отсутствие перепада давления на участке между точками измерения температуры и давления, который мог бы исказить результаты измерения. В головке колонны по нормалям «Дестинорм» эти требования учтены. Температуру измеряют в паровой камере 1 (см. рис. 100), которая достаточно хорошо изолирована; применяют термометр 2 со стандартным шлифом, подвесной термометр или же встроенную термопару. Смачивание термометра исключено благодаря тому, что флегма в колонну возвращается через капилляр 3 и мерный сосуд для флегмы 9, расположенные ниже термометра. Остаточное давление измеряют непосредственно за охлаждаемой ловушкой с большим свободным сечением, поэтому между термометром 2 и манометром отсутствует какое-либо значительное сужение проходного сечения.
Выбор условий испытания эффективности колонн. Разделяющая способность колонны в условиях испытания в первую очередь зависит от нагрузки, которую поэтому следует во время опытов поддерживать строго постоянно. Скорость испарения целесообразно регулировать по перепаду давления в колонне, применяя контактный манометр (см. разд. 8.4.2); мощность обогрева контролируют по амперметру. Перед установлением заданной нагрузки режим работы насадочной колонны доводят до захлебывания с целью улучшения смачиваемости насадки. Для этого увеличивают мощность кипятильника, наблюдая за показаниями контактного термометра, до тех пор, пока в нижней части колонны не начнется процесс захлебывания, который затем распространяется по всей колонне, вплоть до головки. Захлебывание колонны поддерживают в течение примерно 15 мин и затем уменьшают мощность кипятильника, чтобы флегма снова свободно стекала вниз. Этот прием повторяют несколько раз, а затем с помощью контактного манометра устанавливают уровень необходимой нагрузки (см. также [39] к гл. 1).
Менее эффективно «холодное» захлебывание, когда колонну наполняют разгоняемой смесью и после сливания жидкости начинают ректификацию.
158
Рис. 101 иллюстрирует сильное влияние, какое оказывает «горячее» захлебывание на эффективность разделения. Испытания были проведены в колонне Подбельняка диаметром 25 мм с насадкой Хелигрид (см. рис. 274) при нагрузке 1000 мл/ч. Обнаружено, что стационарный режим массообмена в колонне устанавливается только через 8—10 ч, а захлебывание приводит к повышению числа теоретических ступеней разделения на 50% [213].
Предварительное захлебывание особенно благоприятно сказывается на разделяющей способности колонны при использовании насадки из проволочных спиралей. Эффективность же работы насадки, состоящей, например, из колец Рашига размером 4,5 X 6,0 мм, не определяется степенью их предварительного смачивания. Сведения о захлебывании под вакуумом приведены в разд. 5.4.1. Ленц [214] определил нагрузки, соответствующие захлебыванию, и оптимальные нагрузки для колонны с насадкой из стеклянных колец Рашига (5,5х4,0 мм) и из проволочных колец (сталь V 4А, 2 мм) при ректификации эталонных смесей На О—Da О (27%) и н-гептан—метилциклогексан. Диаметр колонны составлял 37,5 мм. Были исследованы числа теоретических ступеней разделения, приходящихся на 1 м рабочей высоты колонны, и перепад давления в колонне при различных скоростях потока паров. Нагрузка при оптимальном режиме для колец Рашига совпадает с нагрузкой при захлебывании колонны, а для насадки из проволочных колец она соответствует 81—88% максимальной нагрузки. Если насадочная колонна работает в так называемом эмульгаци-онном режиме, то интенсивность массообмена может повыситься в 6 раз с одновременным увеличением пропускной способности [214а].
После установления стационарного режима колонны при бесконечном флегмовом числе (примерно через 2 ч) из головки отбирают пробу объемом 0,1—0,2 мл; эту пробу для анализа не используют. Еще через 1 ч из головки и из куба отбирают пробы 0,1—0,2 мл и анализируют их. При этом следует руководствоваться теми же положениями, что и при отборе проб при исследовании фазового равновесия (см. разд. 4.6.3). Продолжают через каждый час отбирать и анализировать пробы, пока не будут достигнуты постоянные составы жидкости, взятой из головки и из куба.
Пробы должны быть по возможности малого объема, чтобы избежать возмущений, препятствующих установлению стационарного режима работы колонны. При этом характерно, что в корот-
Рис. 101.
Влияние предварительного захлебывания на эффективность разделения в колонне (по данным Брауэра с сотр.).
Пунктиром показана зависимость при работе колонны без предварительного захлебывания.
159
ких колоннах стационарный режим восстанавливается быстрее, чем в длинных колоннах с большим числом теоретических ступеней разделения.
При испытании длинных колонн необходимо иногда увеличивать интервал между отборами проб (более 1 ч). Время установления стационарного режима при использовании обычных эталонных смесей лежит в интервале 10—20 ч. Испытания рекомендуется проводить как при уменьшении, так и при увеличении нагрузок [208].
Определение разделяющей способности колонны в рабочих условиях проводят по методике, описанной в разд. 4.10.4, при этом устанавливают число эквивалентных теоретических ступеней разделения.
Расчет числа теоретических ступеней разделения. Если уже получено 3—5 постоянных значений концентраций дистиллята и кубовой жидкости, то можно испытание закончить и приступить к расчету числа теоретических ступеней разделения. Для повышения надежности данных испытание повторяют на другой день при тех же условиях.
При аналитическом определении п полученные значения хЕ (дистиллят) и хв (куб) подставляют в уравнение Фенске (1086), справедливое при v = оо. При графическом определении п применяют метод Мак-Кэба и Тиле (см. разд. 4.7.1). Если испытание проведено в рабочих условиях, т. е. при конечном флегмовом числе, то определяют число эквивалентных теоретических ступеней по методике, изложенной в разд. 4.10.4.
Число теоретических ступеней, соответствующее разделяющей способности колонны в рабочих условиях, можно определить также по уравнению, предложенному Оболонцевым и Фростом [202]:
lg xF-axE = “экв ’g (1-х£)-а(1-х£) (132)
Где a — доля отобранного дистиллята от загрузки куба, моль; хЕ и хр—содержание легколетучего компонента соответственно в дистилляте и в исходной смеси, % (мол.).
Оболонцев и Фрост предлагают следующую методику определения числа теоретических ступеней разделения колонны. После достижения стационарного режима колонны отбирают дистиллят при заданном конечном флегмовом числе до тех пор, пока количество отогнанного дистиллята приблизительно не станет равным массе (или числу молей) легколетучего компонента в первоначально загруженной кубовой жидкости состава хР. Таким путем получают числа эквивалентных теоретических ступеней разделения пэкв/и (см. разд. 4.10.4). При испытании по методу'Оболонцева и Фроста сначала работают в течение 3 ч при v — оо и выбранной нагрузке, затем переходят к режиму с конечным флегмовым числом. Так как пользоваться предложенным ими уравнением трудно, была разработана соответствующая диаграмма [84, 202].
160
4.10.9. НЕКОТОРЫЕ ДАННЫЕ ПО РАЗДЕЛЯЮЩЕЙ СПОСОБНОСТИ НАСАДОЧНЫХ
И ТАРЕЛЬЧАТЫХ КОЛОНН
По опубликованным данным практически невозможно сравнить между собой ВЭТС для различных насадочных колонн или коэффициенты полезного действия для тарельчатых колонн. Это объясняется тем, что испытания проводили с различными эталонными смесями в разных условиях и только в редких случаях приводили аппаратурно-технологические параметры, указанные в разд. 4.10 в качестве безусловно необходимых. Дополнительные трудности возникают из-за того, что чистота применяемых эталонных смесей не всегда была гарантирована, а растворение смазки для кранов в отбираемых пробах часто приводит к искажению результатов. По-видимому, в настоящее время необходимо перейти к стандартным методам испытания, чтобы таким образом обеспечить получение сравнимых данных. В свете последних научных достижений становятся необходимыми новые исследования эффективности важнейших насадочных и наиболее распространенных тарельчатых колонн, учитывающие эти достижения и основанные на использовании таких современных точных методов анализа, как инфракрасная спектроскопия, газовая хроматография и масс-спектрометрия.
В табл. 28 дана приближенная оценка эффективности ректификационных колонн; в ней приведены результаты испытания Таблица 28Б.
Результаты испытаний тарельчатых колонн [215]
Автор конструкции колонны или тип колонии
Кезом
Гроль — Олдершоу Зигварт
Бруун
Грайнер и Фридрихе
Штаге (тарелка с брызго-отбойниками)
Штаге (колпачковая тарелка с открытыми сливными патрубками) Штаге (колпачковая тарелка с брызгоотбойни-ками)
Шмюклер и Фриц Бранд
Колонны со стандартны-'s ми шлифами
Расстояние между тарелками, мм Диаметр колонны, мм
23—25 »#33
25 25
65 50
20 или 40 5
40 45
60
50
35—150 30—120
Общая УС тарелки, мл К. п. д. тарелки, % *
1,0 50
около 1—2 80—90
около 7,5 100—110
0,7 или 1,4 70—100
около 2— 2,5 64—66
70—90
80—95
3,75 75—95
80—90 40—90 40—95
* Указанные значения нельзя безоговорочно использовать для сравнения, они Дают только приближенное представление о коэффициенте полезного действия тарелки (см. разд. 7.3.3.)
6 Э. Крель
161
Таблица 28А.
Результаты испытаний эффективности насадочных колонн
Параметры насадки
Насадочные тела Материал Диаметр, мм Высота, мм Толщина стенки, мм Поверхность насадки в 1 л; м2
Кольца Рашнга Стекло гладкое 2,0 2,0 0,5 2,140
То же 2,0 2,0 0,6 1,680
» 3,0 3,0 0,7 1,320
» 4,0 4,0 0,6 1,109
» 4,5 4,5 0,5 1,382
» 5,0 5,0
» 6,0 6,0
» 6,5 6,5 0,5 1,109
» 10,0 10,0
Фарфор 5,5 5,5 1,1
То же 8,0 8,0 1,1
Глина 10,0 10,0 1,7
Проволочная сетка 3,0 3,0
То же 5,0
» 6,0
Кольца «Прим» Металл 2,2 2,5
Спирали Виль- Стекло 3,0 1,5—3,0 0,4
сона » 5,0 5,0 0,65
» 6,33
Разрезные » 2,1
стеклянные 3,1
спирали » 3,1
» 6,33
Спирали Сталь V2A 1,6 1,6 0,2
То же 2,0 2,0 0,2 4,547
4,0 4,0 0,4 1,489
Шарики Стекло гладкое То же 3,0 4,0 0,900
Стекло матированное 4,0 0,601
Стекло гладкое 6,0
Стекло матированное 7,5 0,450
Стекло гладкое 8,0
Седла Фарфор 4,0 1,500
То же 6,0 1,150
» 8,0
» 10,0 0,720
Условия определения числа теоретических ступеней разделения: а см. [216]; б — шим нагрузкам, большие ВЭТС относятся к большим нагрузкам; г) эталонная смесь к различным давлениям н к средней плотности орошения.
162
Параметры колонны ВЭТС по данным различных исследователей Литература
Диаметр, мм Поперечное сечение, см2 к « со со ст н о о АО О _ со 3 2 UfflS Шульце-Штаге [216 ]а Зигварт [2171° Торманн [213 Iе Майлз с сотр. [1921г Различные измерения
5—7
6—9
24 4,5 600 5,82—9,52
35 9,6 900 И,1 7—12
20 3,1 1250 15,5 8—14
24 4,5 600 5,00—14,3
50 19,6 950 18,2
24 4,5 600 3,69—7,5
24 4,5 600 7,07—11,30
24 4,5 600 7,23—12,80
25 4,9 1000 1,4—2,8 Разд. 7,8
20 3,1 600 4,35
50 19,6 1000 1,4—3,5
3—5
24 4,5 600 8,00—13,35
< 7—12
25 4,9 1067 7—9,2
10 0,8 800 4,8
13 1,3 1260 7,4 4—6
20 3,1 1250 9,5
25 4,9 1067 3,6—4,2
40 12,5 1000 1,4—2,0 Собствен-
ные дан-
ные
24 4,5 600 1,25—3,32 То же
24 4,5 600 1,82—5,00 2—5
! 24 4,5 600 6,00—6,06 5-6
6—7
f 24 4,5 600 5,82—7,22
!, 24 4,5 600 3,31—7,50
ho 7,0 450 4,7—6,3 Собствен-
30 7,0 450 6,0—9,0 ные дан-
|30 7,0 450 7,5—11,4 ные
зо 7,0 450 9,0—12,8
[остальные параметры не указаны; в) меньшие ВЭТС относятся преимущественно к меиь-|я-гептаи — метилциклогексаи и «-додекан — циклогексилцнклопеитаи; ВЭТС относятся
6*
163
насадочных и некоторых наиболее часто применяемых тарельчатых колонн. Различные конструкции насадочных и тарельчатых колонн рассмотрены подробно в разд. 7.8 и 7.3.3.
4.11. ГИДРАВЛИЧЕСКОЕ СОПРОТИВЛЕНИЕ, ПРЕДЕЛЬНАЯ СКОРОСТЬ ПАРОВ
И РАСЧЕТ ОСНОВНЫХ РАЗМЕРОВ КОЛОННЫ
Для ректификации при атмосферном давлении в лаборатории обычно применяют колонны диаметром 10—30 мм, а для ректификации под вакуумом — колонны диаметром 20—50 мм. При очень высоких нагрузках колонны диаметром 50 мм можно применять также для разгонки при атмосферном давлении. Если необходимо работать в режиме, близком к захлебыванию, чтобы обеспечить максимальную разделяющую способность, то следует предварительно точно рассчитать диаметр колонны, при котором можно приблизиться к желаемой нагрузке.
Напомним, что под нагрузкой колонны подразумевают количество паров вещества, проходящее в единицу времени через колонну и конденсирующееся в головке колонны с образованием флегмы и дистиллята. Поскольку насыпная насадка и насадки других видов оказывают сопротивление как поднимающимся парам, так и стекающей вниз жидкости, то нагрузку нельзя увеличивать беспредельно. С увеличением скорости испарения, т. е. нагрузки, растет разность давлений в головке и в нижней части колонны, которую называют перепадом давления в колонне (или гидравлическим сопротивлением колонны). Гидравлическое сопротивление колонны зависит от типа и размеров колонны и ее насадки, рабочего давления ректификации, физико-химических свойств смеси, а также от нагрузки или скорости паров. Перепад давления в колоннах с концентрическим зазором можно вычислить по формуле (188). Данные по гидравлическому сопротивлению колонн с вращающейся насадкой приведены в табл. 30 и 31.
Тарельчатые колонны имеют то преимущество, что их нагрузку можно снижать до очень низких значений, при этом их разделяющая способность даже увеличивается. Насадочные же колонны имеют определенную минимальную нагрузку, при которой еще обеспечивается противоточный массообмен. В промышленности во многих случаях колонны регулируют по перепаду давления во избежание захлебывания и для поддержания оптимальной нагрузки. Этот способ успешно применяют также для регулирования лабораторных установок (см. разд. 8.4.2).
Скорость паров (отнесенную к свободному сечению колонны), при которой в колонне начинает накапливаться жидкость, называют нижней предельной скоростью. Верхней предельной скоростью паров называют такую их скорость, при которой накопление жидкости в колонне достигает такой степени, что в тарельча-164
тых колоннах слой жидкости перебрасывается на вышележащую тарелку, а в насадочных колоннах в нижней части образуется пенящийся слой жидкости, который постепенно поднимается в верхнюю часть колонны.
После фундаментальных работ Маха [219], Барта [220] и Лева [221 ] появилось много статей, в которых были рассмотрены закономерности, связанные с гидравлическим сопротивлением насадочных колонн. При тщательном экспериментальном исследовании взаимосвязи между перепадом давления и разделяющей способностью насадочных колонн Брауэр [208] определил сопротивление насыпной насадки 13-ти различных видов (см. табл. 29) в зависимости от нагрузки по жидкости. Испытания проводили
Таблица 29.
Параметры насадочных тел, использованные Брауэром [208] при измерении гидравлического сопротивления колонн
Тиг насадочных тел Материал Размеры, мм Соотношение диаметров колонны и насадочного тела Свободный объем насадки м*/м8 Удельная поверхность насадки, м*/м8 Длина проволоки или ленты, из которой изготовлено насадочное тело, мм
1. Спирали с разомкнутыми витками V2A (сталь) 3X3X0,45 1 : 12,3 0,777 1802 43
2. Спирали с разомкнутыми витками То же 4X4X0,5 1 : 9,25 0,822 1480 51.
3. Спирали с разомкнутыми витками » 3X3X0,40 1 : 12,3 0,771 .1815 44
4. Спирали с сомкнутыми витками » 2X2X0,3 1 : 18,5 0,661 2680 32
5. Спирали с сомкнутыми витками » 3X3X0,45 1 : 12,3 0,704 1717 60
6. Спирали с сомкнутыми витками V4A 4X4X0,4 1 : 9,25 0,760 1543 100
•7. Кольца с перемычкой (свернутые из ленты) V2A 2X2X0,25 1 : 18,5 0,716 1965 10,5
8. Кольца с перемычкой То же 4X4X0,3 1 : 9,25 0,824 1176 18,7
9. Кольца с перемычкой » 6X6X0,4 1 : 6,16 0,811 946 24,1
10. 11. Кольца Рашига Эллипсы Фарфор Алюминий 5X5 3,7X7,4 1 : 7,4 0,475 0,306 977
12. 13. Шарики Кольца Рашига Стекло Фарфор 2 16X16 1 : 18,5 1 : 2,3 0,339 1786
105
Рис. 102.
Зависимость удельного гидравлического сопротивления насыпной насадки различных типов (см. табл. 29) от плотности орошения (по данным Брауэра).
Прямыми а—а, b—b, с—с отмечены границы гидродинамических режимов [см. уравнение (133)].
в колонне диаметром 37 мм с рабочей высотой 1 м при атмосферном давлении со смесью н-гептан — метилциклогексан. Результаты весьма тщательных измерений, представленные на рис. 102, позволяют сделать следующие выводы:
1) Все кривые похожи одна на другую. Точки излома, ограничивающие различные режимы, лежат на линии изломов а—а, b—b и с—с. 2) Во всех интервалах нагрузок, ограниченных
точками излома, ход кривых, отражающих изменение гидравлического сопротивления, можно описать с помощью уравнения вида:
^Plh—cfn (133)
где: п = 1,0 для области ниже а—а (ламинарный поток паров); п = 1,78 для области между а—а и Ь—Ь (турбулентный поток); п = 2,7 для области между b—b и с—с (турбулентный поток); п = 3,3 для области выше с—с (затопление, пено-образование);
Коэффициент с является функцией свойств паров и жидкости, а также параметров насадки.
Рис. 103 иллюстрирует взаимосвязь между гидравлическим сопротивлением насадки и ее разделяющей способностью. Линейная зависимость числа теоретических ступеней, приходящихся на 1 м рабочей высоты колонны, пуд для насадки из спиралей (см. табл. 29) от гидравлического сопротивления при турбулентном движении паров показывает, что во всем интервале нагрузок для турбулентного режима между переносом количества движения, тепла и вещества в процессе ректификации существует аналогия. Механизм массообмена при этом остается неизменным. Выше линии изломов с—с (см. рис. 103) пар в виде пузырей барботирует через накопившийся слой жидкости, причем процесс массообмена протекает уже по другому закону [203, 206, 208].
Каст [222] учел истинный путь, который проходит двухфазный поток и вместо высоты слоя насадки ввел понятие эффек
166
тивной длины (х7Ук; коэффициент у. является физическим параметром, характеризующим искривленность траектории потока. Каст установил, что его опытные результаты в области перехода от ламинарного потока паров к турбулентному в колоннах с насадочными телами размером до 50 мм, а также данные других авторов находятся в хорошем согласии с полученным им уравнением для расчета гидравлического сопротивления колонны. Тойч [223] вывел полуэмпирическое уравнение для упрощенного расчета перепада давления в колонне (в безразмерной форме); соответствующие исследования были ограничены насадочными телами размером не менее 25 мм. Указана область применимости этого уравнения. Путем обработки опытных данных Гелбе [224], изучавшим работу насадки из спиралей с разомкнутыми витками размером 4x4x0,5 мм в интервале рабочих давлений 10—740 мм рт. ст., Глазер [225] использовал в расчетах безразмерный параметр е:
<«>
где р — плотность паров, кг/м3; g — ускорение свободного падения, м2/с; Нк — рабочая высота колонны, м; ВЕП — высота единицы переноса, м; Др — перепад давления, кгс/м2; Хк — объем жидкости в колонне, м3.
Зависимость параметра е ог важнейших переменных также можно представить в безразмерной форме. Параметр е изменяется в зависимости от нагрузки; эта зависимость позволяет четко проследить взаимосвязь между переносом количества движения и вещества, а также учесть влияние давления.
На основе собственных и литературных данных Райхельт, а также Райхельт и Бласс провели анализ гидродинамических закономерностей, характерных для насадки из колец Рашига и шаров. В результате были получены уравнения для расчета гид-
Рис. 103.
Взаимосвязь между перепадом давления и разделяющей способностью насадки различных типов (см. табл. 29) (по данным Брауэра).
Рис. 104.
Зависимость гидравлического сопротивления насадки из фарфоровых седел размером 4—8 мм от скорости воздуха при плотности орошения водой 40 мл/(см2-с).
167
1,0 1,1 1,2 1,3 1,4 1,5 1,6 w, м/с
Рис. 105.
Зависимость гидравлического сопротивления иасадки из спиралей Вильсона размером 3 мм от скорости воздуха при расходах орошающей жидкости от 200 — 1800 мл/ч.
равлического сопротивления в однофазном потоке, проходящем через насадку из шаров и цилиндров.
Хекманн и Крель [226] исследовали гидравлическое сопротивление фарфоровых седел размером 4, 6 и 8 мм с использованием системы воздух—вода. Рабочая высота колонны составляла 1 м, а диаметр — 22 мм. Опытные данные, представленные на рис. 104, можно использовать только в качестве приближенной оценки перепада давления. Рис. 105 показывает, как сказывается увеличение расхода жидкости на гидравлическом сопротивлении насадки из спиралей Вильсона размером 3 мм. Очень выгодны (малый перепад давления) также кольца из проволочной сетки, как это видно из рис. 106 и 107. На них представлены результаты опытов, проведенных с использованием различных смесей.
Основываясь на опыте, приобретенном при стендовых испытаниях аппаратуры, Нойманн [227] ввел понятие «эквивалентной» скорости воздуха для оценки нагрузки по пару или жидкости.
Рид и Фенске [228] вывели уравнение для расчета перепада давления, которое Брауэр [208] привел к виду
Д<- ° X- wG ' <'®>
где р — абсолютное давление, кгс/м2; Др — разность давлений, кгс/м2; М — молекулярная масса; е0 = (VK — VH)/VK — свободный объем сухой насадки;
168
Ик — объем колонны, соответствующий рабочей высоте, м3; Vh — объем насадки, м3; Vp — объем удержанной жидкости, м3; Р — универсальная газовая постоянная, кгс-м/(кгс-°С); Т — абсолютная температура, К; Нк — высота слоя насадки (рабочая высота колонны), м; g — ускорение свободного падения, м2/с; т]—динамическая вязкость паров,кгс-с/м2; р — плотность паров, кгс-с2/м4; О — удельная поверхность насадки, м2/м3; D — удельный расход паров, м3/(м2 • с).
Сравнение результатов расчета по формуле (135) с экспериментальными данными дает очень хорошее согласие при оптимальном способе укладки насадочных тел.
Кривые на рис. 108 [229], а также данные, приведенные в табл. 30 [200], свидетельствуют о том, что физико-химические свойства перегоняемого вещества также существенно влияют на перепад давления. Данные табл. 30 относятся к ситчатой колонне Олдершоу. Следует указать на то, что в этом эксперименте применяли специально подобранные вещества, которые при температуре кипения имели неодинаковые значения плотности и поверхностного натяжения.
Умхольц и Винкль [230] определили зависимость перепада давления от нагрузки в ситчатых колоннах диаметром 25 мм,
О 2 6 6 8 10 12 16 16 18
Нагрузка, л/ч
Рис. 107.
Зависимость перепада давления, приходящегося иа одну теоретическую ступень разделения, в слое колец из проволочной сетки от нагрузки при остаточных давлениях от 10 до 735 мм рт. ст.
Диаметр колонны 5 см, рабочая высота 1 м, данные Тюркауфа.
Рис. 108.
Зависимость удельного гидравлического сопротивления колонны от скорости паров различных веществ (по данным Штаге):
/ — этанол; 2 — хлороформ; 3 — бензол; 4 — н-гептан; 5 — динзопро-пиловый эфир; 6 — дравннол; 7 — толуол; 8 — этнлацетат.
169
Таблица 30.
Гидравлическое сопротивление реальной тарелки Др ситчатой колонны Олдершоу при ректификации различных веществ
Диаметр колонны 28 мм, 30 реальных тарелок
н-Гептан (^6и°п = = 98,424 °C, рЭД = 0,68368) Мети лци к л ог екс а и (I760 = 101,10 °C, кип р2° = 0,76944) Ж Бензол (№$ = кип = 80,09 °C, = 0,87895) Муравьиная кислота (/760 _ 100 75 »с кип рЭД = 1,2206)
нагрузка, мл/ч Др, мм рт. ст. нагрузка, мл/ч Др, мм рт. ст. нагрузка, мл/ч Др, мм рт. ст. нагрузка, мл/ч Др, мм рт. ст.
1000 1000 1000 0,76 200 0,88
1500 0,50 1500 0,62 1500 0,82 400 0,95
2000 0,53 2000 0,63 2000 0,84 600 1,02
2500 0,55 2500 0,65 2500 0,87 800 1,09
3000 0,57 3000 0,67 3000 0,91 1000 1,18
3500 0,60 3500 0,71 3500 1,00 1125 1,23
4000 0,64 4000 0,77 4000 1,11
4500 0,70 4500 0,85 4280 1,17
5000 0,77 5000 0,93
5230 0,80 5230 0,97
Поверхностное натяжение использованных веществ изменяется в интервале 20— 40 дин/см (на границе с воздухом) при 20 °C.
в которых с шагом 25 мм установлены тарелки с долей свободного сечения 0,162, имеющие различное число отверстий неодинаковых диаметров. Из табл. 31 [192] видно, каким образом влияет рабочее давление ректификации на гидравлическое сопротивление и предельные нагрузки в колонне. В частности, согласно данным, приведенным в табл. 31, насадочным телам в форме шариков соответствует относительно узкий интервал рабочих нагрузок и большое гидравлическое сопротивление, что связано с малой долей свободного объема (0,26) насадки этого типа. Напротив, для насадок «Хели-грид» (см. разд. 7.3.4) имеется очень широкий интервал рабочих нагрузок и большая верхняя предельная нагрузка (см. также [39] к гл. 1).
Для расчета необходимого диаметра колонны следует знать верхнюю предельную скорость паров. На основании чисто теоретических рассуждений Шумахер [231 ] рассчитал константы скорости kn (м/с) для различных типов насадки по следующей формуле:
kn = P«K'n/rff,g или ®П = У kndnS/ Pfl (136)
где Рц — приведенная плотность паров; р^ = Рг/Рж" плотность паров (г/л)/плот-ность жидкости (г/л) (при рабочем давлении), te>n — предельная скорость паров; dH—диаметр насадочного тела, м; g — ускорение свободного падения.
170
При использовании этой формулы предполагают постоянство вязкости паров и жидкости и учитывают влияние на предельную скорость паров типа и размеров насадочных тел, плотности паров и жидкости, но игнорируют влияние вязкости паров и жидкости и поверхностного натяжения жидкости.
Для константы скорости kn установлены следующие значения в зависимости от вида насадки: 0,01 для колец Рашига, 0,021 для колец «Прим», 0,019 для седел, 0,004 для кусковой насадки, 0,0126 для спиралей (по данным автора).
Таблица 31.
Перепад давления и предельные нагрузки для различных насадочных тел при остаточном давлении 20—760 мм рт. ст. в колонне диаметром 25 мм с рабочей высотой 1,07 м
Тип насадочного тела Размер насадочного тела, мм Остаточное давление, мм рт. ст. Перепад давления, мм рт. ст. Предельная нагрузка, г/ч
мин. макс. мни. макс.
«Хели-грид» — 50 5 28 1200 1650
100 5 28 650 1600
150 6 27 550 1100
760 1 27 500 5500
Шарики (неплотная упа- 3,6 20 6 26 450 700
ковка) 50 5 32 500 900
100 6 35 700 1150
150 5 32 1000 1400
760 3 27 125 2500
Шарики (плотная упаков- 3,6 20 3,5 31 380 720
ка) 50 3,5 30 620 900
100 4 30 800 1170
150 4 35 1000 1300
Седла 6,3 20 2 8 450 800
50 1 8 600 1100
150 2 8 900 2200
760 2 800 2600 4800
Стеклянные спирали с тре- 6,3 20 1 5 450 900
мя витками 50 1 5 800 1400
150 1 5 1000 2200
760 1 — 1200 1600
Одинарные стеклянные 6,3 20 2 14 450 900
витки 50 3 14 970 1200
150 3 18 1450 2000
760 2 — 2400 —
171
Рис. 109.
Номограмма Шумахера для определения предельной скорости паров для насадок.
— диаметр насадочного тела.
Для быстрого расчета искомой предельной скорости паров была разработана номограмма (рис. 109), применение которой поясним на следующем примере.
Пусть нужно определить предельную скорость паров при ректификации органической кислоты нормального строения С? при 20 мм рт. ст. (/|®п = = 125,0 °C). Насадочные тела — седла размером 4 мм. Определяем необходимые величины:
kn =0,019
20
рп = -ЧС|а • 130'0,01605 = 0,105 [по формуле (32)] оУо
0^= —= (при 20 мм рт. ст.) = 0,0001165 = 0,1165-10-8
« pF 900 (г/л) v г '
На номограмме рис. 109 необходимые построения для определения предельной скорости показаны пунктирной линией. Через точку пересечения вертикальной линии, соответствующей константе скорости 0,0190, и горизонтальной линии, соответствующей диаметру d = 4 мм, проводят линию, параллельную 172
наклонным линиям сетки. Из точки пересечения этой параллельной линии с вертикальной линией, соответствующей значению = 0,12, проводят горизонталь до вертикальной шкалы предельных скоростей и считывают искомую величину 2,5 м/с.
Конечно, с такой предельной скоростью паров колонна работать не может, поэтому берут 50—70% от найденного значения (60% соответствуют скорости 1,5 м/с).
Усложним поставленную задачу, пусть теперь нужно определить необходимое поперечное сечение колонны для перегонки кислоты С7 нормального строения при 20 мм рт. ст. и нагрузке 400 г/ч. Для расчетов понадобится формула
Р-22 400 (273 -1- 0 760
щп-3600-273р
(137)
где f — поперечное сечение колонны, см2; D — расход паров в колонне, моль/ч; t — температура перегонки, °C; wn — выбранная скорость паров, см/с; р — рабочее давление процесса перегонки, мм рт. ст.
Выполнив простейшие арифметические операции в формуле (137), получим
DT 22 400-760
wnp ’ 3600-273
(138)
При нагрузке D = 400 г/ч = 3,4 моль/ч при = 150 см/с требуется колонна с поперечным сечением:
Отсюда рассчитываем радиус колонны
г = Vf/л = К7,9/3,14 = 1,6 см
(140)
Для ректификации при атмосферном давлении формула (138) приводится к виду
/ =0,022807/к>п (см2)
(141)
Для сравнения подсчитаем, какая предельная нагрузка может быть при перегонке н-гептана в колонне диаметром 30 мм при атмосферном давлении. Предельная скорость паров при работе с насадкой из седел размером 4 мм составит 0,38 м/с. Отсюда
D = /аУп/0,028871 = 7,9-38/(0,0288-371) = 35,5 г-моль/ч = 3550 г/ч = 5200 мл/ч
(142)
То, что такие высокие нагрузки вообще возможны в колонне диаметром 30 мм, доказывают опыты Коллинга [171 ]. Сравнительный расчет был проведен главным образом для того, чтобы показать, насколько существенно увеличивается скорость паров и, следовательно, уменьшается нагрузка при переходе к пониженному давлению.
Автору до сих пор не известны какие-либо специальные методы расчета предельной скорости паров для лабораторных тарельчатых колонн. Уравнения, справедливые для промышленных колонн, в данном случае не могут быть использованы, так как дают большие погрешности. Опыт показывает, что тарельчатые колонны могут быть нагружены на х/3 от нормальной нагрузки насадочных колонн того же диаметра. Это обусловлено тем, что, во-первых, слой жидкости на тарелках и, во-вторых, паровые патрубки со
173
сравнительно узким поперечным сечением приводят к увеличению гидравлического сопротивления.
Вопросы гидродинамики потоков в насадочных колоннах и в зернистых слоях подробно рассмотрел Барт [220], который, в частности, отметил, что насадочные тела с острыми кромками вызывают в два и три раза большее гидравлическое сопротивление по сравнению с насадочными телами округленной формы.
На основании анализа гидродинамических закономерностей однофазных потоков, движущихся в слоях насадки, Дэвид [191] наметил последовательность расчета числа теоретических ступеней разделения в насадочной колонне с кольцами Рашига, имеющими размеры от 8 до 50 мм. Дэвид исходил при этом из той предпосылки, что перепад давления, скорость паров и плотность паров вследствие их сильного влияния на разделяющую способность насадочной колонны должны быть учтены в любой расчетной методике. Исходя из известного уравнения для определения потери напора в трубопроводах, коэффициент трения можно представить в следующем виде:
g = (143)
дапРпЛ
где Др — перепад давления, мм вод. ст.; d„—диаметр насадочного тела, м; шп — скорость паров, отнесенная к свободному сечению колонны, м/с; рп — удельный вес паров, кгс/м3; h — высота слоя насадки, м.
Отношение числа теоретических ступеней разделения, приходящихся на 1 м насадки, пуд к коэффициенту трения £ было принято в качестве базовой величины, не зависящей от критерия Рейнольдса. Эта величина, имеет постоянное значение (0,13) для всех смесей, использованных Дэвидом. Он принимает, что эффективность разделения имеет максимальное значение при верхней предельной скорости паров. Однако в лабораторных колоннах эта скорость не намного превышает ту скорость паров, которая соответствует минимально допустимой нагрузке. Поэтому соотношение
попт = 0,13g (144)
имеет силу также для лабораторных колон, если эффективность разделения максимальна (попт отнесено к 1 м рабочей высоты колонны).
При обобщении экспериментальных данных было получено следующее эмпирическое уравнение для расчета рабочей высоты колонны Нк:
Нк = 1 +(« — 0,13g)(10+ </н)/120 (м) (145)
Уравнение (145) справедливо для флегмового числа v — оо, т. е. для соотношения расходов жидкости и паров равного 1. В результате обработки результатов 115 опытов с использованием системы вода—воздух Бек [244] предлагает новые уравнения для насадочных тел размером 8—80 мм и для колонн диаметром от 150 до 1200 мм;
174
4.12. ТЕПЛОВЫЕ РАСЧЕТЫ
Тепловые расчеты процесса лабораторной перегонки проводят редко, поскольку в данном случае затраты энергии по сравнению с полупромышленными или промышленными установками весьма незначительны. Обычно в лабораториях перегонку проводят при большем или меньшем избытке тепла, а фактическую потребность в электрической энергии регулируют с помощью дополнительных сопротивлений. В лабораторной практике газ до сих пор еще применяют при дистилляции по методу Энглера, при аналитических разгонках, как средство обогрева масляных, песочных бань и бань с металлическими теплоносителями. Применения открытого газового пламени для нагревания избегают при перегонке веществ с высоким давлением паров ввиду возможной опасности перегрева жидкости, растрескивания аппаратуры или взрыва. В настоящее время предпочтение отдают электрическому обогреву при помощи закрытых колбонагревателей или нагревательных устройств, в которых электрическая спираль защищена слоем изоляционного материала. Для достижения невысоких температур применяют инфракрасное излучение (в видимой и невидимой частях спектра), которое обладает всеми преимуществами радиационного обогрева [232]. Применение токов высокой частоты для нагревания в лабораторных условиях находится еще только в стадии проверки.
При периодической перегонке разделяемую смесь, загруженную в куб, сначала нагревают до температуры кипения. При дальнейшем подводе тепла (в соответствии с энтальпией испарения смеси) происходит испарение. Скорость испарения зависит от количества тепловой энергии, подводимой в единицу времени. При непрерывной ректификации часть тепла подводят к исходной смеси уже в теплообменнике для предварительного подогревания исходной смеси. Основная часть тепловой энергии расходуется для нагревания куба.
Обозначим общий расход тепла через Q; тогда уравнение общего теплового баланса для различных видов перегонки без учета тепла, необходимого для подогревания кубовой жидкости до температуры кипения, будет иметь следующий вид:
для дистилляции
Q = Qk = Qa Qn == Окон 4- Qn (146)
для периодической ректификации
Q = Qk = Од 4" Q<b 4" Qn — Qkoh 4" Qn (147)
для непрерывной ректификации
Q — Qk 4" Опит — Од 4" Оф 4~ Qn = Окон 4" Qn (148)
где Qk — количество тепла, подводимое в кубе (колбе); QnHT — количество тепла, подводимое при непрерывной ректификации в теплообменнике предварительного подогрева; 0д—энтальпия дистиллята (в парообразном состоянии); Оф — эн-
175
Рис. 110.
Схема теплового баланса при дистилляции (а), периодической (б) и непрерывной (в) ректификации.
тальпия флегмы (в парообразном состоянии); Qn—тепловые потери; QKOh— количество тепла, отводимое в конденсаторе при конденсации паров и охлаждении дистиллята.
Эти уравнения справедливы при условии, что дистиллят и флегма охлаждены до температуры, при которой смесь поступает в куб. Дополнительные пояснения к приведенным соотношениям даны на рис. ПО.
Чтобы дать представление о затратах тепла при лабораторной перегонке, приведем следующий пример. Пусть необходимо разделить смесь бензол—толуол [50% (масс.) ] при атмосферном давлении с флегмовым числом 2 при нагрузке колонны 0,5 кг/ч и загрузке 2 кг исходной смеси (20 °C). Физико-химические параметры компонентов этой смеси, необходимые для дальнейших расчетов, таковы:
Бензол
°С........................ 80,1
Теплоемкость ср при 50 °C, ккал/(кг-°С)................... 0,43
Энтальпия испарения ДЯу, ккал/кг........................ 93,8
(при 80,1 °C)
Толуол
110,6
0,43
86,5 (при 109,7 °C)
* 1 ккал = 4168,84 Дж, 1 Дж = 2,388- 10-4 ккал.
Принимаем, что испаряется только бензол, хотя в действительности с повышением температуры в кубе испаряется и некоторая часть толуола (вследствие увеличения концентрации толуола 176
при отгонке бензола). Обусловленная этим допущением незначительная погрешность несколько завышает расход тепла, так как энтальпия испарения для толуола меньше, чем для бензола. Тепловые потери оценить весьма трудно; они складываются из тепловых потерь в кубе, головке колонны и, главным образом, в самой колонне. Тепловые потери в кубе в значительной степени можно уменьшить применением подходящего теплоизолирующего материала (см. разд. 7.7). Методы устранения тепловых потерь в самой колонне изложены ниже. Затраты тепла на подра л > смеси до 80 °C составляют г*" 1
QK = в — Л) ср 0п (149) 1
QK=2(80— 20) 0,43 ккал = 51,6 ккал (150) I
+ Тепловые потери (10%) ККал I
56,8 ккал I
Затраты тепла на испарение дистиллята и флегмы составляют L
QK=(E + R)&HV -( Qn (151)1
Qk = (0,5 + 1,0)93,8 ккал/ч = 140,7 ккал/ч (152) 1
, -г 14,0 ккал/ч 1
+ Тепловые потери (10%) т——---’
r v ' 154,7 ккал/ч
где В — количество исходной смеси в кубе, кг; С — температура в гдццс! [Lj-гонки, °C; 4—температура исходной смеси. °C.
Исходя из эквивалентного соотношения между электрической и тепловой энергией
1 кВт = 860 ккал (153)
получим, что для проведения данной разгонки необходим электрический подогреватель с фактической мощностью
154,7/860 = 0,18 кВт = 180 Вт (154)
Расчет поверхности конденсатора обсуждается в разд. 7.4.
Формула (191) может быть использована для расчета греющей поверхности теплообменника для подогрева питающей жидкости при непрерывной ректификации (в этой формуле Q — количество тепла, подводимое к жидкости; F — поверхность теплообмена, Д/ — разность между температурой греющей среды и температурой нагреваемой жидкости). Средняя разность температур вычисляется по формуле (192).
В лабораториях обычно приходится работать со стеклянными приборами; в случае применения стекла в качестве конструкционного материала коэффициент теплопередачи можно принять приблизительно равным 2,5-10-2 ккал/(см2-ч-°C). При использовании обычных круглодонных колб для перегонки при нормальных скоростях испарения поверхность колбы вполне достаточна для подвода необходимого тепла.
Чтобы обеспечить возможно более полное исключение тепловых потерь, в качестве тепловой изоляции колонн используют
177
.Рис. 111.
Зависимость тепловых потерь в изолированной колонне от рабочей температуры.
1 — колонна с вакуумированной рубашкой; 2 — колонна с изоляцией из стекловолокна, толщина слоя 50 мм; 3, 4 — колонны, изолированные магнезией (85%), толщина слоя изоляции 25,4 мм (3) и 50,8 мм (4).
1,2 — данные Роуза [158]; 3, 4 — данные
Юнга [233].
в первую очередь материалы с очень низкой теплопроводностью (см. разд. 7.7), например магнезию, асбест, минеральную вату и стеклянную шерсть [1531. Однако это не всегда полностью достигается (табл. 32).
Юнге [233 ] провел измерения тепловых потерь в колонне с вакуумированной рубашкой и в колонне с тепловой изоляцией из стекловолокна толщиной 50 мм. Результаты этих исследований в зависимости от температуры внутри колонны приведены на рис. 111; на рисунке представлены также данные Роуза [153], взятые из табл. 32.
* Тепловыми потерями в ректификационных колоннах никак нельзя пренебрегать при балансовых опытах. «Дикая» флегма, вызванная тепловыми потерями в колонне, может быть исключена лишь в том случае, если дополнительно обогревать изоляционный кожух. Только адиабатически работающая колонна может давать наилучшие и воспроизводимые результаты.
Тепловые потери в теплоизолированной колонне можно рассчитать по следующей формуле:
'к-2лНкЫ Qn=z 2,3 lg (</H3/rfK)
где К — теплопроводность теплоизоляционного материала, ккал/(м-ч-град); Qn — тепловые потери, ккал/ч; Нк — высота колонны, м; At — разность между Таблица 32.
Тепловые потери в колонне со слоем изоляции из магнезии (85% оксида магнии)
Колонна диаметром 25,4 мм и высотой 30,5 см
(155)
Перегоняемое вещество Разность температур /760 — 25, кип °C Толщина изолирующего слоя, 25,4 мм Толщина изолирующего слоя 50,8 мм
тепловые потери 1 «дикая» флегма, мл/ч тепловые потери «дикая» флегма, мл/ч
кал/ч кал/см2 кал/ч кал/см2
Бензол 55 4 800 20 54 3450 14,3 42
Толуол 85 7 600 31,6 101 5400 22,4 73
п-Цимол 151 13 800 57,2 238 9800 40,7 170
178
температурой внутри колонны и температурой окружающей среды; с/из — наружный диаметр тепловой изоляции, м; dK — диаметр колонны, м.
Пример. Нужно рассчитать тепловые потери в колонне диаметром 25 мм и высотой 5 м. В качестве изоляции использована стеклянная шерсть в виде крошки [X = 0,055 ккал/(м-ч-°C)] толщиной 62,5 мм. Температура внутри колонны составляет 76,6°С, комнатная температура 18°С. По формуле (155) находим:
_ 0,055.6,28-5.58,6 .. , ,
Qn =-----2 31о-6---= 56,5 ккал/ч (156)
Таким образом, несмотря на изоляцию, имеют место тепловые потери (56,5 ккал/ч), которые следует компенсировать дополнительным обогревом во избежание накопления жидкости у стенок колонны.
4.13. ХАРАКТЕРИСТИКА РАЗДЕЛЯЕМЫХ
ВЕЩЕСТВ И ПОЛУЧЕНИЕ КРИВЫХ РАЗГОНКИ
При лабораторной перегонке наряду с температурой кипения определяют также такие характеристики дистиллята, как плотность. показатель преломления, температуры затвердевания и плавления, а иногда также молекулярную массу и иодное число (рис. 112). Точную характеристику продукта можно получить, измеряя несколько Показателей (особенно при аналитических разгонках). Фракционный анализ дистиллята только по температуре кипения в большинстве случаев приводит к ошибочному заключению [234].
В последнее время для идентификации фракций все шире привлекают инфракрасную спектроскопию, масс-спектроскопию, а также газовую хроматографию. Для анализа все чаще применяют проточные приборы, обеспечивающие непрерывность контроля.
Физико-химические константы разделяемых веществ обычно берут из справочников и руководств (см. в [235]), однако к этим данным следует относиться весьма критически. Часто опубликованные данные значительно расходятся между собой. Даже для таких веществ, как бензол и 1,2-дихлорэтан, указанные значения физико-химических констант расходятся между собой на величину, которой нельзя пренебречь. Обычно отдают предпочтение более поздним данным, относительно которых можно предположить, что они определены с максимально возможной точностью. Если на диаграмму разгонки нанести теоретические значения констант чистых веществ (см. рис. 84), то оценка полученных результатов значительно упростится.
К сожалению, в литературе не всегда можно найти все необходимые значения физических констант, и требуется либо пересчитать физические константы для других условий, либо рассчитать их заново.
Фундаментальный труд Рида и Шервуда [235] посвящен физическим свойствам газов и жидкостей и методам их расчета. Методы расчета термодинамических параметров газов и жидкостей весьма удачно систематизированы также и в книге Хехта с сотр. [236 ].
179
Показатели преломления в разных справочниках часто указываются для различных температур, поэтому не исключена возможность применения интерполяции. Для членов гомологического ряда веществ графическая зависимость показателя преломления от числа атомов углерода для различных температур, как показал Штаге [237], имеет вид равносторонней гиперболы. Используя эту закономерность, можно корректировать соответствующие диаграммы (рис. 113). Следует учитывать также, что отклонение температуры на 1 °C приводит к изменению показателя преломле; ния в среднем на 5 единиц в четвертом знаке после запятой, поэтому необходимо очень точно регулировать температуру в ячейке рефрактометра, по возможности с точностью до 0,1 °C. Очень удоб-
Рис. 112.
Диаграммы разгонки бензиновой фракции.
180
1&30
1,kW
е W
1,390
1,380
1,310 .
10 15 20 25 30
пс
Рис. ИЗ.
Зависимость показателя преломления карбоновых кислот нормального строения от числа атомов углерода для различных температур.
ные номограммы для расчета различных физических констант чистых веществ приведены в работах Харбанда [2381, а также Орличека и Пелля [239].
При проведении перегонки при атмосферном давлении необходимо постоянно следить за барометрическим давлением. Отклонения порядка 20 мм рт. ст. могут привести, например, к из
менению температуры кипения бензола на 1 °C. Даже если рабочий барометр точно откалиброван по прецизионному барометру, необходимо вводить дополнительную поправку, учитывающую температурную деформацию столбика ртути и шкалы. С помощью этой поправки результаты измерений приводят к 0 °C, так как согласно определению единица давления 1 мм рт. ст. соответствует температуре 0 °C. Номограмма Хойсслера (240] позволяет без дополнительных расчетов сразу определить скорректированное значение барометрического давления (рис. 114). Номограмма основана на зависимости:
6кор=6[1 — (а — ₽Н1 (мм рт. ст.) (157)
где 6кор — скорректированное барометрическое давление; Ь — отсчитанное барометрическое давление; а — коэффициент линейного расширения ртути (0,000182 на 1°С); Р — коэффициент линейного расширения шкалы (для латуни — 0,000019, для стекла — 0,000008); t — комнатная температура, °C.
В соответствии с правилами калибровки стеклянных термометров (см. разд. 8.2.1) термометр погружают в калибровочную жидкость до деления шкалы, соответствующего измеряемой температуре. Если столбик ртути не погружен полностью в жидкость или пар, температуру которых измеряют, то следует внести поправку на выступающий столбик ртути. Для измерения средней температуры выступающего участка ртутного столбика рядом : с рабочим термометром устанавливают вспомогательный термометр. Скорректированные показания рабочего термометра определяют по формуле:
| ^кор = 1 + ka {t — ^ср) (158)
[где t — показание рабочего термометра; k — кажущийся коэффициент объемного расширения ртути в стекле (для термометров из йенского стекла № 16 k = Р= 0,000157, а йенского стекла № 2954 k = 0,000163); а — длина выступающего ктолбика ртути, выраженная в градусах; <Ср — средняя температура выступаю-|цего столбика ртути, показываемая вспомогательным термометром.
181
Номограмма Коуля [2411 построена на основе зависимости (158) при k = 0,000156. Применительно к перегонке при атмосферном давлении номограмма дополнительно позволяет определить температуру кипения точно при 760 мм рт. ст. (обычно для удобства сравнения результаты пересчитывают относительно этого давления).
Корректировка температуры кипения базируется на зависимости:
= (Ь — 760) dt/dp (159)
где dtldp — производная температуры кипения от рабочего давления в мм рт. ст. (зависит от природы вещества). .
Комбинирование уравнений (158) и (159) приводит к соотношению, позволяющему пересчитывать температуру кипения для 760 мм рт. ст.
К =- [ka (t — t0) ] — (b — 760) dt/dp (160)
Номограмма, приведенная на рис. 115, облегчает приведенный выше расчет. Этой номограммой пользуются следующим образом.
Сначала вычисляют t — t0, затем полученную на шкале А точку соединяют с соответствующей точкой на шкале а. Затем точку пересечения на шкале Б соединяют со значением барометри-
Рис. 114.
Номограмма Хойсслера для корректировки показаний барометра.
182
Рис. 115.
Номограмма Коуля для корректировки показаний термометра.
ческого давления (в мм рт. ст.) на шкале В. Точка пересечения последней прямой со шкалой dtldp дает скорректированное значение t, соответствующее полному погружению в жидкость термометра и давлению 760 мм рт. ст.
Пример. Допустим, что при перегонке под атмосферным давлением температура в головке колонны соответствует точке кипения фенола и составляет t = = 179,8 °C. Средняя температура выступающего ртутного столбика оказалась равной /ср = 30°С (она измерена вспомогательным термометром, шарик которого подведен к середине непогруженного участка ртутного столбика). Длина выступающего столбика соответствует а = 40 °C, барометрическое давление 750 мм рт. ст.; для фенола dtldp = 0,045.
Находим t— tcp— 149,8 °C. Соединим точку 150 на шкале А с точкой 40 на наклонной шкале а, и на шкале Б получим точку пересечения 0,95. Это и есть поправка на столбик ртути. Эту точку соединяем с точкой 750 на шкале В и получаем окончательную поправку ~Н,4°С в точке пересечения линии 0,045 шкалы dtldp с прямой, соединяющей шкалы Б и В. Таким образом, скорректированная температура равна (179,8 °C + 1,4 °C) = 181,2 °C (см. рис. 115).
Аналогичную методику для уточнения температур кипения 1ри 760 мм рт. ст. предлагает Рекхард [57], который определил
183
Выход дистиллята, °/q
Рис. 116.
Ступенчатая диаграмма непрерывной разгонки сырой смеси жирных кислот.
6
Mff 8 10 1?. /4 1618 20
пс
Рис. 117.
Столбиковая диаграмма распределения постоянно кипящих фракций по числу атомов углерода для сырой смесн жирных кислот.
групповые константы с в уравнении (18) (разд. 4.4) для 400 органических веществ. Данный способ применим также и к многокомпонентным смесям.
Для записи значений измеряемых величин в лабораторных условиях очень удобно пользоваться печатными бланками (карты опытов). С помощью записей в этих бланках значительно упрощается построение диаграммы разгонки. Диаграмму разгонки можно строить либо по возрастанию выхода дистиллята (см. ниже рис. 118), либо по возрастанию температуры (см. рис. 84, 112). В то время как первый способ еще применяют в промышленности для анализа многокомпонентных смесей (нефть, бензин), все большее распространение получает второй способ. Согласно этому способу на оси ординат откладывают значения температуры кипения, а на оси абсцисс — выход дистиллята. Для повышения точности отсчета шкалу температур обычно строят в более крупном масштабе, чем шкалу выхода дистиллята. Целесообразно также (если при этом не снижается наглядность) наносить на диаграмму разгонки все измеренные величины, в том числе температуру куба, и кривую изменения рабочего давления. После того как на основании результатов измерений идентифицированы определенные фракции, на диаграмме для большей наглядности проводят утолщенные вертикальные линии, разграничивающие отдельные фракции. На рис. 84 приведен пример разделения головного погона жирных кислот в результате ректификации с большим числом тео
184
ретических ступеней при 20 мм рт. ст. Для бинарных смесей удобна также такая форма диаграммы, для построения которой на оси у откладывают значения концентрации дистиллята, а на оси х — выход дистиллята (см. рис. 90).
Для оценки результатов непрерывной ректификации, в частности смесей гомологов, большую пользу может оказать построение ступенчатой (идеализированной) диаграммы [242] (рис. 116). На ее основе можно построить столбиковую диаграмму фракционного состава смеси (рис. 117). Наглядные симметричные диаграммы Майер—Грольмана и Веселовского [243] также позволяют быстро проанализировать результаты разделения, полученные при разгонке по Энглеру или при аналитической ректификации многокомпонентных смесей, например моторных топлив, сланцевых масел, смол. По этому методу на ось у наносят значения температуры кипения, а на ось х (вправо и влево от оси у) — выход дистиллята в процентах от общего количества по фракциям, укладывающимся в температурные интервалы не уже 10 °C. На диаграмме получают площади (их обычно заштриховывают), напоминающие по форме репу или луковицу и дающие наглядное представление о результатах разгонки. Дополнительно слева на симметричной диаграмме приводят ряд чисел, соответствующих количествам дистиллята (в %), которые были получены с момента начала разгонки до определенной температуры; справа на диаграмме наносят числа, показывающие выход дистиллята (в %) для определенных температурных интервалов. На диаграмме разгонки (рис. 118, б) значение 180 °C соответствует верхнему температурному пределу бензиновой фракции, а 325 °C — верхнему температурному пределу фракции среднего масла.
Рис. 119-
Симметричная диаграмма разгонки обычного (а) и специального (б) дизель-юго топлива.
185
На рис. 118 дано сравнение обычной и симметричной диаграмм разгонки для сланцевого масла.
Основные преимущества симметричной диаграммы разгонки заключаются в том, что эти диаграммы позволяют быстро сравнивать количество кубового остатка (27%) с общим количеством дистиллята и по ним легко установить температуру, при которой начинается термическое разложение (по утолщению площади диаграммы при температуре около 320 °C). Кроме того, путем наложения диаграмм, нанесенных на кальку, можно быстро сравнить результаты различных разгонок.
Даже рядом изображенные симметричные диаграммы площадей, построенные для различных разгонок, дают быстрый и надежный способ сравнения полученных результатов. На рис. 119 сравниваются две симметричные диаграммы разгонки для обычного и специального дизельного топлива.
4.14. ПРИМЕРНАЯ ПОСЛЕДОВАТЕЛЬНОСТЬ
РАСЧЕТА ПРОЦЕССА РАЗДЕЛЕНИЯ
Ниже рассмотрена рабочая схема решения задач разделения, приведенная также в табл. 4.
4.14.1. ПЕРИОДИЧЕСКАЯ РЕКТИФИКАЦИЯ
ПРИ АТМОСФЕРНОМ ДАВЛЕНИИ
В лабораторию регулярно поступают относительно большие количества смеси (около 10 л в неделю) бензол — н-гептан, которую нужно разделить методом периодической ректификации. Дистиллят должен содержать 99% (мол.) бензола, а кубовый остаток не более 2% (мол.) бензола.
Характеристика компонентов разделяемой смеси:
Бензол н-Гептан
Содержание, % (мол.) . . 60 40
Молекулярная масса . . 78,11 100,20
у760 °C ^КИП’ . . 80,1 98,42
Плотность (при 20 °C), г/см3 . . . . . 0,87895 0,68368
Относительная летучесть 1,77
1. Предварительная оценка необходимого числа теоретических ступеней разделения. При атмосферном давлении разность температур кипения компонентов смеси равна 18,3 °C. При условии идеального поведения смеси необходимо либо 9 теоретических ступеней разделения (расчет по диаграмме на рис. 72), либо 16 теоретических ступеней [расчет по формуле (96) ]. Как показано ниже в п. 3, исходная смесь не является идеальной, поэтому необходимое число теоретических ступеней разделения должно быть увеличено.
186
Пример ясно показывает, что по разности температур кипения можно получить только грубо приближенную оценку для п.
2. Рабочее давление разгонки. Разделение целесообразно проводить при атмосферном давлении, поэтому отпадает необходимость выбора оптимального рабочего давления ректификации. Следует только учесть, что температура кипения должна быть скорректирована относительно давления 760 мм рт. ст. (см. рис. 114, 115); при этом необходимо внести поправку на выступающий ртутный столбик.
3. Расчет числа теоретических ступеней разделения и флегмового числа. Кривая равновесия системы бензол — н-гептан известна (см. табл. 26, разд. 4.10.3), она асимптотически приближается к диагонали [при увеличении концентрации до 100% (мол.) 1.
Возьмем за основу величины, указанные в разд. 4.10.3, и построим кривую равновесия [диаграмма размером 50x50 см с делениями в % (мол.)].
С помощью диаграммы равновесия пар — жидкость определим условия ректификации, как это описано в разд. 4.7.1. Рабочая линия процесса ректификации для укрепляющей части колонны [хв = 60% (мол.) и хЕ = 99% (мол.)] пересекает ось ординат в точке у = 32. Из (78) следует
/мнн = 99/32 — 1 = 2,09
Выберем рабочее флегмовое число v = 9, и по (75) получим
У =99/10 =9,9
Рабочая линия, проведенная через у = 9,9 и хЕ = 99,0 для рабочего флегмового числа v = 9, служит базой для графического изображения ступеней разделения между этой линией и кривой равновесия. Для начала разгонки находим число теоретических ступеней п = 25.
Концу разгонки соответствует дистиллят состава хЕ = 99% (мол.) и кубовая жидкость концентрацией 2% (мол.). При этих условиях рабочая линия пересекает ось ординат в точке у = 3, поэтому
^мнн = 99/3 — 1 = 32
Определяем, что этому рабочему числу соответствует 25 теоретических ступеней; берем за основу v = 50, откуда получаем у — 99/51 =1,94
Соединим точки у = 1,94 и хЕ = 99,0. Между новой рабочей линей и кривой равновесия строим ступенчатую линию. Как и сле-овало ожидать, получаем п = 25 теоретических ступеней.
Таким образом, получены следующие параметры ректификации: исло теоретических ступеней разделения п = 25; флегмовое :исло в начале ректификации v = 9; флегмовое число в конце ектификации v = 50.
187
4. Определение нагрузки, количества дистиллята и времени установления рабочего режима. Прежде всего пересчитаем концентрацию бензола из % (мол.) в % (об.) (см. разд. 3.4) по формуле (15):
78,11-06.0,879
78,11.0,6-0,879+ 100,2.0,4-0,684 “ /о ^°°о
Таким образом, около половины загрузки куба останется в кубовом остатке. Объем жидкости, соответствующий удерживающей способности (УС) колонны (около 100 мл), следует отобрать как промежуточную фракцию, и при следующей разгонке ее загружают в колбу вместе с исходной смесью.
Если требуется получать 200 мл/ч дистиллята, то при рабочем флегмовом числе v — 9, соответствующем началу разгонки, нагрузка колонны составит 2000 мл/ч.
Если нагрузку колонны постоянно поддерживать на уровне 2000 мл/ч, то в конце разгонки при флегмовом числе v = 50 можно отбирать только 2000/51 40 мл/ч дистиллята. Для отбора
дистиллята в конце разгонки со скоростью 200 мл/ч потребовалось бы увеличить нагрузку колонны до 200-51 = 10 200 мл/ч. На практике, это, конечно, неосуществимо, так как при повышении нагрузки в 5 раз колонна не обеспечит требуемой эффективности разделения, соответствующей 25 теоретическим ступеням. При постоянной же нагрузке по мере обеднения кубовой жидкости легколетучим компонентом флегмовое число нужно постепенно увеличивать с 9 до 50.
Параметры периодической разгонки, обеспечивающие заданное разделение, можно рассчитать по уравнению Биллета [135]. Метод основан на столь громоздких вычислениях, что для ознакомления с ним следует обратиться к оригиналу.
В заключение определим, какой порядок имеет время установления стационарного режима, т. е. время, в течение которого устанавливается состав дистиллята, равный 99% (мол.) (см. разд. 4.8.3). Для этого нужно знать общую УС колонны. Для приближенной оценки (см. разд. 4.10.5) можем принять, что в насадочных колоннах УС составляет 3 мл на одну теоретическую ступень разделения. Примем, что общая УС колонны равна 75 мл.
По формуле (123) вычисляем фактор интенсивности массообмена (R = 2000/3600 = 0,55 мл/с):
I =25-0,55/75 =0,18 с’1
Подставив найденное значение в формулу (124), получим приближенное значение пускового времени для начальных условий ректификации:
тп =26а/(0,18-2) =676/0,36 = 1877 с « 30 мин
Таким образом получаем (при условии достаточного подвода тепла в кубе): нагрузка колонны — 2000 мл/ч; скорость отбора дистиллята в начале разгонки при v = 9 — 200 мл/ч н пусковое время в начале разгонки — 30 мин.
188
5. Расчет размеров колонны. Для расчета размеров колонны, обеспечивающей нагрузку 2 л/ч, прежде всего необходимо знать скорость потока паров, предельную для выбираемой насадки. Пусть нужно использовать насадку из фарфоровых седел размером 4 мм. Ход расчета предельной скорости паров подробно описан в разд. 4.11. Приведенная плотность паров составляет:
РЛ=Рп/Рпс=2>7/87«,95 = 3,07-Ю-з
Величину рп рассчитывают j по формуле (32):
рп = (78,11 -760/353,35) 0,01605 = 2,7 кг/м3 * * 6
Пользуясь номограммой, приведенной на рис. 109, находим, что предельная скорость паров дапр для заданных условий при значении константы k = 0,019 (для седел) составит 0,49 м/с. Она может быть вычислена по формуле (136):
аупр = КО,019-0,004-9,81/0,00307 = 0,49 м/с
Исходя из того, что рабочая скорость паров составляет 60% от предельной скорости, находим, что дапр = 0,3 м/с (или 30 см/с). Далее по формуле (141) вычисляем необходимую площадь поперечного сечения колонны:
/=0,0228-22,5-353,35/30= 6,0 см2
Нагрузка колонны равна D = 2000 мл/ч = 1758 г/ч = 22,5 моль/ч.
Окончательно устанавливаем, что диаметр колонны равен 30 мм (/ = 7,06 см2), при этом удельная нагрузка составит 285 мл/(см2-ч). Для седел размером 4 мм при указанной удельной нагрузке можно принять, что ВЭТС = 8 см (см. табл. 28Д). Таким образом, колонна должна иметь следующие размеры:
Внутренний диаметр ..................... 30 мм
Рабочая высота ....................... 2000 мм
Общая высота (включая стандартные шлифы) ............................... 2150 мм
Методика тепловых расчетов приведена в разд. 4.12. Количество тепла, необходимое для нагревания смеси до точки кипения,
вычисляют по формуле (149), а для испарения дистиллята и флегмы — по формуле (151). Поверхность теплообмена в конденсаторе
рассчитывают по уравнению (191).
Отводимое из конденсатора тепло можно определить по формуле (155); пример из разд. 4.12 поясняет ее использование.
6. Испытание колонны на эффективность (см. разд. 4.10.8) нельзя провести с применением разделяемой смеси. Даже если содержание легколетучего компонента в кубе довести до 2%
(мол.), для обогащения дистиллята до 99% (мол.) при v = оо потребуется всего только 18 теоретических ступеней разделения. Испытание обычно проводят, используя смесь н-гептан — мети лциклогексан при нагрузке 2 л/ч. Если в этих условиях не бу-
189
дет достигнуто 25 теоретических ступеней разделения, то высоту колонны следует соответственно увеличить. Значение ВЭТС, равное 8 см для седел размером 4 мм, при удельной нагрузке 285 мл/(см2-ч), выбрано только ориентировочно.
Установка должна быть хорошо изолирована. Колонну снабжают заранее рассчитанным компенсационным обогревательным устройством (см. разд. 7.7.3). Применение автоматической головки колонны (см. разд. 7.5.3) позволяет проводить перегонку почти без наблюдения за установкой. В головке колонны целесообразно установить контактный термометр, связанный через реле с сигнальным звонком (см. разд. 8.2.2). Контактный термометр настраивают на температуру кипения бензола. Как только последует сигнал, начальное флегмовое число, равное 9, следует увеличить до 20. Точно так же флегмовое число увеличивают до 50.
Нагрузку целесообразно регулировать по гидравлическому сопротивлению с помощью контактного манометра (см. разд. 8.4.2). В зависимости от количества исходной смеси в качестве куба можно использовать подвесной сосуд из йенского стекла (см. разд. 7.6.1) или круглодонную колбу емкостью от 4 до 10 л, обогреваемую с помощью колбонагревателя (см. разд. 7.7.1).
О других возможностях автоматизации сообщается в гл. 8; о сборке установок см. также гл. 9.
4.14.2. НЕПРЕРЫВНАЯ РЕКТИФИКАЦИЯ
Способы расчета процесса непрерывной ректификации изложены в разд. 4.7.2 [пример — разделение смеси жирных кислот Св—С7 состава 81% (мол.) Св при 20 мм рт. ст., хЕ = 96%, хА ~ 0,5% ] и в гл. 9 (пример — разделение многокомпонентной смеси жирных кислот С*—С8 при 20 мм рт. ст.). В разд. 5.2.2 описана аппаратура и способы проведения непрерывной ректификации.
4.14.3. ВАКУУМНАЯ ПЕРЕГОНКА
Расчет вакуумной ректификации следует в том же порядке, что и атмосферной ректификации. Дополнительно в расчет условий процесса вакуумной перегонки входит: 1) выбор оптимального рабочего давления разгонки (разд. 4.6.2 и 4.10.6); 2) построение кривой равновесия идеальной смеси для заданного рабочего давления (разд. 4.6.1, табл. 10); 3) определение зависимости объема паров от давления (разд. 4.11); 4) расчет диаметра труб и герметичности аппаратуры (разд. 5.4.1); 5) измерение и регулирование давления (разд. 8.3).
4.15. ИСПОЛЬЗОВАНИЕ ЭВМ ПРИ РАСЧЕТЕ КОЛОНН
Применение электронных вычислительных устройств в практике лабораторной и пилотной ректификации несомненно эффективно, промышленная же ректификация с ее разнообразными проблемами регулирования сегодня уже не мыслима без использования вычислительной техники. Благодаря автоматизации технологических процессов в настоящее время появилась возможность управлять многостадийными процессами разделения. Важнейшие
190
критерии для сравнения различных управляющих машин при автоматизации лабораторных установок рассмотрены Блаттом и Фляйс-снером [259].
Различают цифровые вычислительные машины (ЦВМ) и аналоговые вычислительные машины (АВМ), а также их комбинацию в виде гибридной вычислительной системы [2481. ЦВМ выдает результаты в виде цифровых таблиц, в А^ВМ результаты выводятся в виде изменяющихся во времени напряжений, которые различными способами могут быть изображены в графической форме. Подробные сведения о методах математической обработки с помощью ЭВМ можно найти в специальной литературе [248, 249].
Практически все технические расчеты процесса перегонки можно проводить с помощью ЭВМ. Уже применение настольных вычислительных машин дает возможность определить давление паров, параметры фазового равновесия и оптимальные условия для получения максимального числа теоретических ступеней разделения, а также рассчитывать флегмовые числа. Применение ЭВМ типа IBM 7040, например при исследовании работы насадочных колонн в переходном режиме, обеспечивает выигрыш во времени благодаря быстрому определению состава вещества (в мольных долях) на основе данных по плотности и показателям преломления [162].
Разработано большое число программ для расчета термодинамических параметров систем, состоящих из нескольких фаз [109]. Для расчета фазового равновесия многокомпонентных смесей предложена обширная программа на языке Фортран [891.
Процессы дробной перегонки (дистилляция по Энглеру, аналитические разгонки по методу Американского общества по испытанию материалов, см. разд. 7.2), применяющиеся, главным образом, для контроля качества продукции, сопряжены, как показал Рот [250], с очень сложными термодинамическими закономерностями. Соответствующую математическую модель процесса можно рассчитать, используя, например, ЦВМ типа ZRA 1.
Приняв за основу метод Мак-Кэба и Тиле, Шубринг [2511 разработал программу расчета процесса разделения бинарных смесей, предназначенную для ЭВМ IBM 705. Допустив вполне практически приемлемые упрощения, в соответствии с которыми мольная энтальпия испарения не зависит от состава смеси, а энтальпия растворения равна нулю, с помощью этой программы можно рассчитать число теоретических ступеней разделения для двухкомпонентных смесей, как идеальных, так и неидеальных, в том числе азеотропных смесей. С помощью перфокарт в имеющуюся программу закладывают данные по равновесию, концентрации питающей жидкости, дистиллята и кубовой жидкости. Время решения одной задачи составляет от 15 с до 5 мин. Напечатанная таблица, полученная на ЭВМ, дает для каждой задачи последовательность возможных значений числа теоретических ступеней разделения в зависимости от флегмового числа или от
191
Рис. 120.
Упрощенная схема, поясняющая процедуру расчета числа теоретических ступеней разделения на ЭВМ IBM 705 (по данным Шубринга [251]).
отношения количества образующихся паров к количеству кубового продукта. Упрощенная блок-схема, представленная на рис. 120, поясняет ход расчета, а на рис. 121 приведена итоговая диаграмма, показывающая зависимость числа теоретических ступеней от флегмового числа для непрерывного разделения бинарной смеси. Применяя метод номограмм для отражения взаимосвязи между и vn, можно на одной и той же диаграмме представить зависящие друг от друга параметры укрепляющей и исчерпывающей частей колонны [2511.
Силей [252] применил ЭВМ для расчета оптимальных параметров лабораторной колонны Олдершоу диаметром 31,8 мм с 12 реальными тарелками в исчерпывающей части и 10 реальными тарелками в укрепляющей части. На ЭВМ Electric KDF 7 (Англия) были проанализированы 280 процессов разделения. В качестве эталонной смеси использовали смесь метилциклогексан— толуол. При этом за расчетное число теоретических ступеней разделения принимали то значение, которому соответствовала минимальная погрешность. Были изучены возможные погрешности, возникающие при измерении состава смеси, при определении положения и наклона рабочей линии, а также погрешность данных по равновесию.
Рис. 122 иллюстрирует изменение общей относительной погрешности для интервала флегмовых чисел 1,0—1,3 в исчерпыва-192
ющей части колонны. Форма кривых для укрепляющей части колонны аналогична.
Эти кривые располагаются на графике тем выше, чем больше флегмовое число; наименьшие значения п, при которых кривые определены, изменяются от 1,0 до 0,7.
Использование ЭВМ необходимо при точном расчете процесса разделения многокомпонентных смесей в ректификационных колоннах [264]. О возможности математического моделирования предварительной разгонки сырой нефти сообщают Киршбах, Френ-цель и Штранкмюллер [265]. Ляйбниц и Шулер [253] описывают процедуру расчета процесса разделения идеальных многокомпонентных смесей при использовании ЭВМ ZRA 1. Вопросы расчета фазового равновесия в неидеальных многокомпонентных растворах рассмотрены Нагелем с сотр. [254]. Аналоговые вычислительные машины широко применяют при технологических расчетах процессов массо- и теплопередачи [255]. В последние годы были усовершенствованы, главным образом, те методы, которые невозможно реализовать без помощи ЭВМ [6]. Имеются в виду прежде всего так называемые матричные методы расчета Ванга и Хенке [256], Саржента и Мертага [257], а также Стэйнт-хорпа с сотр. [258].
Рис. 121.
Итоговая диаграмма разделения (251].
Условия процесса: а = 1,5; Xg ~ 99,9% (масс.); х% = 65%, х& — 0,1%
Рис. 122.
Относительная суммарная погрешность определения числа теоретических ступеней в укрепляющей части колонны прн разных флегмовых числах Эталонная смесь метилциклогексан—толуол.
7 Э. Крель
193
При исследованиях гидравлического сопротивлёйий, а Также массо- и теплопередачи в неподвижных слоях насадки Брауэр и Мьюс [260] разработали программу на алгоритмическом языке Фортран, предназначенную для расчета перепада давления и высоты слоя с учетом структуры насыпной насадки.
Вагнер и Бласс [262 ] описывают метод расчета процесса ректификации в тарельчатой колонне, реализуемый на гибридной ЭВМ; в качестве примера для расчета выбрана система дихлорэтан—толуол. Метод позволяет рассчитать изменение во времени составов дистиллята и кубовой жидкости при разделении бинарных смесей по заданным рабочим параметрам процесса при известной кривой равновесия.
Теоретические и практические вопросы, связанные с исследованием динамических характеристик промышленных ректификационных колонн, рассмотрены Кёлером и Шобером [264].
5
Обычные методы перегонки
После проведения ориентировочных расчетов процесса разделения решается задача его технологического и аппаратурного оформления. При выборе рационального метода перегонки руководствуются такими показателями, как имеющееся количество исходного сырья, желаемая производительность колонны и качество дистиллята. При этом следует учитывать физико-химические свойства разделяемых веществ, а также аппаратурные и энергетические возможности.
5.1. КЛАССИФИКАЦИЯ МЕТОДОВ ПЕРЕГОНКИ ПО КОЛИЧЕСТВУ
ПЕРЕРАБАТЫВАЕМОГО ПРОДУКТА
Размеры дистилляционной и ректификационной колонны должны находиться в точном соответствии с количеством перерабатываемого вещества. При необходимости можно, конечно, большое количество сырья перерабатывать и в небольшой колонне, затрачивая на это много времени; в другом крайнем случае при проведении процесса в слишком большой установке увеличиваются потери продукта и кубовые отходы. В лабораториях часто требуется разделять смеси в количествах, измеряемых миллиграммами, и наоборот, необходимо перерабатывать от 5 до 10 кг/ч сырья с применением полупромышленных методов. В связи с проблемами масштабного перехода от малых аппаратов к большим все большее значение приобретают исследования процессов дистилляции и ректификации на пилотных установках.
5.1.1. МИКРО- И ПОЛУМИКРОПЕРЕГОНКА
В препаративной микротехнике обычно работают с количествами вещества менее 0,1 г. Разделение смеси при загрузке до 5 мл методами перегонки целесообразно отнести к области микротехники, а до 50 мл — к области полумикроперегонки. При этом следует принципиально различать дистилляцию (простую перегонку), фракционную перегонку и ректификацию. Штаге и Гем-мекер [1 ] указывают на эффективность разделения смесей в микроколичествах в совокупности методами перегонки и газовой хроматографии. Для более полного использования газовых хрома-
Т
195
Рис. 123.
Прибор Боденхаймера для микро дистилляции:
1 — конденсационная трубка; 2 — насадка из стеклянного порошка; 3 — капилляр с пробой жидкости; 4 — нагревательный блок с термометрами.
Рис. 124.
Горизонтальная колонка Янцеиа для микроректификации.
тографических методов часто возникает необходимость в предварительном разделении смесей путем перегонки. Авторы дают превосходный обзор по ректификационным колоннам различных типов, обсуждают конструктивные усовершенствования, а также рассматривают средства автоматизации процесса разделения. Штаге [1а] подробно описывает современные приборы для микроперегонки.
Для дистилляции веществ в количестве от нескольких миллиграмм до 1,0 г применяют дистилляционные капилляры, трубки с шарообразными расширениями и маленькие колбы. Колонны с очень низкой удерживающей способностью по жидкости, например полые колонны, колонны из концентрических трубок (щелевые трубчатые колонны) и колонны с вращающейся лентой применяют для разделения 1—25 мл вещества; при объеме исходной смеси от 25 до 50 мл можно также использовать насадочные и тарельчатые колонны.
С помощью прибора, описанного Боденхаймером [2], можно проводить фракционную перегонку нескольких миллиграмм жидкой смеси (рис. 123) и одновременно измерять температуру кипения смеси с точностью 1—2 °C. Трубка 1, один конец которой сужен до диаметра 1 мм, помещена в нагревательный блок 4. Температуру измеряют двумя стеклянными термометрами. Исследуемую пробу жидкости помещают в запаянный конец капилляра 3, остальной объем капилляра 3 заполняют стеклянным порошком 2. Выступающая из нагревательного блока расширенная часть трубки 1 имеет шлифованную изнутри поверхность молочно-белого цвета.
Температуру нагревательного блока равномерно увеличивают со скоростью 4—6 °С/мин. В момент приближения к точке кипения на стенках конденсационной трубки наблюдается запотевание, и при достижении точки кипения стекло в этом месте стано-196
вится прозрачным. По длине смоченного (прозрачного) участка можно оценить количество дистиллята, если перед этим провести калибровку с эталонными веществами. Чтобы выделить более мелкие фракции, нагревают то место трубки, где сконденсировалась первая фракция, при этом ее пары конденсируются в трубке 1 на большем расстоянии от нагревательного блока, для чего конец трубки охлаждают влажной фильтровальной бумагой.
Горизонтальный ректификатор Янцена (рис. 124) позволяет проводить ректификацию образцов массой от 1 до 100 мг. Смесь загружают в капиллярную трубку длиной 1200 мм и диаметром 5,5—5,7 мм [3]. В нагревательном цилиндрическом кожухе создают Перепад температур в 30 °C и с помощью тока водорода обеспечивают перенос вещества в направлении понижения температуры. При этом легколетучие компоненты перемещаются дальше, чем труднолетучие. Прибор предназначен специально для разделения высококипящих веществ. Рис. 125 иллюстрирует результаты разделения смеси додекановой и тетрадекановой кислот. Фишер [За 1 описывает новый усовершенствованный автоматический прибор такого типа.
Для разделения смеси в количествах от 0,05 до 0,5 мл обычно применяют трубку с шарообразным расширением (например, как на рис. 126). Эту узкую (диаметром 5—7 мм) тонкостенную трубку заполняют следующим образом. Шарик нагревают, и после охлаждения разделяемая жидкость благодаря образовавшемуся разрежению втягивается в трубку; жидкость можно загружать также с помощью капиллярной пипетки. Техника перегонки в такой трубке, особенно способом обогрева, подробно описана Бертеле и Хумелем [36].
56
52
48
44
40
10 20 30 40 50 60 70
Длина трубни, см
Рис. 125.
Результат разделения смеси додекановой и тетрадекаиовой кислот в го* ризоитальной колонке Янцеиа [3 ].
Рис. 126.
Прибор Бабкока для микродистилляции под вакуумом из трубки с расширениями.
197
Рис. 127.
Прибор для микроперегонки 00100.
Прибор имеет перегонную микроколбу емкостью 5 мл и приемные пробирки емкостью 6 мл (6 шт.) или 2 мл (9 шт.).
Изготовитель: фирма Юхайм, Золингген.
Рис. 128.
Прибор Креля для мнкроперегонки (от 1 до 10 мл смеси) при атмосферном давлении и под вакуумом:
/ — капельницы; 2 — проволочные сетки.
Изготовитель: народное предприятие «Комбинат технического стекла», Ильмен ау.
В микромасштабе можно проводить даже вакуумную перегонку. На рис. 126 показан прибор Бабкока [4], с помощью которого получают три фракции 1, 2 и 3. Чтобы собрать некоторое количество дистиллята, не прерывая процесса перегонки, в микроприборах применяют поворотные вакуумные приемники (рис. 127).
Любое количество фракций дистиллята можно отобрать при микроперегонке в приборе Креля (рис. 128). Этот прибор применяют для перегонки как при атмосферном давлении, так и под вакуумом. Его можно видоизменять в зависимости от количества исходной смеси. Так, при 1—2 мл вещества целесообразно работать с полой трубкой. При необходимости можно вставить сверху в качестве брызгоотбойников одну или две конические проволочные сетки, которые несколько улучшают разделение. Если исходное количество вещества составляет 3—5 мл, то на стеклянной перфорированной перегородке размещают стеклянные кольца Рашига или спирали размером 4x4 мм. При 5—10 мл вещества можно применять спирали размером 2x2 мм, насыпанные на коническую проволочную сетку.
Сначала ректификацию проводят при закрытом кране тонкой регулировки (о = оо) до тех пор, пока в системе не установится постоянная температура. Затем приступают к работе при любой 198
рабочей нагрузке и любом флегмовом числе, применяя капельницы 1 и кран для регулирования этих параметров. Благодаря таким приемам можно получать точные и воспроизводимые результаты анализов малых проб вещества по температурам кипения.
В отличие от полых трубок (см. разд. 7.3.1) колонна Вигре обладает большей орошаемой поверхностью за счет большого числа заостренных глубоких вмятин, кроме того в средней части она имеет ввод для питающей смеси. Шрадер и Рицер [5 ] использовали такую колонну в качестве дефлегматора в приборе для микроперегонки (рис. 129). Он содержит плоскодонную колбу 1, которая обеспечивает постоянную поверхность испарения, и приемник 3 со стеклянными трубками 2 емкостью 0,1 мл. Дистиллят стекает к трубкам 2 по стеклянной нити 4. Разумеется, прибор можно применять только для разделения высококипящих веществ, так как в нем не предусмотрен водяной холодильник. Более вместительный прибор Кленка [6] также имеет только воздушное охлаждение (рис. 130). Колонка длиной 8,5 или 13 см со спиральной металлической лентой длиной 120 мм и вакуумированной рубашкой обеспечивает хорошее разделение при очень малом количестве смеси.
Как насадочные, так и тарельчатые колонны вследствие их значительной удерживающей способности по жидкости можно применять только для проведения полумикроперегонки (см. разд. 4.10.5). Причем следует стремиться к тому, чтобы при
Рис. 129.
Прибор Шрадера—Рицера для микроперегонки с колонкой Вигре: 1 — плоскодонная колба; 2 — стеклянные трубки; 3 — приемник; 4 — стеклянная нить.
Рис. 130.
Прибор Кленка со спиральной вставкой для микроректификации более 4 мл вещества.
Рис. 131.
Прибор Гульда с сотр. для молекулярной микродистилляции по методу «холодного пальца» :
1 — «холодный палец»; 2 — Кольцевой выступ; 3 — выемка; 4 — капилляры; 5 — приемник.
199
Таблица 33.
Параметры колонок для аналитической полумикроректификации
Колонна Внутренний диаметр, мм Длина, см га £ л о, га ч « 2 > Объем фракции, мл 1 Нагрузка, мл/ч Скорость отбора дистиллята, мл/ч Флегмовое число ВЭТС, см УС колонны, мл
Колонна с вращающейся лентой 5 90 100—150 0,5 10—30 0,5—1,5 40 1,1 1,9
Колонна «Гипер-кол» 8 60—90 20—100 0,5— 1,0 20—30 0,5—1,5 40 0,7 48
Колонна с узкой кольцевой щелью 8 60 10—50 0,5 20—30 0,2—0,5 100 0,9 3,2
заданном исходном количестве вещества размеры колонны были бы по мере возможности минимальными. В качестве насадочных тел при полумикроперегонке можно применять спирали размером от 1,6 до 2 мм (см. разд. 7.3.2 и 7.8).
Для тонкого разделения веществ в очень малых количествах более эффективны щелевые трубчатые колонны (см. разд. 7.3.1) и колонны с вращающейся лентой (см. разд. 7.3.5). Колонны обеих разновидностей также широко применяют Для аналитических разгонок и вакуумной микроректификации высококипящих веществ [7].
Винтерс и Динерштайн [8 ] описывают устройство, состоящее из восьми полумикроприборов для аналитической разгонки; устройство имеет центральную измерительную ячейку.
Технические характеристики некоторых приборов для аналитической полумикроперегонки приведены в табл. 33 [8].
Шпигельберг [9] сообщает о разделении эфирных масел в колонне с вращающейся лентой при 4—13 мм рт. ст. Примеры разделения высокомолекулярных органических соединений в щелевой трубчатой колонне приведены Янценом и Витгертом [3], а также Фишером [7].
Молекулярную дистилляцию (см. разд. 5.4.4) в микромасштабе можно проводить либо по методу «холодного пальца», либо по принципу падающей пленки, т. е. в тонком слое. На рис. 131 [10] показан прибор, основанный на методе «холодного пальца». Разделяемую смесь загружают в выемку 3; пары конденсируются на «холодном пальце» 1. Дистиллят скапливается у кольцевого выступа 2, откуда по каплям стекает во вращающийся приемник 5 с капиллярами 4.
Для перегонки веществ в количествах менее 4 г Брегер [11] предложил оригинальный прибор, с наружной поверхностью
200
испарения и внутренней поверхностью охлаждения (рис. 132). Этот небольшой прибор с длиной рабочего участка 185 мм имеет ту особенность, что он может вращаться вокруг горизонтальной оси (проходящей через его середину). Благодаря этому исходную смесь, а также и дистиллят можно многократно (при различной мощности обогрева) испарять из падающей пленки. Перед началом перегонки смесь дегазируют. Прибор Пашке с сотр. [12] предназначен для микроперегонки приблизительно 0,5 г вещества (рис. 133). Исходным веществом пропитывают цилиндрик 1 из стеклянной ваты, который подвешивают на крючке. Прибор вакуумируют, затем включают систему обогрева кожуха 2. Количество отогнанного вещества определяют по шкале 3.
Опыт показывает, что нагревание малых колб при микроперегонке сопряжено с трудностями. В колонне часто наступает захлебывание. Поэтому диаметр колонны должен составлять не менее 6—8 мм. Весьма удобен для нагревания малых колб микроколбо-нагреватель, в котором используется инфракрасное излучение в невидимой части спектра. Применение инфракрасного излучения позволяет без особых затруднений проводить вакуумную ди-
Рис, 132.
Прибор Брегера для молекулярной микродистнлляции меиее 4 г вещества (принцип «падающей пленки»):
1 — участок измерения температуры; 2 — нагревательный кожух; 3 — колба для исходной смеси; 4 — приемник для дистиллята; 5 — конденсатор; 6 ~ обогреваемая поверхность.
Рис. 133.
Прибор Пашке с сотр. для молекулярной микродистилляции 0,5 г вещества: 1 — цилиндрик из стеклянной ваты; 2 — нагревательный кожух; 3 — шкала.
201
стилляцию, так как в этом случае кипение жидкости происходит более равномерно и без «бросков».
Пфайл [13] дает обстоятельный обзор препаративной микротехники, в котором также описаны приборы для микроперегонки. В этой связи следует указать на микровесы Айгенбергера [14], с помощью которых можно сравнительно быстро определить плотность вещества при очень малых количествах пробы. Более простая установка сконструирована Клазеном [15].
5.1.2. АНАЛИТИЧЕСКАЯ РЕКТИФИКАЦИЯ
Цель аналитической ректификации состоит в том, чтобы из двух или многокомпонентной смеси извлечь отдельные компоненты с возможно более высокой степенью чистоты. Степень чистоты устанавливают путем определения физических констант вещества, например коэффициента преломления, плотности, точки затвердевания или плавления, а также молекулярной массы. Так как обычно не известно, какие компоненты и в каких количествах содержатся в разделяемой смеси, то анализ с применением ректификации следует проводить периодическим способом. Для аналитической ректификации применяют колонны с достаточным числом теоретических ступеней разделения (в разд. 4.7—4.12); головка колонны должна обеспечивать точное регулирование нагрузки и флегмового числа. Чтобы получить точное представление о количественном соотношении разделяемых компонентов необходимо, чтобы промежуточная фракция была как можно меньше. Промежуточной фракцией является количество дистиллята, которое отбирают между фракциями сравнительно чистых (или весьма чистых) компонентов. По мере отбора промежуточной фракции в ней постепенно уменьшается содержание легколетучего компонента (см. рис. 56). Количество загрузки выбирают исходя из содержания того компонента, который необходимо выделить и который находится в исходной смеси в минимальном количестве. Далее необходимо стремиться к тому, чтобы отношение
Общая УС , т = _-----------------------------
Доля выделяемого компонента в загрузке
было по возможности минимальным, что позволит уменьшить количество промежуточной фракции. О благоприятном влиянии УС колонны при этом не может быть и речи, поскольку при аналитической ректификации работают с высоким флегмовым числом (см. разд. 4.7.1). Поэтому для аналитических разгонок следует выбирать колонны малого диаметра, так как отрицательное влияние общей УС колонны растет одновременно с увеличением числа теоретических тарелок.
По данным Штаге [16], наименьшая из выделяемых фракций должна иметь объем по меньшей мере в десять раз превышающий общую УС колонны, применяемой для разделения. Это означает, 202
например, что при общей УС, равной 120 мл, объем загрузки должен составлять 12000 мл при выделении в чистом виде компонента, содержание которого в исходной смеси составляет 10%. Это относится обычно только к разделению близкокипящих компонентов, когда требуется очень большое флегмовое число, а степень чистоты выделяемых компонентов приближается к 100%.
Наряду с насадочными колоннами для проведения аналитических работ особенно пригодны высокоэффективные щелевые трубчатые колонны, колонны с вращающейся лентой, а также колонны с регулярной насадкой, обладающие очень низкой общей УС (см. разд. 7.3.1, 7.3.4 и 7.3.5).
При разделении многокомпонентных смесей, когда исходная смесь имеется в достаточном количестве, сырую смесь предварительно разделяют на отдельные фракции путем расширительной дистилляции (см. разд. 5.4.2) или же методом обычной непрерывной ректификации (см. разд. 5.2.2). При этом к эффективности разделения не предъявляют высоких требований, а объем отдельных фракций определяют заранее путем предварительной пробной разгонки (см. разд. 7.2) сырья. Фракции, полученные при грубом разделении, затем дополнительно разделяют на исходные компоненты путем аналитической ректификации, используя то благоприятное обстоятельство, что соотношение т при этом значительно уменьшается.
В качестве примеров практического применения метода аналитической ректификации многокомпонентных смесей следует указать на работы Кюнхансса с сотр. [17] по разделению легкого масла бурого угля, работы Проффта и Бухманна [18] по получению хинолиновых оснований, а также ректификационное разделение смеси синтетических аминов жирного ряда, проведенное Фюрстом и Шветликом [19]. Приборы «Дестинорм» применяют, в частности, при разделении смесей аминовых кислот минерального происхождения путем ректификации сложных метиловых эфиров соответствующих ацетиламиновых кислот [20] и при разделении буроугольных смол для определения их состава [21 ]. Гутвассер и Мюллер [22] разработали специальный пленочный испаритель для фракционной перегонки масляных жирных кислот при остаточном давлении 0,1 мм рт. ст. Мэир с сотр. [23] показал, что рациональным сочетанием обычной перегонки, азеотропной ректификации, адсорбционных методов и метода зонной плавки можно добиться четкого разделения керосина на отдельные компоненты.
Нередко бывает так, что имеющейся исходной смеси недостаточно для выделения из нее в чистом виде компонентов, содержащихся в незначительном количестве. В этих случаях нужно применять колонны, имеющие особенно низкую УС по жидкости, например роторные колонны, и в особенности колонны с вращающейся лентой (см. разд. 7.3.5). При использовании насадочных или тарельчатых колонн за счет повышения флегмового числа
203
при появлении промежуточной фракции добиваются того, чтобы в данных условиях разделения размер этой фракции был по возможности наименьшим. Ситчатые и колпачковые колонны (см. разд. 7.3.3) редко применяют для аналитической ректификации. Однако по сравнению с насадочными колоннами они имеют то преимущество, что для них можно определить градиент концентраций в верхней части колонны путем отбора и анализа проб с верхних тарелок. Например, если концентрация легколетучего компонента на третьей тарелке сверху начинает снижаться, то можно сразу же увеличить флегмовое число. Таким путем добиваются малого размера промежуточных фракций.
При аналитической ректификации используется также следующий способ разделения: сначала концентрируют компоненты в сравнительно большой колонне, затем ректификацией во второй меньшей колонне (с малой УС) получают чистые вещества. Для очень трудных вариантов тонкой аналитической ректификации, когда для разделения требуется более 100 теоретических ступеней разделения, целесообразно применять щелевые трубчатые или многотрубчатые колонны (см. разд. 7.3.1), а также колонны с насадкой из проволочных спиралей или из проволочных сеток (см. разд. 7.3.4). В щелевых трубчатых колоннах при работе в условиях вакуума достигают значений ВЭТС ниже 5 мм [7].
При отсутствии высокоэффективной насадки (ВЭТС ль 2 см) нередко из-за ограниченной высоты помещения возникают трудности с получением необходимого числа теоретических ступеней разделения. В помещении высотой 3,5 м при рациональном конструктивном оформлении установки можно разместить колонну с рабочей высотой около 2,5 м.
В этом случае наилучшие результаты можно получить, если работать с двумя последовательно включенными колоннами, как это показано на рис. 134. Такой прием хорошо себя оправдал, в частности при аналитической ректификации оснований буроугольных смол [241. Дистиллят первой колонны непрерывно подают в нижнюю часть второй колонны; вторая колонна может иметь меньшие размеры. Нагрузки и флегмовые числа для обеих колонн также могут различаться. Необходимо только следить за тем, чтобы количество жидкости в кубе сохранялось постоянным. Этот комбинированный способ работы имеет то преимущество, что вторая ректификация (если она необходима) происходит одновременно с первой. В то время как содержание легколетучего компонента в кубовой жидкости первой колонны значительно уменьшается [примерно с 50 до 1 % (мол.)], концентрацию кубовой жидкости второй колонны поддерживают постоянной в возможно более узких границах [около 90—95% (мол.)], постепенно повышая флегмовое число в первой колонне. Таким образом, во второй колонне поддерживается почти стационарный режим непрерывной ректификации, преимущество которого можно использовать (см. разд. 5.2.2). В кубе второй колонны накаплива-204
ются высококипящие компоненты, так что этим способом можно получить очень маленькую промежуточную фракцию. После прекращения подачи питающей жидкости в первую колонну кубовую жидкость второй колонны можно переработать периодической ректификацией или проанализировать содержание в ней легколетучего компонента.
Для аналитической ректификации следует обязательно применять такую приставку или головку, которая обладает возможно меньшим «мертвым» объемом. Под «мертвым» объемом понимают
Рис. 134.
Схема установки из двух последовательно соединенных колони по нормалям «Дестинорм» .
205
методу деления потока паров или
Рис. 135.
Диаграмма разгонки смеси растворителей: 1 ~ дистилляция по Зиглеру; 2 — пробная ректификация; 3 — четкая ректификация.
количество жидкости, которое задерживается в приставке или в головке колонны и не принимает участия в противоточном массообмене. Автоматические головки колонн, работающие по жидкости (см. разд. 7.5.3), имеют
то важное преимущество, что они практически не создают «мертвые» объемы. Следует также иметь в виду, что возможно загрязнение дистиллята за счет растворения в нем смазки крана. Это обстоятельство обычно не принимается во внимание, хотя может привести к существенным ошибкам. В связи с этим разработаны устройства, обеспечивающие отбор дистиллята без его контакта со смазкой для кранов (см. разд. 4.6.3). В последнее время в автоматических головках колонны (см. разд. 7.5.3) применяют магнитные клапаны, наилучшим образом отвечающие этим требованиям. При аналитической ректификации большое внимание уделяется достижению наиболее высокой степени чистоты выделяемого продукта. В связи с этим следует рассмотреть вопрос о том, что лучше: непрерывно отбирать дистиллят как определенную долю от всего образующегося конденсата или работать с периодическим отбором дистиллята, когда конденсат поочередно либо полностью направляют в колонну как флегму, либо полностью отбирают в качестве дистиллята. В последнем случае флегмовое число определяется отношением времени подаа^^щегм^г^лительности отбора дистиллята. При работе с
средняя концентрация легколетучего компонента в дистиллят^ полученном
^ДДДДДЦ^^^Д/иднак^оыл^усИМш^^^^^^^^^^^» более полное разделений
смеси достигается, когда дистиллят отбирают
а не через каждые 30 с [25 ]. Если нужно поддерживат^^^геевы-' сокие значенг^Фл^^овоп^шсла, как например при ректификации изотопов |ИИИИМЩДДУго по соображениям технического порядка работают (см. разд.
5.1.4). —....... ... ‘ ----
Очевидно, анализы смесей по температурам их кипения можно также выполнять и при разделении их в простых дистилляционных приборах, если разность температур кипения компонентов относительно велика или если нужно определить только температурные пределы кипения. Соответствующая аппаратура описана в разд. 7.2. В этом случае очень трудно получить правильную кривую
206
разгонки, как это иллюстрирует рис. 135 на примере анализа смеси растворителей. Как видно из рисунка, начало кипения смеси по приведенным кривым не совпадает. Это объясняется тем, что при дистилляции по Энглеру (см. разд. 7.2) начало кипения принято регистрировать в момент отрыва первой капли от трубки конденсатора. При проведении же пробной ректификации (рис. 135) с той же первоначальной загрузкой куба сначала работают при бесконечном флегмовом числе до установления стационарного режима. В этом случае значительно точнее улавливается начальный момент кипения, которому в действительности соответствует 36 °C, как это было установлено для данной смеси методом четкой ректификации. При дистилляции по Энглеру кипение начинается при 59 °C, а при пробной ректификации — около 45 °C.
Значительная затрата времени на аналитическую четкую ректификацию (например, 120 ч для разделения нефтяной фракции с интервалом кипения от —30 до +260 °C) послужила стимулом для моделирования процесса ректификации с использованием специальной газовой хроматографической аппаратуры [261. При этом получаются опытные значения концентраций, которые сравнимы с результатами разделения в ректификационной колонне с числом теоретических ступеней разделения 100. Указанным способом можно анализировать как сырые нефти, так и нефтяные фракции соединений с числом атомов в углеродной цепи от 1 до 40. «Прибор для одновременной аэрографии и ректификации» с помощью небольшого встроенного компьютера позволяет получать кривые «температура кипения — концентрация [% (масс.)]». Площади под этими кривыми непрерывно интегрируются и подсчитанные значения через каждые 10 с регистрируются самописцем. На анализ указанной выше нефтяной фракции (от —30 до +260 °C) требуется всего лишь около 1 ч [27].
5.1.3. ПРЕПАРАТИВНАЯ И НАРАБОТОЧНАЯ ПЕРЕГОНКА
После идентификации всех компонентов смеси методом аналитической ректификации обычно ставится очередная задача — получить эти компоненты в достаточно больших количествах. Если разность температур кипения велика и нет необходимости в тонком разделении, то для наработки продукта вполне можно обойтись простыми дистилляционными приборами (рис. 136, а), собранными из стандартных деталей. На рис. 136, б показана модификация прибора с колонной. Эти приборы, разумеется, работают с неизмеряемой «дикой» флегмой. Для улучшения воспроизводимости результатов используют приставку или головку колонны, обеспечивающую точную регулировку нагрузки и флегмового числа (см. разд. 7.5). Приемник дистиллята Аншюца и Тиле (см. рис. 136), предназначенный для работ под вакуумом, можно при-
207
страница zuo
При работе с низкими флегмовыми числами средняя концентрация легколетучего компонента в дистилляте, полученном с периодическим отбором, всегда меньше, чем при непрерывном отборе. Однако было установлено, что при очень большом флегмовом числе (порядка 100) более полное разделение смеси достигается, когда дисстилят отбирают через каждый 1 час, а не через каждые 30 с. [25]. Если нужно подержать более высокие значения флегмового числа, как например при ректификаии изотопов (V= 1000-10000) то по соображениям технического порядка работают с периодическим отбором дистилята (см. разд. 5.1.4).
Рис. 136.
Приборы из стандартных деталей для дне тилляции (а), для ректификации (б).
менять также и при ректификации под атмосферным давлением. Кроме того, при высоких нагрузках можно также проводить простую перегонку с дефлегмацией (см. разд. 5.2.3) и так называемую расширительную перегонку (см. разд. 5.4.2).
“ б Лабораторные установки обыч-
но снабжены стандартными шлифами, поэтому диаметр колонны обычно не превышает размера, при котором можно еще применять шлиф NS45. Полупромышленные колонны из стекла, снабженные сферическими, коническими или плоскими шлифами, имеют наибольший номинальный диаметр около 150 мм. Для пилотных установок колонны изготавливают из стекла, керамического материала или металла, причем при выборе материала следует учитывать вероятность коррозии. Номинальные диаметры пилотных колонн лежат в интервале от 100 до 400 мм.
Штаге [281 дает обзор установок, применяемых для препаративного разделения в лабораториях и на опытных производствах. Хампель [29], занимавшийся проблемой получения ультрачистых растворителей, пришел к выводу, что наряду с экстракцией перегонка по-прежнему является самым эффективным методом очистки растворителей (свыше 99,9%). Высокой степени очистки достигают при ректификации спиртов, простых эфиров, бензола, пиридина и ряда углеводородов. Например, содержание примесей в четыреххлористом углероде может быть доведено до 10“4% и менее.
5.1.3.1. Полупромышленные установки
Размеры полупромышленных аппаратов должны соответствовать ожидаемой нагрузке н необходимой разделяющей способности (см. разд. 4.11). Стандартные детали и узлы для полупромышленных установок обычно изготавливают со шлифами NS10, NS14,5, NS29 и NS45. Непрерывно действующие установки, рассчитанные на более высокие нагрузки, описаны в разд. 5.2.2, 5.4.2 и 5.4.5. Для ректификации при нагрузках 20 л/ч была, в частности, разработана полупромышленная установка согласно нормалям «Де-стинорм», в комплект которой входят колонны диаметром 50, 70 и 90 мм. Она пригодна для работы как при атмосферном давлении, так и под вакуумом (остаточное давление до 1 мм рт. ст).
208
На рис. 137 дана схема полупромышленной установки непрерывного действия, собранная из стандартных деталей. При сборке полупромышленной установки часто копируют схемы промышленных установок.
Детали колонн снабжены коническими шлифами, причем для установки термометров и патрубков для жидкостей используют конические шлифы NS 14,5; в опасных участках предусматривают более подвижные сферические шлифы размером 35 мм (см. разд. 3.1).
Для обеспечения тонкого регулирования количества дистиллята головку колонны (см. рис. 137) снабжают многоходовым краном; флегмовое число можно контролировать с помощью градуированных мерников флегмы и дистиллята. Охлаждающий змеевик, вмонтированный в паровую трубу, обеспечивает возможность работы с дефлегмированием паров. Количество полученной при этом флегмы можно рассчитать по изменению температуры охлаждающей воды или же измерить с помощью градуированной бюретки. Аналогичную головку применяют в полупромышленных установках также при автоматическом регулировании флегмового числа по принципу деления потока жидкости (см. рис. 312).
При непрерывной ректификации питающую жидкость предварительно подогревают во вспомогательном теплообменнике, выполненном в виде U-образной трубки (рис. 138). При частично закрытом кране 1 уровень жидкости в теплообменнике поднимается до соединительной трубки 2; в этот момент начинается рециркуляция жидкости. В ходе рециркуляции смесь может нагреваться до 250 °C.
На рис. 137 показана только принципиальная схема установки. Степень автоматизации установки определяется ее насыщенностью контрольно-измерительной и регулирующей аппаратурой (см. гл. 8).
Для периодической разгонки в качестве куба обычно применяют круглодонные колбы емкостью лишь до 10 л; для разделения сырья в больших количествах используют подвесные кубы (см. рис. 316) или металлические кубы с плоскими или сферическими шлифами, на которые сверху устанавливают остальные стеклянные приборы. Целесообразно использовать куб из стали V2A емкостью 25 л, снабженный греющей баней и электронагревателями мощностью по 3 кВт с трехступенчатым регулированием (см. разд. 7.7.2). В связи с этим следует отметить, что одно из преимуществ непрерывной ректификации как раз и состоит в том, что даже при нагрузках до 20 л/ч можно использовать кубы емкостью всего 2—5 л (см. разд. 7.6.1).
Тарельчатая колонна Креля (рис. 139) диаметром 90 мм, предназначенная для использования в полупромышленной установке, выполненной по нормалям «Дестинорм», имеет встроенные устройства для перетока жидкости, как и в промышленных колоннах. Благодаря этим устройствам протекаемый в такой колонне
209
^Рис. 137.
Схема непрерывно действующей полупромышленной установки по нормалям «Дестинорм» при нагрузке до 20 л/ч. Изготовитель: народное предприятие «Комбинат технического ” *
стекла», Ильменау.
Рис. 138.
Подогреватель питающей жидкости для колонн непрерывной ректификации:
1 — край; 2 — соединительная трубка. Изготовитель: народное предприятие «Комбинат технического стекла», Ильменау.
Рис. 139.
Тарельчатая колонна Креля с внутренними перетоками (диаметр 50, 70 и 90 мм).
Изготовитель: народное предприятие «Завод технического стекла Штюцербах».
процесс ректификации более соответствует промышленному. Краны для отбора проб и термометры могут быть установлены на любой тарелке, поэтому в ходе ректификации можно следить за изменением концентрации и температуры по высоте колонны (прежде всего в ее верхней части).
Колонны с колпачковыми тарелками «Лабодест» конструкции Штаге (см. разд. 7.3.3) также имеют промежуточные перетоки, они изготавливаются с номинальными диаметрами до 100 мм. Зависимость коэффициента полезного действия тарелки от ее конструкции рассмотрена в разд. 7.3.3.
5.1.3.2. Пилотные установки
Для изготовления пилотных ректификационных установок * в качестве конструкционного материала обычно применяют стекло. Этот материал имеет то существенное преимущество, что при проведении ректификации можно визуально контролировать гидродинамические процессы, протекающие в колонне и других частях установки. К тому же заводы технического стекла в каталогах предлагают большой выбор стандартных деталей с плоскими шлифами и с трубками, присоединительные концы которых снабжены кольцевыми выступами со сферическими или плоскими торцами [165]; благодаря этим стандартным деталям значительно облегчается сборка установок для перегонки в промышленных условиях. Обычно применяют боросиликатное стекло, отличающееся химической стойкостью, а также стойкостью к воздействиям повышенных и быстро изменяющихся температур (см. разд. 7.1). Ниже приведены дополнительные сведения [301 относительно технических изделий из стекла «Разотерм» (тип стекла 320 по стандарту TGL 7209), выпускаемых народным предприятием «Йенским заводом стекла Шотт».
Толщина стенок в 5—8 мм обеспечивает достаточную механическую прочность стеклянных приборов. Кроме того, низкое значение коэффициента линейного расширения стекла «Разотерм» (а = 3,3 10 6) обуславливает стойкость к изменениям температуры.
Сборку пилотных установок проводят чаще всего по агрегатному принципу, применяя стандартные элементы, например трубы с наружными кольцевыми выступами на концах, фасонные части труб, теплообменники, круглодонные колбы, цилиндрические сосуды, колпачковые колонны с номинальными диаметрами 200, 400 и 500 мм (рис. 140), царги для колонн, запорные органы, дозировочные насосы, а также контрольно-измерительные и регулирующие приборы. Уже выпускаются стандартные и типовые установки для обработки воды путем дистилляции и ионного обмена, вакуумные испарители циркуляционного типа производитель-
* См. примечание на с. 11.
211
Рис. 140.
Ректификационная установка из стекла «Разотерм» с колпачковой колонной диаметром 400 Мм.
Изготовитель: народное предприятие «Йенский завод стекла Шотт», Иеиа.
Рис. 141.
Вакуумный циркуляционный испаритель производительностью 100 л/ч (по воде).
Изготовитель: народное предприятие «Йенский завод стекла Шотт», Иена.
ностью от 5 до 100 л/ч по дистилляту (рис. 141), установки для регенерации растворителей. Кроме того, выпускаются и специальные типовые узлы, например автоматические делители флегмы и клапаны с автоматическим приводом. Применение стандартных узлов из разных материалов дает возможность соединять стеклянные колонны с металлическими или фарфоровыми кубами в том случае, когда для проведения периодической ректификации требуются более вместительные кубы, чем изготавливаемые из стекла (до 200 л) (см. разд. 7.7.2).
Применение стандартных прямых, фасонных и переходных участков труб, а также запорных органов обеспечивает бездефектный монтаж замкнутых трубопроводных систем [165]. Диаметры выпускаемых труб от номинального размера 15 мм ступенчато увеличиваются до 500 мм. Прямые участки труб, которые в настоящее время изготавливаются длиной до 3 м, а также фасонные части соединяют с помощью накидных фланцев, при этом сферические 212
или плоские торцы труб, снабженных на концах кольцевыми выступами, допускают угловое смещение до 3°. Предусмотрены также различные соединения специального назначения (винтовые соединения для труб с заплечиками, фланцевые соединения для гладких труб).
Показанные на рис. 140 и 141 ректификационные установки (габаритная высота до 14,5 м) снабжены парогенератором с нагревателями. Испарительная способность одного нагревателя (с плоской спиралью), имеющего номинальный размер 200 или 300 мм, составляет 20 л/ч воды при давлении насыщенного пара 2 ати. Достоинством этих установок является возможность увеличения охлаждающей и греющей поверхности при необходимости. При расчете теплообменника коэффициент теплопередачи k можно принять равным 400 ккал/(м2-ч-°С) при нагревании жидкостей паром (через стенку). При конденсации паров в холодильнике принимают k =300 ккал/(м2 ч °С), а при охлаждении жидкостей k = 150 ккал/(м2 ч °C).
Стеклянные детали, трубопроводы и пилотные установки фирмы Квикфит герметичны при работе под вакуумом. Концы труб снабжены конусными переходами; при номинальных диаметрах труб от 15 до 225 мм — сферические шлифы. Толщины стенок лежат в интервале 2—10 мм. Конструкционные элементы фирмы Квикфит могут быть дополнительно защищены покрытием из полиэфиров, стойким при температуре до 150 °C [1651.
Проявляется тенденция к созданию стандартной аппаратуры для перегонки, в том числе циркуляционных испарителей и сборных конструкций из стеклянных приставок и кубов, выполненных из других материалов. Эта тенденция распространяется также и на автоматизированные пилотные установки непрерывного действия с электромагнитными делителями флегмы, регуляторами уровня и расхода (рис. 142). Применением различных вентилей из стекла можно значительно упростить обслуживание подобных установок. Такие вентили подробно описаны в разд. 7.2.1.
Для автоматизации пилотных установок из стекла дополнительно требуется регулирование систем нагревания кубовой и питающей жидкости (см. разд. 7.7.2); кроме того, необходимы приборы для регулирования флегмового числа (см. разд. 8.4.1 и рис. 142) и скорости испарения (см. разд. 8.4.2). Для измерения и дозирования расхода газа и жидкости в пилотных установках были разработаны специальные типовые приборы из стекла (см. разд. 8.6).
Для создания устройств, предназначенных для дозирования агрессивных жидкостей (температура до 100 °C, кинематическая вязкость жидкостей до 30 сСт и плотность до 2 г/см3), особенно рекомендуется комбинирование стекла и политетрафторэтилена. На «йенском заводе стекла Шотт» разработан ряд бессальниковых дозировочных насосов (рис. 143), используемых в комплекте со стандартным приводом РАЕ 32/50 (насосный завод «Зальцведель»),
213
Рис. 142.
Рис. 143.
Головка дозировочного насоса из стекла с тефлоновым сильфоном.
Предназначена для установки на специальном приводе РАЕ 32/50 (стандарт TGL 17-747907). Изготовитель:’ народное предприятие «Иенскнй завод стекла Шотт», Иена.
Рис. 144.
Схема газового проходного вентиля с условным проходом 15 мм.
Изготовитель: народное предприятие «Иенскнй завод стекла», Иена.
Установка для непрерывной вакуумной ректификации (высота 7,2 м, ширина 1,9 м) с колонной диаметром 150 мм:
1,7 — холодильники (0,2 м2); 2 — кипятильник (0,5 м2); 3 — теплообмен* ник для питающей жидкости (0,14 м2); 4 — дефлегматор (1,4 м3); 5 — магнит; 6 — регулятор флегмы.
Изготовитель: фирма «Стеклянная техника», Висбаден, Шнрштайи.
Номинальные расходы лежат в пределах 63 и 650 л/ч при максимальном напоре от 3,5 до 2,5 кгс/см2. Повышенной коррозионной стойкостью обладает также газовый проходный вентиль с условным заводом (рис. 144). Корпус вентиля выполнен из стекла, мембрана и основание мембраны — из политетрафторэтилена. Вентиль закрывают поворотом шпинделя на 230°.
214
Для разделения агрессивных веществ, разрушающих стекло, предназначены перегонные аппараты, изготовленные из фарфора (рис. 145). Наименьший куб имеет емкость 50 л; номинальный диаметр колонны не менее 50 мм.
Фирма Подбильняк (Чикаго) поставляет автоматизированные пилотные ректификационные установки серии «ректификатор» четырех типов, предназначенные для разделения в периодическом или непрерывном режимах (рис. 146). Установки фирмы Подбильняк имеют следующую характеристику:
емкость куба ................
диаметр колонны .............
рабочая высота колонны . . . рабочее давление ............
рабочая температура . , . .
45—450 л
76—304 мм
1,5—3,0 м
от 3 мм рт. ст. до 136 ат
от —40 до +350 °C
В колоннах преимущественно применяется насадка «Хели-пэк» или «Окта-пэк» (см. разд. 7.8). Предусмотрен обогрев электрический, паровой или с применением теплоносителя «Даутерм».
Рис. 145.
Полупромышленная периодическая ректификационная установка из фарфора Изготовитель: народное предприятие «Керамический завод Хешо-Кала».
Рис. 146.
Автоматизированная пилотная ректификационная установка Подбильияка Изготовитель: фирма Подбильияка, Департамент обрабатывающей промышленности, Франклин парк, Иллинойс, США.
215
Карпентер и Хельвиг [31 ] описывают типовые детали и узлы, применяемые в пилотных установках. Жордан в книге [32], в которой рассмотрены полупромышленные опытные химические установки, в первую очередь обсуждает проблемы переноса опытных результатов на промышленные установки.
Часто приходится решать вопрос о том, что можно ли вместо проведения исследований на пилотных установках ограничиться применением чисто расчетных методов, основанных на масштабном переходе от малых аппаратов к большим. Однако для ректификационного разделения веществ еще нет методов для достаточно точного математического описания процесса с учетом всех решающих факторов. Поэтому опытно-промышленные испытания по-прежнему остаются важнейшим источником сведений, необходимых для масштабного моделирования [33]. В первую очередь это относится к насадочным колоннам, для которых гидродинамические характеристики газового и жидкостного потоков играют особую роль (см. разд. 4.2). Кроме того, для оценки стоимости ректификационных колонн с целью уменьшения капиталовложений необходимо знать зависимость разделяющей способности и перепада давления от нагрузки. Эту зависимость для большинства колонн до сих пор нужно устанавливать экспериментально. Чтобы можно было сравнивать различные колонны, для их испытаний следует подбирать одинаковые смеси и испытания проводить в одинаковых условиях (см. разд. 4.10 и 4.11).
5.1.3.3. Получение дистиллированной воды
Типичным примером наработочной перегонки в лабораторных условиях является получение дистиллированной воды, осуществляемое чаще всего по непрерывному способу.
Аппараты для дистилляции воды обычно работают при постоянном уровне жидкости в перегонном кубе. Это достигается благодаря тому, что предварительно нагретая в конденсаторе вода поступает в куб при непрерывном отводе ее избытка, что и обеспечивает регулирование уровня. Благодаря применению кварцевого стекла гарантируется высокая степень чистоты дистиллята (см. рис. 147).
Большое число предложенных конструкций дистилляторов указывает на то, что до сих пор еще не найден наиболее экономичный способ их работы. Поэтому для более экономичного использования тепла переходят на внутренний электрообогрев и предусматривают достаточный слой тепловой изоляции. Производительность лабораторных аппаратов лежит в интервале 0,5— 2,1 л/ч. Производительность от 40 до 50 л/ч обеспечивают широко применяемые дистилляционные аппараты, изготовленные из меди, никелированные (до зеркального блеска) или оцинкованные внутри. Они прочны и при хорошей теплоизоляции имеют высокий коэффициент полезного действия.
216
Шоттом и др. разработан автоматический аппарат для дистилляции воды с электрическим обогревом, выполненный из дюралюминия 50 (рис. 148). Отдельные модели этого прибора дают от 10 до 70 л/ч дистиллированной воды с электропроводностью менее 0,5 мкСм при условии, что аппарат подпитывают полностью обессоленной водой. Народное предприятие «Йенский завод стекла Шотт» выпускает дистилляционные установки с электрическим и паровым обогревом производительностью от 8 до 110 л/ч. Бидистилляторы с электрическим обогревом имеют производительность 8 и 24 л/ч. Для них специально разработан погружной нагреватель THQ 6 (номинальная мощность 6 кВт).
Представляет интерес аппарат с эффективным использованием тепла, работающего по принципу «per descensum» (нисходящего потока) (см. рис. 36). Такой аппарат имеет к. п. д. 95%. Аналогично работает прибор Кюлльманна [35], в котором получают дистиллят без какой-либо примеси газа; с помощью дополнительной приставки можно одновременно получать одно- и двукратно дистиллированную воду (рис. 149).
Бидистиллятор 1600 Зельнера [36] очень экономичен и дает от 1,3 до 1,6 л/ч продукта (рис. 150). Тепловая энергия к нему
Рис. 147.
Непрерывный дистиллятор «Пюратор» из кварцевого стекла для получения однократно дистиллированной воды (2 л/ч):
1 — перегоииый куб; 2 — внутренний нагреватель; 3 — дистилляционная приставка; 4 — теплообменник; 5 — трубка для измерения уровня; 6 — головка с колпаком и капельницей.
Изготовитель: народное предприятие «Кварцевое стекло», Берлин.
Рис. 148.
Автоматический дистиллятор воды нз дюралюминия 50 производительностью 10 — 70 л/ч.
А — охлаждающая вода; Б — газ для пневмоавтоматики; В — отработанная вода; Г — газовая защита; Д — исходная вода.
Изготовитель: фирма Шотт, Майнц,
217
Рис. 149.
Аппарат Кюлльманна для одновременного получения одно* и двукратно дистиллированной воды.
Изготовитель: фирма Кройцер—Крайлер, Мюнхен.
Рис. 150.
Дистиллятор воды 1600 Зельнера Изготовитель: фирма Шотт, Майнц.
подводится только на первой испарительной ступени, на которой из водопроводной воды получается монодистиллят. Вторичное выпаривание монодистиллята на второй испарительной ступени проводят при 30—40 °C под вакуумом, создаваемым водоструйным насосом. При этом используется теплота конденсации первичных паров, благодаря чему экономится электроэнергия. Водопроводная вода, необходимая для работы водоструйного насоса, служит первоначально в качестве охлаждающей среды. Часть воды после прохождения конденсатора и насоса используется для питания первой испарительной ступени аппарата. При том же потреблении энергии, что и для простой дистилляции, и при минимальном расходе водопроводной воды получают бидистиллят высокой степени чистоты.
Весьма полезна предварительная очистка питательной воды в ионообменном фильтре, как например в аппарате для дистилляции воды, выполненном из инертного по отношению к воде кварца фирмы «Хереус-Кварцшмельце» (Ханау). В последнее время применяют горизонтальные цилиндрические перегонные кубы. Разбрызгивание кипящей воды (брызгоунос) и образование 218
аэрозоля в этих аппаратах сведено к минимуму благодаря тому, что паровые пузыри движутся по кратчайшей траектории, а зеркало жидкости достаточно велико [36а].
5.1.4. РАЗДЕЛЕНИЕ ИЗОТОПОВ
Химические элементы, встречающиеся в природе, большей частью являются смесями изотопов. Их можно назвать многоизотопными элементами, в то время как изолированные изотопы представляют собой собственно чистые элементы. Изотопы элемента занимают одно и то же место в периодической системе элементов, поэтому они обладают одинаковыми зарядами, но разными массами атомных ядер. Чистые элементы 12С и 13С различаются, например, только тем, что в первом случае атомное ядро содержит шесть, а во втором случае — семь нейтронов. Атомные массы, которыми оперируют в химии, представляют собой усредненные значения атомных масс отдельных изотопов.
Изотопы были впервые открыты в радиоактивных веществах. Большая часть нерадиоактивных элементов также содержит определенное число стабильных изотопов. В табл. 34 приведены некоторые стабильные изотопы, которые могут быть выделены, главным образом, путем ректификации [37].
В органической химии для метки молекул используют прежде всего тяжелые изотопы элементов С, Н, О и N. Вплоть до углерода отсутствуют подходящие радиоактивные изотопы с достаточно большим периодом полураспада, поэтому стабильные изотопы 2Н, 13С, 18О и 15N имеют особенно большое значение для химических исследований, а также в биологии, медицине и геологии.
Элементы с нечетным зарядом ядра обычно состоят лишь из одного или двух изотопов с нечетными массовыми числами (за исключением Н, Li, В и N). Элемент Hg имеет семь стабильных изотопов (массовые числа 196—204) с относительной разностью в атомных массах порядка 0,5%. Все процессы разделения, направленные на обогащение и получение изотопов, основаны на использовании различия атомных масс, обусловливающем их неодинаковые свойства, поэтому относительная разность атомных масс является показателем степени трудности разделения. Точно также относительная естественная концентрация изотопов в исходном сырье имеет важное значение для процесса разделения. Из элементов, указанных в табл. 34, соотношение естественных концентраций изотопов особенно благоприятно в смесях 10В— nB, 20Ne—22Ne и 35С1—37С1.
Для обогащения или полного разделения изотопов применяют методы диффузии, термодиффузии, электролиза и обменные реакции. Обогащение можно также проводить с помощью методов осаждения и центрифугирования. Ректификационные методы разделения применяют для получения изотопов 4Не, D, 10В, 13С, 15N, 18О, 22Ne, 37С1 и 36Аг. Обстоятельный обзор методов получе-
219
Таблица 34.
Стабильные изотопы, относительная разность их атомных масс и их относительная естественная концентрация
Элемент Атомный номер Массовое число Относительная разность атомных масс, % Относительная естественная концентрация, %
Н (D) 1 1 100 99,9844
2 0,0156
Не 2 3 33,3 1,3-10~4
4 99,9999
В 5 10 10 18,83
11 81,17
С 6 12 8,35 98,9
13 1,1
N 7 14 7,15 99,62
15 0,365
О 8 16 «О—17О 6,25 99,757
17 1?О—18О 5,9 0,039
18 «О—18О 12,5 0,204
Ne 10 20 2’Ne—21Ne 5,0 90,51
21 21Ne—22Ne 4,75 0,28
22 20Ne—22Ne 10 9,21
S 16 32 32S_33S 313 95,06
33 33s—3«s 3,03 0,74
34 3«s—”S 5,9 4,18
36 0,016
Cl 17 35 5,7 75,4
37 24,6
Ar 18 36 ”Ar—38Ar 5,55 0,307
38 звДг—«Ar 5,25 0,060
40 з’Аг—«Ar 11,11 99,633
ния и применения стабильных изотопов дают Бродский [38] и Ветцель [39], они рассматривают специальные проблемы ректификации изотопов. Все большее значение приобретает комбинация химического изотопного обмена и ректификации, получившая название «обменной ректификации» [40]. Анализ стабильных изотопов проводят преимущественно методом масс-спектрометрии [39]. Разработаны также другие методы анализа изотопов, которые описали Мерсеа [41 ], а также Мюллер и Мауэрсбергер [42].
При обогащении стабильных изотопов методом ректификации в качестве сырья используют, главным образом, газы; лишь дейтерий и 18О получают из воды. Соотношения давлений паров для подобных смесей изотопов указаны в табл. 35. Разделение всех смесей, за исключением соединения бора ВС13, требует, разумеется, значительных затрат на охлаждение. Кроме того, для достижения обычной степени разделения смесей изотопов за исключением изотопов гелия и водорода требуется более 500 теоретический ступеней разделения. Кун с сотр. [43], применив большое число теоретических ступеней разделения, определил относительную летучесть для соединений изотопов с температурами кипения 80 °C.
Таблица 35.
Соотношения давлений паров для смесей изотопов
Смесь Температура, К Р°/Р° а л т Литература
h2-d2 18,65 3,6 [48]
3Не—«Не 2,19 5,4 [48]
иВС13—10ВС13 260 1,003 [52, 53]
UBF3—10BF3 170 1,01 [53]
12С16О—13CleO 68,2 1,010 [48]
«(у «О—12С18О 68,2 1,007 [48]
12С2Н4—12Ci3CH4 120 1,0014 [54]
169,3 1,0019
12С2Н8—12С13СНв 130 1,0022 [54]
165 1,0006
12СН4—13СН4 97,55 1,0099 [54]
111,8 1,0070
14N2—14N15N 63,3 1,006 [48]
««N2—««N15N 71,5 1,005 [55]
UNi’O—15N16O 117 1,03 [55]
UNi’O—14№8O 120 1,04 [50]
leQi’O—18O18O 63,14 1,01286 [49]
74,05 1,00912
20Ne—22Ne 24,6 1,043 [48]
ЗвДг_4°Аг 83,8 1,006 [48]
128Хе_136Хе 161,4 1,000 [48]
Р® — давление паров соединения, содержащего легкий изотоп, р® — давление па-ров соединения, содержащего тяжелый изотоп.
221
5.1.4.1. Разделение изотопов низкотемпературной ректификацией
Методы низкотемпературной ректификации (см. разд. 5.3.1) обычно применяют для разделения смесей изотопов Н—D, 10В— ПВ, 12С—13С, а также изотопов инертных газов — гелия, неона и аргона.
Благодаря большой разности атомных масс и больших различий в давлениях насыщенных паров изотопов получение дейтерия из газовой смеси Н2—D2 путем низкотемпературной ректификации теоретически представляется сравнительно простым. Селлерс и Аугуд [441 подробно и систематически изучили проблемы низкотемпературной ректификации систем HD—Н2, 16О18О—1вО1вО, а также О2—N2. Разделение проводили в колпачковой колонне высотой 27 м. Позднее исследования низкотемпературной ректификации смеси Н—D были проведены Тиммерхаусом с сотр. [45]. Для разделения использовали пилотную установку с колонной диаметром 150 мм, содержащую ситчатые тарелки, и определили ряд параметров для данной системы, важных с точки зрения разделения. Небольшую лабораторную колонну для ректификации смеси Н2—HD описал Вайссер [46].
В настоящее время для промышленного производства тяжелой воды применяют крупномасштабные установки [47]. Значительные трудности аппаратурного характера возникают при разделении газовых изотопных смесей. Поэтому лабораторное получение изотопов при температуре кипения жидкого азота и жидкого воздуха пока еще слишком дорого. Однако если ректификационную установку присоединить к промышленной установке для получения кислорода из жидкого воздуха, то концентрирование изотопов 36Аг, 18О и 16N может оказаться очень экономичным [48, 49]. По-видимому, очень выгодна низкотемпературная ректификация NO при одновременном получении 16N и 18О [501, а также ректификация СО при концентрировании 13С [511.
Методом низкотемпературной ректификации в колонне с 130 теоретическими ступенями разделения Клузиус и Мейер [48] ежесуточно обогащали 15 л аргона до концентрации 0,6% 36Аг (вместо 0,307% в природном аргоне). Для этого применяли насадочную колонну высотой 3 м, изготовленную из латунной трубки с внутренним диаметром 12 мм. Насадка состояла из проволочных спиралей размером 2 Х2 мм, выполненных из нержавеющей стали. На рис. 151 показана схема специально для этой цели изготовленного перегонного куба емкостью 250 мл и конденсатора, охлаждаемого жидким азотом. Бевилогуа с сотр. [164] сообщает о получении изотопов 22Ne и 20Ne, а также о концентрировании 21Ne ректификацией при 28 К:
В полупромышленной колонне диаметром 12 мм и высотой 12 м, заполненной металлическими кольцами диаметром 1,2 мм (рис. 152), Мюленпфордт получал в год 4,5 кг BF3 с 83% 10В.
222
Колонна на верхнем конце имеет- сборник для разделяемого вещества, а на нижнем конце — участок для отбора обогащенного вещества, что обусловлено более низкой летучестью 10BF3. Необходимым условием высокой разделяющей способности является равномерная подача жидкости BF3 в верхний конец колонны. Такая подача обеспечивалась специально разработанным дозировочным приспособлением.
а 6
Рис. 151.
Перегонный куб (а) и конденсатор (б) установки низкотемпературной ректификации для обогащения88 Аг:
/ — платиновый впай; 2 — нагреватель; 3 — перегонная колба для жидкого аргона; 4 —- место отбора; 5 — сильфон из томпака; 5 — насадка; 7 — латунная трубка; 8 — место спая стекла и металла; 9 — сосуд из пнрекса; 10 — верхний и нижний патрубки для отбора; 11 — медная сетка; 12 — резиновая трубка; 13 — двойная стенка из латуни; 14 — высоковакуумирован* ная полость; 15 — сифон для жидкости; 15 — патрубок для отвода паров азота;
17 — трубка из мельхиора; 18 — термометр для измерения температуры жидкого азота; 19 — сосуд с жидким азотом; 20 — свинцовая дробь; 21 — медная пластина.
Рис. 152.
Установка низкотемпературной ректификации для концентрирования 10BF3 конструкции Мюленпфордта:
I.— испаритель с электрообогревом; II — колонна для очистки сырья от SiF4 и HF; III — колонна с насадкой; IV — двойной конденсатор для паров BF3; V — резервуар для исходной жидкости с электрообогревом; VI — вспомогательный конденсатор; VII — конденсатор для паров этилена; VIII - буферный резиновый баллон; IX — резервуар для жидкого воздуха; 1, 2 — патрубки для присоединения к диффузионному насосу; 3,4 — линия для присоединения манометра; 5 — буферная емкость, заполненная водородом; 6 — линия для подачи этилена и водорода; 7 — линия подачи исходного BF3; 8 — линия для отвода обедненной фракции BFS; 9 — линия для отвода обогащенной фракции 10BF3; 10 — линия для отвода загрязнений.
223
5.1.4.2. Получение D и 180 путем ректификации воды
В данном разделе рассмотрен метод получения D и 18О путем ректификации природной воды при атмосферном и пониженном давлениях.
Водород существует в виде трех изотопов: ГН — водород, 2Н — дейтерий и радиоактивный 3Н — тритий. Тритий встречается в природе в ничтожных количествах.
Кислород существует в виде трех стабильных изотопов: 16О, 17О и 18О. Теоретически возможны следующие модификации воды (без учета трития 3Н):
|2Н1вО2Н| 1Н1«О2Н
2Н17О2Н Щ1’02Н
|*Н18О1Н| 2Н1«О2Н Щ18О2Н
Наибольший интерес представляют тяжелая вода D216O(2H2ieO) и вода 1Н218О, обогащенная изотопом кислорода.
, Физико-химические свойства D2O и Н2О следующие [37, 56 ]:
D2O Н2О д
Плотность pf°, г/см3 . . . 1,1059 0,9982 0,1077
Температура плавления, °C 3,82 0,00 3,82
Температура кипения, °C 101,42 100,0 1,42
Температура, при которой
жидкость имеет максималь-
ную плотность, °C .... 11,6 4,0 7,6
Динамическая вязкость, сП 1,260 1,009 0,251
Поверхностное натяжение,
дин/см 67,80 72,75 4,95
Показатель преломления
«20 nD 1,32844 1,33300 0,00456
Как видно из приведенных данных, разность температур кипения D2O и Н2О составляет 1,42 °C, а разности значений плотностей и температур, соответствующих максимумам плотности, очень малы.
Исходным сырьем для получения тяжелой воды является природная вода, содержащая 0,0146 % (ат.) D (1 : 6850). В дождевой воде концентрация D несколько выше, а именно 0,0200% (ат.) (1 : 5000) [56]. Поэтому природную воду, подвергаемую ректификации, можно рассматривать как смесь низкокипящего компонента Н2О (100 °C) и высококипящего компонента D2O (101,42 °C).
224
Поскольку в природной воде наряду с Н2О и D2O содержатся молекулы HDO, относительная летучесть компонентов данной смеси а' определяется выражением:
ХНРО + 2хРгО / 2уНгО ~Ь -УНРО
2хНгО + ХНРО J ^HDo+^D.O
(161)
В соответствии с работой Юри [57 ] а' можно выразить также через отношение давлений паров чистых компонентов:
а = ]/ Рщо/РРгО (162)
Это уравнение выведено на основе предположения, что давление паров HDO является средним геометрическим значением давлений паров Н2О и D2O, а константа равновесия реакции
Н2О 4-D2O^2HDO
(163)
в паровой фазе равна 4,0. Рис. 153 иллюстрирует зависимость а' от температуры, полученную Киршбаумом [56].
Аналитическим путем установлено, что число теоретических ступеней разделения смесей Н2О и D2O минимально при возможно меньшем рабочем давлении и, следовательно, при низких температурах. Однако при проведении ректификации в таких условиях трудно конденсировать пары дистиллята, и производительность колонны значительно ограничена из-за большой нагрузки по пару. При использовании тарельчатых и насадочных колонн имеют место такие потери напора и допустимая скорость
потока паров, которые существенно увеличивают длительность процесса ректификации и делают данный метод разделения экономически невыгодным. Вследствие этого при проведении подобной ректификации обычно идут на компромисс, работая при давлении 50—125 мм рт. ст. Значения а' при температурах 40—60 °C,
р,мм ргп.сгп.
соответствующие этому интервалу давления, лежат в пределах 1,06— 1,05.
При ректификации природной воды графический метод Мак-Кэба и Тиле неприменим для расчета числа теоретических ступеней разделения, так как с одной стороны кривая равновесия очень полога, а с другой — рабочие концентрации D2O очень близки к предельным значениям, а именно: х0 = 0,000146 D2O
Рис. 153.
Зависимость а' = [/Л/'н2оА,Е>гО от температуры.
8 Э. Крель
225
(ат. доли) — исходная концентрация; хк0;, = 0,998 D2O — конечная концентрация; а' = 1,059 (при 40 °C и 55,3 мм рт. ст.).
Для данного примера уравнение Фенске дает в первом приближении следующее значение минимального числа теоретических ступеней разделения при работе с бесконечным флегмовым числом:
«мин = Г 1g^°-!(1~Хо] / 1g а' 1 - 1 = 308 (164)
Необходимое число теоретических ступеней разделения при оптимальной скорости отбора кубового продукта составляет —700 [58]. Следует иметь в виду, что в данном случае нужным компонентом является высококипящее вещество D2O, которое при периодической ректификации накапливается в кубе, а при непрерывной перегонке выводится из него. В качестве дистиллята Получается вода с концентрацией D2O менее 0,0146% (ат.).
Согласно работе Куна [59] эффективность элементарного акта разделения подобной идеальной системы изотопов, имеющая место при однократном частичном испарении жидкой смеси, определяется соотношением:
При данной температуре величина 6 характеризует бинарную смесь, поскольку она не зависит от значения исходной концентрации х0 (в интервале 0,99—10*3 мол. долей). Величину 6 называют параметром разделения. Зная давления паров чистых компонентов и предположив, что тепловой эффект смешения этих компонентов равен нулю, 6 можно рассчитать по уравнению:
Р°/Р° = *в (166)
где р® и — давление паров чистого низкокипягцего и высококипящего компонента соответственно.
Если относительная разность давлений насыщенных паров компонентов мала, то значение 6 можно определить по следующим формулам:
МРи-рЖ (167)
6==а — 1 (168)
Для смесей с тепловым эффектом смешения равным нулю, к которым относится большинство смесей изотопов, 6 можно приближенно рассчитать по уравнению:
6= 1о,7 ДТ /Тр (169)
где ДТ — разность температур кипения двух компонентов при атмосферном давлении; Тр — температура кипения низкокипящего компонента при рабочем давлении ректификации.
226
В табл. 36 приведены значения параметра разделения 6 для бинарных смесей, полученные в предположении, что оба компонента подчиняются правилу Трутона и зависимости их теплот парообразования от температуры примерно одинаковы.
Эффективность ректификационной колонны с определенным числом теоретических ступеней разделения п, возникающую при совмещении большого числа элементарных актов разделения, определяют по формуле:
,1,~Г|1<>н) =епа (170)
х0/(1—х0)
При этом необходимое число теоретических ступеней разделения, требующееся для конкретного процесса ректификации, рассчитывают по уравнению:
(‘/о
Это уравнение соответствует известному уравнению Фенске (1 086) при v = оо.
Минимальное флегмовое число для заданного процесса разделения можно определить в первом приближении по формуле: ^мин = Z/T = (%кои — Хо)/[6хо (1 — х0)] (172)
где Z — количество жидкости, циркулирующей в кубе (соответствует флегме)! т — количество отбираемого кубового продукта (соответствует отбираемому дистилляту).
При отборе кубовой жидкости, обогащенной высококипящим изотопом, максимально возможный относительный выход кубового продукта оценивают по уравнению:
(т/2)макс = Х()6 (1 — Хо)/(%кон — ^о) (173)
Максимальному выходу кубового продукта, определяемому выражением (173), соответствует бесконечное число теоретических ступеней разделения. Поэтому действительное количество отбираемой фракции должно быть несколько меньшим.
Таблица 36.
Параметр разделения 6 и относительная летучесть а для смесей изотопных соединений
Разделяемая смесь Рабочая температура, °C Разность температур кипения» °C Параметр разделения д Относительная летучесть а
H2O-D2O 70 1,6 0,05 1,05
Н216О—Н218О 70 0,165 0,005 1,005
12СС14—13СС14 35 0,036 0,00125 1,00125
C3SC14—Сз7С14 35 —0,003 0,00010 1,0001
8*
227
t/Z
Xo , 7о(мОЛ.)
Рис. 154.
Зависимость числа теоретических ступеней разделения от относительного выхода кубового продукта т/Z, полученная Куном (значение х^ и *кон в мольных долях):
1 — х0 = 0,002, ткон = 0,015, б =- 0,0045; 2 — = 0,002, *Кон = 0,020,
б = 0/0045; 3 — х0 = 0,002, xRoH = 0,020, б = 0,0065.
Рис. 155.
Зависимость необходимого числа теоретических ступеней разделения от концентрации D2O в исходной смеси при v = «.
Давление: 760 мм рт. ст. (100 °C) и 100 мм рт. ст. (50 °C); *кон — = 99,8% (мол.).
Необходимое число теоретических ступеней разделения п, требующееся для конкретного процесса ректификации при конечном флегмовом числе [или при относительном выходе кубового продукта, меньшем чем (т/Х)макс ], можно определить по диаграмме, полученной Куном (рис. 154).
При стационарном отборе кубовой фракции величину п рассчитывают по формуле:
<174>
На рис. 154 приведены в качестве примера зависимости необходимого числа теоретических ступеней разделения от относительного выхода кубового продукта, полученные для процесса обогащения воды стабильным изотопом 18О.
Из рассмотрения кривой 3 видно, что для повышения концентрации изотопа 18О с 0,2% (мол.) до 2% при а = 1,0065 необходимы следующие условия:
t/Z=10~6, «=388; т/Z = Ю'1, «=409; t/Z=2-10-«, «=437; t/Z=5.10- «=567.
Другие методы расчета числа теоретических ступеней разделения и удобные номограммы приведены в работах Хубера [61] и Хильтбруннера с сотр. [62].
Кун с сотр. [60] при получении D2O сначала обогащал природную воду путем электролиза до содержания D2O 1,0% (мол.) 228
и только потом применял ректификацию. В промышленности используют также и обратный путь. На рис. 155 показаны зависимости необходимого числа теоретических ступеней разделения от концентрации D2O в исходной смеси, полученные при бесконечном флегмовом числе и хкон = 99,8% (мол.) для температур 50 °C (100 мм рт. ст.) и 100 °C (760 мы рт. ст.). Материальный баланс двухступенчатого аппарата Куна наглядно показан на рис. 156, из которого видно, что относительный выход кубового продукта составляет на первой ступени 1 : 1270, а на второй-—1 : 78.
Сложности, возникающие в лабораторных условиях при такой скорости отбора кубовой жидкости, затрудняют аппаратурное оформление установки. Поясним детали процесса получения D2O на примере ректификационной установки Куна [59] (рис. 157). Исходная смесь (природная вода), находящаяся в сосуде 1, испаряется и через обогреваемые снаружи капилляры 2 равномерно поступает в трубчатые колонны, заполненные насадкой из проволочной сетки. Расход паров контролируют по показаниям дифференциального манометра 3, подсоединенного к емкостям 4 и 5. Нижние части трубчатых колонн в зоне куба 10 подогревают водя-
Рис. 156.
Схема материального баланса двухступенчатой установки Куна для получения D2O.
Высота разделяющей части 530 см (ступень 1) и 680 см (ступень 2), ВЭТС = = 1,8 см (ступень 1) и 1,7 см (ступень 2), давление в головке колонны 120 мм рт. ст. (ступень 1) н 60 мм рт. ст. (ступень 2).
Рис. 157.
Принципиальная схема ректификационной установки Куна с трубчатыми колоннами для получения D2O:
1 — сосуд для исходной смеси; 2 — обогреваемые капилляры; 3 — дифференциальный манометр; 4, 5 — емкости; 6 — конденсатор; 7 — буферная емкость; 8 — сборник конденсата; 9 — барометрическая труба; 10 — куб; 11 — приемная камера; 12 — клапан; 13 — приемник кубового продукта; 14 — испаритель.
229
Зависимость относительной летучести для смеси Hgieo—Hgl8o от температуры По данным: О — Достровского; Ливлнна и Времена; Мак-Вильямса, Пратта, Делла и Джойса; Д — Вэхла и Урея; □ — Ризен-фельда и Чаига; X — Жаворонкова, Уварова и Севрюговой.
Рис. 159.
"Схема металлической ректификационной установки для обогащения воды изотопом Н21вО.
1 —• основной трубопровод для подачи воды; 2 — трубопровод-для подачи дистиллированной воды; 3,4 — основные конденсаторы; 5 — переточная труба; 6 — линия для исходной смеси; 7 — дефлегматор; 8 — магнитный клапан; 9 — линия к вакуумному насосу; 10 — маностат; 11 — дифференциальный манометр; 12 — манометр кипятильника; 13 — ректификационная колонна; 14 — расходомер; 15 — контрольная головка; 16 — штуцер для отвода продукта; 17 — медная труба (диаметр 25,4 мм) между кипятильником и колонной; 18 — кипятильник; 19 — сборник; 20 — штуцер для выхода сточной воды.
ним паром, поступающим из испарителя 14. Расход греющего пара выбирают таким образом, чтобы обеспечивалось испарение всей жидкости, находящейся в этих частях колонн. Через капилляры, размещенные внизу трубчатых колонн, в камеру 11 отбирают пары путем создания в ней некоторого разряжения. Скорость отбора должна соответствовать выбранному относительному выходу кубового продукта. Посредством клапана 12 осуществляют контроль за расходом отбираемых паров, которые затем конденсируют в расположенном ниже холодильнике. Конденсат стекает в приемник кубового продукта 13. Пары, поднимающиеся по колоннам, выводят через штуцер, присоединенный к емкости 5, и полностью конденсируют в конденсаторе 6. Конденсат, поступивший в сборник 8, насосом подают в сосуд 4, откуда часть его отводят по барометрической трубе 9 в точном соотношении с количеством исходной воды, подаваемой в колонны через капилляры 2. Для работы под вакуумом вакуумный насос присоединяют к сосуду 7 и к штуцеру, размещенному над приемником кубового продукта 13.
Производительность данной установки при разделении природной воды достигает 1,5 кг/ч. Ректификацию проводят в две ступени. На первой ступени воду обогащают от 1 до 50% (мол.) D, 230
а на второй — от 50 до 99,8% D. Нагрузку поддерживают примерно^на 10% ниже нагрузки, соответствующей пределу захлебывания. При заполнении трубчатых колонн кольцами Рашига из проволочной сетки ВЭТС составляет 1,7—1,8 см. Применение насадочных колонн обусловлено высоким коэффициентом поверхностного натяжения воды, что затрудняет равномерное распределение жидкости по стенкам полых трубчатых колонн. Обеспечение равномерного распределения жидкости по насадке является основной проблемой при получении тяжелой воды (см. разд. 4.2).
Соотношение содержаний изотопов кислорода в естественных условиях примерно составляет 1вО : 17О : 18О = 2500 : 1 : 5. При ректификации природной воды кубовая жидкость обогащается изотопом 18О. Как видно из рис. 158, относительная летучесть компонентов смеси Н21вО—Н218О примерно на порядок ниже, чем для смеси Н2О—D2O. При 100°C (760 мм рт. ст.) а составляет около 1,004, а при 50 °C (ПО мм рт. ст.)- 1,007. Данный график
взят из работы Достровского и Равива {53 ]. Значительный разброс опытных данных частично объясняется трудностями экспериментального определения коэффициента а. Сравнительно высокая концентрация изотопа 18О в природной воде, равная 0,204% (ат.), упрощает условия проведения ректификации.
При работе на двухступенчатой ректификационной установке Кун достигал обогащения воды до 90% (ат.) 18О. Достровский с сотр. [53], применяя комбинированную установку, получал повышение концентрации 18О до —95% и 17О до —2,0%. Предварительное обогащение от 0,2% до 1,6% 18О проводили в 10 параллельно включенных колоннах (диаметр каждой колонны 100 мм). При ректификации на многоступенчатом каскаде из колонн (диаметр колонн от 30 до 100 мм) с расходом исходной смеси 800 мл/ч при относительном выходе кубового продукта 1,37-Ю"2 конечная концентрация 18О достигала до 99,8%. Наибольшее обогащение 17О получалось в средней части каскадной установки, в которой концентрация 17О примерно составляла 10%. В указанных процессах особенно хорошо себя зарекомендовала насадка Диксона в виде колец Рашига из фосфористо-бронзовой сетки 100 меш. с S-образными перемычками [63].
На рис. 159 показана схема непрерывно работающей одноступенчатой ректификационной установки, использованной автором для обогащения природной воды изотопом 18О до концентрации 5,8%. Испытания, проведенные на этой установке при 300 мм рт. ст., позволили с большой точностью определить значение а при данном давлении и температуре 76 °C, оказавшееся равным 1,0068 [64]. Уваров с сотр. [54, 65], работая на непрерывно действующей при атмосферном давлении ректификационной колонне диаметром 52 мм, заполненной на высоту 9,5 м насадкой из треугольных спиралей размером 2,0X1,6 мм, добился обогащения 18О от 3 до 24,5 %. ВЭТС составляла около 1 см. Более вы-
Рис. 160.
Металлическая ректификационная установка для непрерывного получения Н218О.
сокое обогащение тяжелой воды изотопом кислорода путем ректификации получил Сташевский [70].
На рис. 160 показана установка непрерывного действия, снабженная медной колонной диаметром 76,2 мм и высотой 9,14 м. Насадка выполнена в виде колец Рашига из проволочной сетки диаметром 1,6 мм. Установка автоматизирована и снабжена специальными аппаратами для электролиза кубовой жидкости, обогащенной 18О. Выделяющийся в виде газа изотоп 18О вступает в реакцию с водородом, также образующимся при электролизе. В качестве конечных продуктов получают таким образом Н218О и D216O.
Если при обычной ректификации время установления стационарного режима лежит в интервале от нескольких минут до 24 ч, то при ректифика-
ции изотопов этот период может длиться несколько недель и даже месяцев. Формулы для определения данного периода времени, представленные в ранних работах, не являются удовлетворительными. Кун с сотр. [59] предложил
Таблица 37.
Результаты простого и двойного концентрирования
Вещество ”эфф Параметры
верхняя часть колонны (13С)
*b,/*b2 (Рл~Рт>+т + ~ + °C
Хлороформ 610 0,62 —8-Ю-4 +0,02
Четыреххлористый углерод 80 0,90 —1,3-10-* +0,033
Метанол 140 0,97 —2 • 10~4 +0,0055
Бензол 80 0,98 —2,5-10~4 +0,006
и рт — давление паров вещества, содержащего легкий и тяжелый изотоп соответст изотопа в кубе и в головке колонны соответственно; /л и tT — температура кипения вещест
232
для расчета времени установления стационарного режима установок с многотрубчатыми и насадочными колоннами (времени пуска) следующую формулу:
>в| <I7S’
где Н — удерживающая способность колонны по жидкости; Z — расход исходной смеси.
Другие сведения по вопросу о получении D и 18О можно найти в работах Джексона и Пигфорда [67], Розена [68] и Бродского [38]. В книге Лондона и Кохена [69] рассмотрена общая теория разделения изотопов. В ней содержатся также сведения об оптимизации условий разделения с использованием каскада ректификационных колонн, которая играет важную роль в промышленной ректификации, в том числе при разделении изотопов [71 ].
5.1.4.3. Получение других изотопов
В настоящем разделе приведены дополнительные примеры использования ректификации для концентрирования изотопов. Относительная разность молекулярных масс ДМ и естественная концентрация наиболее благоприятны у изотопов С и С1:
12С 98,9% „ зьС1 75 4о/
Д/И=8,35% Д/И = 5,7%
1»С 1,1% 37С1 24,6% 0
Кун [59], проводя ректификацию в многотрубчатой колонне, обладающей эффективностью около 250 теоретических ступеней разделения, добился увеличения концентрации 13С на 7%. Концентрацию определяли с помощью масс-спектрометра. При этом был установлен интересный непредсказуемый факт, что тяжелый изотоп углерода 13С накапливается в низкокипящей фракции, а изотоп 37С1 — в высококипящей фракции. Подобный эффект
изотопов при ректификации
процесса
куб (”С1) куб (•’ О)
*B/*BS (Рл-Рт)/Рт 'л~ + °С *b/*b2 (рл-рт)/рт t„-t, °C л т
1,06 1,003 + Ю“Б +3-10-4 —0,003 —0,001 1,50 3-10-" —0,07
венио, при температуре ректификации (34,6 °C); и xg2 — концентрация выделяемого ва, содержащего легкий и тяжелый изотоп соответственно.
233
возникает и при разделении смеси изотопов бора 10В—ПВ. Баерчи и Кун [72] показали, что причиной более высокой летучести тяжелого изотопа является вклад тепловых колебаний его молекул в дисперсионное взаимодействие.
Было установлено, что при ректификации СС14 происходит двойное разделение изотопов. Относительная летучесть смеси 12С—13С примерно составляет 1,00125, что соответствует разнице температур кипения около 0,036 °C (см. табл. 36). Эффект накопления 13С в верхней части колонны наблюдался также при ректификации хлороформа, бензола и метанола. При ректификации хлороформа, кроме того, происходит концентрирование 37С1 в кубе, а при ректификации метанола в кубе накапливается 18О (табл. 37) [43].
Траузер с сотр. исследовал возможность использования молекулярной дистилляции и дистилляции на «коротком пути»* для концентрирования изотопа лития. Он разработал одно-и многоступенчатую установку и определил, что относительная летучесть разделяемой смеси изменяется на первой ступени в пределах от 1,052 до 1,064 при изменении температуры в интервале от 535 до 627 °C. Подобным образом можно разделять и изотопы ртути.
5.2. КЛАССИФИКАЦИЯ МЕТОДОВ
ПЕРЕГОНКИ ПО СПОСОБУ
ПРОВЕДЕНИЯ ПРОЦЕССА
В лаборатории процессы дистилляции и ректификации проводят как при периодическом режиме работы колонны, так и при непрерывном режиме. Необходимость повышения пропускной способности установок приводит к применению полунепрерывных и непрерывных методов работы. В специальных случаях можно применять метод парциальной конденсации, который при правильном использовании обладает определенными преимуществами по сравнению с другими методами перегонки.
5.2.1. ПЕРИОДИЧЕСКАЯ И ПОЛУНЕПРЕРЫВНАЯ перегонка
Под периодической перегонкой понимают процесс разделения некоторого количества смеси, однократно введенного в куб. Различают простую перегонку и фракционную разгонку. При простой перегонке весь дистиллят собирают в одном приемнике. В результате этого процесса исходная смесь разделяется на две части: дистиллят и кубовый остаток. Дистиллят, полученный при фракционной разгонке, состоит из отдельных порций (фракций). Основное отличие периодического процесса от непрерыв-
* Определение этого термина см. в разд. 5.4.4. — Прим. ред. 234
Рис. 161.
Установка с колонной я подогревателем исходной смеси для полунепрерывной вакуумной ректификации.
ного заключается в том, что в периодическом процессе в результате отбора дистиллята непрерывно изменяется состав и количество кубовой жидкости.
Эффективность использования тепла и производительность обычной лабораторной установки можно повысить, применяя полунепрерывный метод работы. От периодического метода он отличается тем, что в куб по мере отбора дистиллята добавляют исходную смесь. Кубовая жидкость обогащается высококипящей фракцией. Исходную смесь подают непосредственно в куб или несколько выше него. При этом данную смесь предварительно подогревают до температуры, близкой к температуре кипения (см. рис. 138). Размер выбранного куба должен соответствовать общему количеству разделяемой смеси и заданному объему высококипящей фракции.
Полунепрерывный метод перегонки применяют в тех случаях, когда нужно отделить низкокипящую фракцию или сконцентрировать смесь, содержащую растворенные твердые вещества. В последнем случае непрерывной подачей исходной смеси предотвращают загустевание или даже затвердевание кубовой жидкости. По истечении определенного промежутка времени подачу исходной смеси прекращают и содержимое куба разделяют периодической перегонкой или выводят в качестве кубового остатка. Непрерывный отбор кубовой жидкости невозможен, поскольку с исходной смесью в куб поступает низкокипящая фракция, которая предварительно должна быть отделена. На рис. 161 показана ректификационная установка полунепрерывного действия с колонной и подогревателем исходной смеси в виде сосуда с обогревающим кожухом.
5.2.2. НЕПРЕРЫВНАЯ РЕКТИФИКАЦИЯ
В отличие от периодической и полунепрерывной ректификации при непрерывном методе работы исходную смесь непрерывно подают в колонну через штуцер, расположенный между укрепляющей и исчерпывающей ее частями (рис. 162). После пуска установки все условия проведения процесса ректификации остаются постоянными. Исходную смесь, предварительно подогретую до температуры, равной температуре жидкости в колонне в месте ввода исходной смеси, разделяют до заданного соотношения на дистиллят и кубовый продукт, которые имеют постоянный состав.
235
Рис. 162.
Вакуумная ректификационная установка непрерывного действия с устройством для подачи исходной смеси (по нормалям «Дестииорм»):
1 — устройство для подачи исходной смеси; 2 — пульт управления; 3 — подогреватель исходной смеси; 4, 8 — мерные бюретки; 5 — термостатированный сосуд для исходной смеси; 6 — головка колонны с мерной бюреткой; 7 — штуцеры для присоединения к вакуумной линии; 9 — укрепляющая часть колоииы; 10 — тройиик для ввода исходной смеси; 11 — исчерпывающая часть колонны; 12 — контактный манометр; 13 — измеритель расхода жидкости, поступающей в куб; 14 — пробоотборник кубовой жидкости; 15 — куб; 16 — нагреватель куба.
Удерживающая способность колонны по жидкости также стабилизируется при постоянных разности температур и градиенте концентраций (см. разд. 4.7.2).
Основное преимущество непрерывной ректификации состоит в том [28], что разделяемая смесь находится в мягких температурных условиях. Кроме того, при непрерывной работе часто удается достигнуть производительности лабораторной установки, такой же, как и для полупромышленных установок периодического действия. Непрерывно работающие лабораторные установки производительностью 10—20 кг/сут можно использовать для получения различных продуктов, например термически нестойких фармацевти-
ческих препаратов, для отгонки растворителей и т. д. Пропускная способность лабораторных установок составляет 0,5— 5 л/ч. Сильно агрессивные вещества, вызывающие коррозию металлической аппаратуры, обычно разделяют в стеклянных установках непрерывного действия. На основе опытных данных, полученных с использованием таких установок, с достаточной степенью надежности можно разрабатывать полупромышленные и промышленные установки из фарфора, технического стекла или металла.
Другое преимущество непрерывной ректификации состоит в том, что при стационарном режиме работы колонны получают дистиллят и кубовый продукт постоянного состава. Кроме того, затраты тепла оказываются существенно ниже, чем в периодическом процессе. При работе на лабораторных ректификационных установках обычно легко обеспечивают постоянство состава исходной
236
смеси, являющееся необходимым условием для непрерывного процесса. Точный состав разделяемой смеси можно предварительно установить путем аналитической ректификации или посредством пробной перегонки (см. разд. 7.2).
5.2.2.1. Области применения непрерывной ректификации
По основным областям применения непрерывную лабораторную ректификацию можно разбить на следующие группы.
1) Опытная ректификация с применением промышленных методов. Эта ректификация используется при разработке процессов разделения в полупромышленном или промышленном масштабе, для сравнительной ректификации, моделирующей промышленный процесс, и для демонстрационных исследований в учебных целях.
2) Предварительное разделение на фракции больших количеств веществ (около 50—100 л). Этой ректификацией пользуются для отделения низкокипящей фракции от высококипящего остатка и для выделения среднекипящей фракции.
3) Наработочная ректификация при производительности установки до 20 кг/сут (см. разд. 5.1.3). С помощью этой ректификации разделяют термически нестойкие смеси, например эфирные масла, ароматические вещества, выделяют чистые вещества из смесей, очищают сырье и растворители, получают фармацевтические препараты.
В разработке промышленных ректификационных установок отчетливо видна тенденция к переходу от периодических методов разделения к непрерывным. Это заставляет и исследовательские лаборатории заниматься данной проблемой.
Методы разделения, реализуемые в крупных установках, необходимо предварительно разрабатывать в лаборатории при одинаковом аппаратурном оформлении процесса. Экспериментальным путем можно быстрее решить поставленную задачу, чем посредством расчетов, особенно при исследовании разделения многокомпонентных смесей. Очевидно также, что лабораторные исследования — это наиболее экономичный путь исследований, так как проведение опытов с использованием промышленных установок требует значительно больших затрат материалов, энергии и времени. Вследствие введения принципа сборки лабораторной установки из отдельных стандартных деталей появилась возможность с помощью лабораторной аппаратуры в значительной мере воспроизвести промышленную установку и благодаря этому смоделировать (конечно, в уменьшенном масштабе) процесс разделения. Таким образом, на основе лабораторных исследований можно проектировать полупромышленные и промышленные установки.
До настоящего времени после разработки метода разделения в лаборатории ставилась задача найти путь перехода от лаборатор
237
ного способа разделения к промышленной установке. Появление сборных комбинированных установок в значительной степени исключило такой путь *. Исследования по ректификации, описанные в разд. 5.2.2.4, проведены с учетом особенностей промышленных установок. Подобные лабораторные установки позволяют также исследовать процессы перегонки, уже внедренные в промышленность, с целью изучения причин нарушений в работе больших установок путем сравнительной и модельной ректификации [73]. Можно также уточнить конструктивные (геометрические) параметры промышленных ректификационных колонн, предназначенных для точного фракционирования изотопов, посредством опытной ректификации в промышленном масштабе. С помощью специальных расчетных программ можно найти распределение концентраций по высоте испытуемой ректификационной колонны, находящееся в хорошем соответствии с опытными данными. На основании этого можно определить оптимальные конструктивные (геометрические) параметры промышленных колонн (см. разд. 4.15; [73а]).
Для разделения бинарных смесей с большой разностью температур кипения преимущества непрерывного метода работы особенно очевидны. На сравнительно простых установках можно проводить ректификацию непрерывно в течение продолжительного времени, достигая сравнительно высокой производительности в условиях работы как при атмосферном давлении, так и под вакуумом. В установке, состоящей из двух ректификационных колонн, можно непрерывно разделять тройную смесь (см. разд. 4.9). Известно, что для разделения смеси, состоящей из п компонентов, необходимо п— 1 колонн. Однако, смеси с числом компонентов больше трех вследствие значительных аппаратурных затрат следует разделять не за один проход, а прежде выделить две или три фракции из нескольких компонентов, после чего эти фракции периодически разделить на чистые вещества.
Если разделяемая смесь имеет большой высококипящий остаток, то сначала следует отделить его в пленочной ректификационной колонне (см. разд. 5.4.3), поскольку высокомолекулярная фракция исходной смеси может легко разлагаться или полимеризоваться при ректификации в обычных условиях. Такое предварительное разделение в мягких температурных условиях дает воз-
* Следует, однако, иметь в виду, что эффективность ректификационной колонны существенно зависит от ее геометрических размеров и лабораторные данные не могут быть в полной мере использованы для разработки промышленной установки. Моделирование ректификационной аппаратуры является предметом специальных исследований. Необходимо также учитывать, что процесс разделения, проводимый в установках из металла, часто отличается от процесса, проте-каемого в стеклянной аппаратуре, особенно при разделении термически нестойких веществ. Вследствие этого необходимо одновременно проводить исследования термической стойкости компонентов исходной смеси в присутствии материала, из которого предполагается изготавливать промышленную установку. — Прим, ред.
238
Исходная смесь С4—С30
4 остаток > С20
4 непрерывная пленочная ректификация
4
Ci—С2о ' 4
непрерывное самопроизвольное испарение
4 4
фракция Cj—С4 С4—С20
4 Г“
периодическая ректификация непрерывная ректификация
I
4 4
непрерывная ректификация непрерывная ректификация
периодическая периодическая периодическая периодическая
ректификация ректификация ректификация ректификация
Рис. 163.
Схема разделения многокомпонентной смеси Ct—С20
можность значительно повысить выход высококипящих компонен-тов. Низкокипящую фракцию можно отделить от основной части методом расширительной перегонки (см. разд. 5.4.2), позволяющей вести процесс с высокой скоростью (до 3 л/ч). С помощью этих двух вышеописанных методов можно выделять основную часть смеси, свободную от высококипящего остатка, а затем фракционировать ее на обычной ректификационной установке непрерывного действия. Схема разделения многокомпонентной смеси Сг—С20 наглядно поясняет подобный процесс (рис. 163).
5.2.2.2. Теоретические расчеты
Расчет процессов периодической или непрерывной ректификации предполагает определение следующих параметров: рабочего давления (атмосферного или вакуума), требуемого числа теоретических ступеней разделения, минимального флегмового числа, температуры исходной смеси, количества затрачиваемого тепла.
Обычно перед непрерывной ректификацией для определения ее основных параметров проводят периодическую пробную перегонку (или еще лучше аналитическую ректификацию). Рекомендации по выбору давления приведены в разд.5.4. Требуемое число
230
Рис. 164.
Зависимость к. п. д. тарелок от нагрузки при циклическом методе работы с тремя различными периодами цикла (1—3) и непрерывном методе (4) [76 ].
теоретических ступеней разделения и минимальное флегмовое число рассчитывают по методикам, описанным в разд.4.7.2, а рабочую температуру исходной смеси выбирают как можно ближе к температуре кипения. Необходимые размеры ректификационной ко-
лонны вычисляют по формулам, представленным в разд. 4.11, тепловые расчеты проводят по методике, изложенной в разд.4.12. При расчете диаметра колонны принимают во внимание, что в исчерпывающей части колонны количество стекающей жидкости больше, чем в укрепляющей части, вследствие подачи исходной смеси. Если количество дистиллята невелико (около 10—20% от количества исходной смеси), то рекомендуется выбирать колонну с диаметром исчерпывающей части большим, чем получается по расчету, исходя из нагрузки укрепляющей части колонны.
Следует указать на возможность проведения процесса ректификации циклическим методом, исследованным Гельбиным [74]. Например, Каннон [75] предложил подавать пар в ректификационную колонну циклически с периодом 3 с, для этого на паропроводе, соединяющем испаритель с колонной, устанавливают соответствующее регулирующее устройство. Мак-Виртер и Ллойд [76] для реализации циклического метода разделения применяли тарельчато-насадочную колонну, на пяти тарелках которой размещались небольшие слои насадки. При разделении этим методом смеси метилциклогексан—толуол они добились значительного повышения производительности ректификационной колонны. Были определены оптимальный период цикла и характер зависимости нагрузки от времени. Из графика, приведенного на рис. 164, отчетливо видно, что к.п.д. тарелок со слоями насадки при циклическом методе работы значительно выше, чем при непрерывном процессе.
5.2.2.3. Аппаратура для непрерывной ректификации
В лабораторных и промышленных ректификационных установках непрерывного действия используют в основном насадочные и тарельчатые колонны. Тарельчатые колонны, изготовленные из стекла, применяют для специальных целей, в том числе для разделения агрессивных веществ.
240
В 1931 г. Бурстин и Винклер [77 ] разработали установку с трубчатой колонной для непрерывной дистилляции при атмо-s' сферном давлении и под вакуумом, изготовленной из чугуна и никелевой стали (рис. 165). Установка предназначена для разде-? ления кубовых остатков перегонки сырой нефти. Она оказалась 1 пригодной также и для дистилляции легко разлагающихся и пе-нящихся жидкостей, поскольку в ней использован принцип пле-L ночной дистилляции. По конструктивному выполнению установка полностью соответствовала промышленному агрегату, поэтому Г все опытные данные, полученные при работе на ней, можно ИС-S. пользовать при разработке промышленного процесса.
i Коллинг и Трамм [78] описали регулирующее устройство для
f‘ лабораторной колонны непрерывного действия, обеспечивающее I автоматическое ведение процесса ректификации. На рис. 166 показана схема установки Коллинга и Трамма, предназначенной !для непрерывной ректификации при атмосферном давлении.
О вакуумной ректификационной установке непрерывного действия, выполненной по нормалям «Дестинорм» (см. рис. 162) сообщалось в разд. 5.2.2. На рис. 167 показана непрерывно дей-s ствующая ректификационная установка типа «Лабодест», пред-
, назначенная для работы при атмосферном давлении и под ваку-
' умом с остаточным давлением до 20 мм рт. ст. Ректификационная
колонна снабжена стеклянными колпачковыми тарелками с отра-« жательными перегородками для пара (см. разд. 7.3.3), обладаю-» щими к. п. д. от 80 до 100%. Подобные тарелки аналогичны по
Рис. 165.
Установка Бурстина—Винклера для непрерывной дистилляции с обогреваемой трубчатой колонной:
1 — напорный бак; 2 — игольчатый клапан; 3 — стеклянный шар для измерения расхода исходной смеси; 4 — штуцеры для присоединения к вакуумной линии; 5 — холодильники; 6 — карманы для термометров; 7 — металлический блок; 8 — газовые горелки; 9 — приемники дистиллята; 10 — дефлегматор; // — кожух с изоляцией из асбеста.
241
Рис. 166.
Автоматическая установка Коллиига— Трамма для непрерывной ректификации при атмосферном давлении:
1 — реле; 2 — контактный манометр; 3 — сосуд для холодной смеси; 4 — электромагнитные клапаны; 5 — электрические контакты реометра; 6 — клапан; 7, 13 — капилляры; 8 — реометр; 9 — линия сжатого воздуха; 10 — контактные термометры; 11 — термометр; 12 — холодильник полной конденсации; 14 — игольчатые клапаны; 15 — приемник дистиллята; 16 — кран для слива дистиллята; 17 — переливной патрубок; 18 — обогревающий кожух; 19 — теплоизолирующий кожух; 20 — электронагреватель.
Рис. 167.
Непрерывно работающая ректификационная установка Штаге модели «Ла-бодест» с колонной из стекла, содержащей колпачковые тарелки с отражательными перегородками для пара:
1 — насос; 2 — циркуляционный испаритель; 3 — предохранительное уст-стройство; 4 — регулятор давления с дифференциальным манометром; 5, 8 — исчерпывающая и укрепляющая части колонны с обогревающими кожухами; 6 — подогреватель исходной смеси; 7 — сосуд для исходной смеси; 9 — головка колонны; 10 — конденсатор; 11 — расширительный сосуд; 12 — буферный сосуд;. 13 — холодильник дистиллята; 14 — коллектор с кранами; 15 — охлаждаемая ловушка; 16 — шкаф управления; 17 — сменный приемник дистиллята; 18 — расходомер жидкости, стекающей из колонны; 19 — холодильник кубового продукта; 20 — промежуточный приемник кубового продукта.
Изготовитель: фирма Фишера «Лабораторная и исследовательская техника», Бонн — курорт Годесберг.
242
принципу работы промышленным тарелкам, поэтому на основе экспериментальных данных, полученных с использованием этой колонны, можно непосредственно разработать промышленную установку. Данная ректификационная установка особенно пригодна для демонстрационных целей, поскольку она позволяет визуально наблюдать за ступенчатопротекающим процессом массообмена. Установка поставляется с колоннами высотой 600— 1300 мм, диаметром 30—120 мм и числом тарелок 5—28 при расстоянии между тарелками в колонне 35—150 мм. Следует отметить также, что циркуляционный испаритель по конструктивному выполнению аналогичен промышленному аппарату. Все основные измерительные и регулирующие приборы сосредоточены в специальном шкафу управления.
Таким образом, на примере данной ректификационной установки отчетливо видна тенденция к созданию лабораторных и пилотных установок, наиболее приближенных к промышленным. Возможность сборки установок из серийных деталей и узлов, разработанных Штаге (серия приборов «Лабодест»), предприятием Фишера «Лабораторная и исследовательская техника» (Бонн— курорт Годесберг) и различными другими фирмами, способствовали упрощению конструктивного оформления установок.
Необходимо отметить также приборы, выполненные по нормалям «Дестинорм», выпускаемые народным предприятием «Комбинат технического стекла» (Ильменау), и детали для ректификационных установок, изготовляемые Народным предприятием «Завод стекла Шотт» (Иена). Поскольку данные узлы и детали стандартизованы, при выходе из строя какого-либо элемента установки можно быстро заменить его. При дальнейшей разработке серийных деталей и аппаратуры ректификационных установок Необходимо более широко моделировать их промышленные образцы.
5.2.2.4. Примеры непрерывной лабораторной ректификации
В лабораторных ректификационных установках непрерывного действия, описанных выше, можно разделять жидкие смеси как при атмосферном давлении и под вакуумом, так и при небольших избыточных давлениях (см. разд. 5.4.5). Кривые разгонки продуктов непрерывной ректификации смеси жирных кислот С4 — С]9, представленные на рис. 168, показывают достигаемую'при этом степень разделения.
При использовании регулирующих устройств на ректификационной установке, показанной на рис. 162, можно непрерывно разделять на основные компоненты смеси фенолов. На рис. 169 приведены результаты, полученные на первой стадии разгонки, при которой выделяют о-крезол и смесь м- и п-крезолов. Для аналитического контроля было отобрано 120 проб; определяли плотность кубового продукта и температуру затвердевания дистиллята. Как видно из диаграммы, в течение 22 ч работы значения температур исходной смеси, дистиллята и кубовой жидкости, а также их физико-химических свойств изменялись незначительно. Вакуум в системе регулировали с помощью автоматического стенда с вакуумным насосом (см. разд. 8.3).
243
Выход дистиллята, °/о(масс.) Рис. 168.
Диаграммы пробной разгонки продуктов непрерывной лабораторной ректификации смеси жирных кислот С4—С8 (а) и С10 — С1а (б).
Результаты непрерывной ректификации смеси фенолов.
Давление 30 мм рт. ст.; продолжительность испытаний 22 ч;
Дистиллят: смесь фенол—о-крезол; Кубовый продукт: смесь м- и га-кре-зол —ксиленол—высококипящий остаток. /, 2, 3 — температура кубовой жидкости, исходной смеси и дистиллята соответственно; 4 — плотность кубовой жидкости; 5 — температура затвердевания дистиллята.
Таким же способом непрерывно ректифицируют сырой фенол с температурой затвердевания 37,0 °C, получая при флегмовом числе 10 и давлении 60 мм рт. ст. чистый фенол (99,2%) с температурой затвердевания 40,3 °C. Высококипящий продукт непрерывно выводят из куба, поэтому его время пребывания в кубе значительно меньше, чем при периодическом методе работы. Благодаря этому существенно уменьшается термическое разложение получаемого вещества.
Применение непрерывного метода при азеотропной и экстрактивной ректификации, расширительной и пленочной перегонке, а также молекулярной дистилляции подробно рассмотрено ниже (см. разд. 5.4.2, 5.4.3, 5.4.4 и 6.2).
5.2.2.5 Выведение ректификационных установок на стационарный режим
Работа отдельных регулирующих устройств ректификационных установок рассмотрена в разд. 5.2.2.3, здесь обсуждены дополнительно некоторые вопросы.
Для уменьшения времени установления стационарного режима работы разделяемая смесь, вводимая в куб 15 (см. рис. 162), в момент подачи ее в колонну должна иметь состав, соответствующий составу кубового продукта. Для обеспечения хорошего смачивания насадки жидкость, введенную в куб, необходимо предвари-244
тельно перегнать периодическим методом, отбирая дистиллят ожидаемого состава. Затем в тройник 10 подают исходную смесь, которая предварительно нагревается до требуемой температуры в подогревателе 3. С помощью мерных бюреток 4 и 6 устанавливают соответственно расход исходной смеси и флегмовое число. Нагрузку укрепляющей части ректификационной колонны, зависящую от скорости подачи исходной смеси, дополнительно регулируют с помощью контактного манометра 12. В соответствии с изложенным в разд. 4.7.2 установка должна работать таким образом, чтобы расходы отбираемых дистиллята и кубового продукта соответствовали расходу исходной смеси. Поэтому краны на мерных бюретках головки колонны 6 и приемника кубового продукта 8 необходимо устанавливать в таких положениях, чтобы в единицу времени через них проходили соответствующие количества жидкости.
В качестве примера приведем оптимальные параметры процесса непрерывного разделения смеси бензол—толуол, содержащей 20% (об.) бензола. При подаче 500 мл/ч исходной смеси необходимо отбирать 100 мл/ч дистиллята и 400 мл/ч кубового продукта. При флегмовом числе 2 нагрузка должна быть равна 300 мл/ч. Время выхода установки на стационарный режим составляет от 0,5 до 1 ч, что определяется колебанием температур в головке и кубе колонны (см. рис. 169) *. После того как температура подогрева исходной смеси отрегулирована, установка работает с постоянными технологическими параметрами, и необходимое обслуживание установки ограничивается лишь контролем за расходами исходной смеси и отбираемых продуктов, а также наблюдением за показаниями контрольно-измерительных приборов.
5.2.3. РАЗДЕЛЕНИЕ МЕТОДОМ ПАРЦИАЛЬНОЙ КОНДЕНСАЦИИ
Под парциальной конденсацией (дефлегмацией) понимают частичную конденсацию движущихся паров. Ее проводят в специально предусмотренной для этого части колонны—дефлегматоре. Самопроизвольно дефлегмация может происходить при утечке тепла от стенок колонны в случае неудовлетворительной теплоизоляции. Ее называют также краевой конденсацией (рис. 170а).
Метод ступенчатой конденсации паров в дефлегматоре, состоящем из системы труб, при понижающейся температуре охлаждения позволяет разделять и получать фракции компонентов с большой разницей температур кипения (рис. 1706). Примеры расчетов для подобной чистой частичной конденсации, при которой образующийся конденсат сразу же выводится из системы, представ-
* Указанный период времени выхода на стационарный режим не относится к процессам ректификации изотопов и близкокипящих веществ, для которых пусковой период значительно больше. — Прим. ред.
245
Рис. 170.
Дефлегматор (/) с колонной (2) для краевой конденсации (а) н прибор для ступенчатой парциальной конденсации (б) Приборы Штаге модели «Лабодест», тип III.
лены в работе Зигварта [81 ]. Степень разделения при частичной конденсации можно значительно увеличить, обеспечивая
противоточное движение потоков пара и жидкости [82]. При ректификации пары, выходящие из колонны, должны быть разделены в заданном соотношении, вытекающем из задачи разделения. В промышленности дефлегматор часто применяют для получения флегмы, а отбираемый дистиллят конденсируют в продукционном конденсаторе. Таким образом используют укрепляющее действие дефлегматора для концентрирования высококипящей фракции *. Метод парциальной конденсации применяется прежде всего для разделения газовых смесей в технике глубокого холода. Отто [83 ] сообщает о проведении парциальной конденсации в вертикальных трубах для разделения бинарных смесей.
Эффективность дефлегматора зависит от природы разделяемой смеси [84] и обычно не превышает одной теоретической ступени разделения. Преимущество дефлегматора заключается прежде всего в его незначительной удерживающей способности по жидкости. Ниже приведены два примера, иллюстрирующие эффективность дефлегматоров при разделении смесей этанол—вода и бен-ьол—толуол:
Этанол— Веизол— вода толуол
Концентрация низкокипящего компонента перед
дефлегматором, % (мол.).................... 67
Концентрации низкокипящего компонента после дефлегматора, % (мол.)..................... 78 80
Флегмовое число................................ 5 5
Число теоретических ступеней разделения . . 3 1
* В современных промышленных установках парциальные конденсаторы применяют редко, так как это затрудняет регулирование работы ректификационных колонн и делает последние более громоздкими из-за необходимости сооружении крупногабаритных дефлегматоров. Кроме того, укрепляющее действие дефлегматора становится относительно малым в колоннах с большим числом теоретических ступеней разделении. — Прим. ред.
246
Разделяющую способность дефлегматора можно рассчитать по формуле Фабуса:
1g О-ви + 1) =^-Г(а1б-7- + 1й4^-) (176)
где уви — внутреннее флегмовое число, равное отношению количества конденсата (моль/ч) к количеству входящего пара (моль/ч); уг — концентрации низко-кнпищего компонента в паре на входе в дефлегматор (мол. доли); — кон-центрация ннзкокипищего компонента в ларе на выходе из дефлегматора (мол. доли).
Значения уг и у2 можно определить из этого выражения с помощью специально разработанной номограммы [85]. Применительно к многокомпонентной смеси Фишер [86] предложил уравнение для расчета концентрации паров, выходящих из дефлегматора, и состава конденсата. Герман [87] попытался разрешить некоторые сложные проблемы парциальной конденсации. Исследования проводили на промышленных парциальных конденсаторах, однако полученные результаты применимы также к лабораторным и пилотным дефлегматорам. Дальнейшая разработка методов определения разделяющей способности парциальных конденсаторов проведена Трестером [88].
При аналитической и препаративной перегонке в лаборатории обычно проводят процесс с полной конденсацией паров. Метод парциальной конденсации используют только при проведении сравнительной ректификации, аналогичной промышленному процессу. В этом случае дефлегматор устанавливают в верхней части колонны (см. рис. 170а). Преимущество метода с полной конденсацией паров состоит в том, что этим методом сравнительно просто разделять конденсат в определенном соотношении, в то же время устанавливать постоянной скорость подачи флегмы с помощью дефлегматора очень затруднительно, поскольку даже незначительные колебания расхода и температуры охлаждающей воды вызывают изменение составов флегмы и паров дистиллята, а также их количеств. В промышленности скорость подачи флегмы при перегонке методом парциальной конденсации обычно не измеряют, а регулируют степень охлаждения дефлегматора по температуре в головке колонны. Количество образующейся флегмы рассчитывают приблизительно, измеряя расход и температуру охлаждающей воды на входе и выходе дефлегматора с учетом удельной теплоты испарения дистиллята. Поскольку в промышленности обычно работают с одними и теми же продуктами, такой метод вполне пригоден. Однако при разделении многокомпонентной смеси определение количества подаваемой флегмы подобным образом становится слишком неточным.
Для лабораторных исследований с целью моделирования промышленного процесса, основанного на парциальной конденсации, можно применять разработанную автором специальную головку колонны для частичной конденсации, выполненную”по нормалям «Дестинорм» (рис. 171). Поднимающиеся пары частично конденси-
247
6
Рис. 171.
Головка колонны для парциальной конденсации с капилляром для счета капель по нормалям «Дестинорм»:
1 — теплоизолирующие полуцилиндры из стекловолокна; 2 — воронка для приема дистиллята; 3 — дефлегматор; 4,5 — термометры; 6 — конденсатор; 7 — перфорированная воронка; 8 — кран; 9 — капилляр для счета капель дистиллята; 10, 12 — мерные бюретки для дистиллята и флегмы соответственно; 11 — приемная бюретка для дистиллята.
Рис. 172.
Ректификационная колоииа с тремя парциальными конденсаторами [92].
руются в дефлегматоре 3. Образовавшийся конденсат через приемную воронку 2 отводится в мерную бюретку 12 с капилляром для счета капель, позволяющую в любой момент времени точно измерять скорость подачи флегмы. Участок головки между приемной воронкой и ректификационной колонной изолируют полуцилиндрами 1 из стекловолокна. Температуру охлаждающей воды на входе и выходе дефлегматора измеряют термометрами 4 со стандартными шлифами. Пары, не сконденсированные в дефлегматоре, полностью конденсируются в конденсаторе 6. Конденсат стекает в перфорированную воронку 7 и при открытом кране 8 проходит через капилляр для счета капель 9 в мерную бюретку 10. Между мерной 10 и приемной 11 бюретками можно дополнительно установить холодильник.
Головки для частичной конденсации применяют в основном при проведении сравнительной ректификации, поскольку разделяющая способность дефлегматора сравнительно невелика, и точное регулирование флегмового числа в лаборатории сопряжено со значительными трудностями. Метод ступенчатой парциальной конденсации преимущественно используют для предварительного разделения смесей компонентов с большой разницей в температурах кипения, и прежде всего для отделения воды или другой низкокипящей фракции. В этом случае колонну заменяют системой труб со встроенными конденсаторами и обеспечивают понижение температуры охлаждения потока паров по ходу его движения (см. рис. 1706). С помощью метода парциальной конденсации можно 248
сравнительно быстро и точно разделить, например, смесь жирных кислот С4—С20 на фракции С!—С4, С4—С9 и С9—С20. В промышленности этот метод применяют для разделения низкокипя-щих углеводородов с четырьмя и менее атомами углерода.
По данным Юнге [89] правильно проведенная парциальная конденсация в колонне может повысить ее разделяющую способность. При этом имеется в виду краевая конденсация на стенках колонны, обусловленная потерями тепла, нарушающими адиабатичность работы аппарата. Аналогичные способы ступенчатого разделения описаны также Трение [90]. Однако Кун [91] на основе обширных теоретических исследований и расчетов показал целесообразность проведения частичной конденсации именно в верхней части колонны.
Вебер [92] установил, что при разделении методом парциальной конденсации можно использовать ректификационные колонны с меньшим диаметром верхней части (рис. 172). Возможность уменьшения объема верхней части колонны обусловлена возрастанием в ней концентрации низкокипящего компонента и снижением требуемого флегмового числа. При этом поперечное сечение колонны следует уменьшать в соответствии с ростом концентрации. Кроме того, необходимо устанавливать промежуточные дефлегматоры, пропускная способность которых снижается по ходу движения потока паров. Фойгт [93 ] на основе теоретических исследований показал, что разделительную способность ректификационной колонны можно существенно повысить, если отводить тепло не от определенных участков колонны, а от всей ее поверхности. Метод парциальной конденсации позволяет обогащать пары низкокипящим компонентом и, следовательно, не пригоден, например, для обогащения стабильных изотопов, являющихся в основном высококипящими компонентами. В этих случаях необходимо, наоборот, подводить тепло к стенкам исчерпывающей части ректификационной колонны, чтобы уменьшить ее пропускную способность по жидкости (см. разд. 5.1.4; [93а, б]).
5.3. КЛАССИФИКАЦИЯ МЕТОДОВ ПЕРЕГОНКИ ПО ТЕМПЕРАТУРЕ ПРОЦЕССА
Нормальной температурной областью для проведения процессов ректификации считают интервал от 20 до 250° С. Если температуры кипения разделяемых веществ лежат ниже комнатной температуры, то проводят низкотемпературную ректификацию с использованием специальных хладоагентов для конденсации паров дистиллята. Процессы перегонки, протекающие при 250—400° С, относят к высокотемпературной ректификации. Возможна также изотермическая перегонка, при которой температуру в кубе поддерживают постоянной, а изменяют рабочее давление.
249
5.3.1. НИЗКОТЕМПЕРАТУРНАЯ РЕКТИФИКАЦИЯ
Смеси низкокипящих углеводородов и газов На, Na, Оа и СО можно разделять путем перегонки как при атмосферном давлении с применением специальных хладоагентов, так и при повышенном давлении. Если разделение проводят при повышенном давлении, то стремятся повысить температуру головки колонны до такого значения, чтобы можно было использовать обычные охлаждающие средства (см. разд. 5.4.5). Из-за того, что для перегонки под давлением необходима более сложная аппаратура, чаще применяют лабораторные и пилотные установки низкотемпературной ректификации. Методика проведения низкотемпературной ректификации разработана очень подробно. Созданы полностью автоматизированные установки для проведения низкотемпературной ректификации в интервале от —190 до 20° С. В этих установках применяют как насадочные, так и полые спиральные колонны. Во многих случаях отбираемые пробы дистиллята и кубового продукта анализируют методом газовой хроматографии (см. разд. 5.1.2).
Низкотемпературную ректификацию используют для очистки газов, а также как сравнительную ректификацию, аналогичную промышленному процессу. Это относится прежде всего к очистке отходящих промышленных газов без концентрирования в них водорода и, главным образом, к очистке природного газа, например выделение гелия и азота из природного газа, что по-прежнему является трудной проблемой.
Основные исследования газовых смесей, содержащих наряду с газами жидкие низшие углеводороды, проведены Подбильня-ком [94]. Он разработал колонну для точного фракционирования с насадкой из проволочных спиралей, навитых с малым шагом (так называемая насадка «Хэли-грид», которую применяют при обычной перегонке, см. разд. 7.3.4). В обзоре способов низкотемпературной ректификации Гроссе-Етрингхауз [95] рассмотрел технику проведения этих процессов, использовав экспериментальные данные Вустрова [96 ]. В этом обзоре также указано, что пробы дистиллята и кубовой жидкости следует отбирать очень тщательно с использованием полуавтоматических и автоматических устройств для моментального и непрерывного отбора. Процесс разделения следует проводить следующим образом. Сначала с помощью жидкого азота (—195,8° С) отделяют несконденсировавшуюся часть паров и анализируют её на аппарате Орса. Конденсирующуюся часть исходной смеси необходимо освободить от СОа, На и NH3 в промывном аппарате и сконденсировать. Для ректификации применяют насадочную колонну с посеребренным высоко вакуумированным кожухом; колонна снабжена спиралью, компенсирующей температурные напряжения. Дефлегматор с конической трубой припаивают (рис. 173) или присоединяют с помощью шлифов.
Дефлегматор представляет собой сосуд с изолирующей вакуумированной рубашкой, который предназначен для загрузки 250
хладоагента. Колонна и дефлегматор заполнены насадкой из металлических спиралей (сталь V2A) размерами 2x2x0,2 мм. Установка в значительной степени автоматизирована. Она снабжена такими регулирующими устройствами, как манометр с автоматическим регулированием давления и приспособление для стабилизации температуры в холодильнике.
Шток и Гауптшайн, а также Драун [97] предложили очень удобные устройства для поддержания постоянной температуры в холодильнике головки колонны, основанные на использовании
Рис. 173.
Колонна Гроссе—Етрингха-уза для низкотемпературной ректификации с припаянным дефлегматором.
Рис. 174.
Схема весов Штока для измерения плотности газа.
Рис. 175.
Установка Коха—Гильберата для низкотемпературной ректификации:
1 — спиральная колонна; 2 — кран на шлифе; 3 — термометр; 4 — конденсатор; 5 — капилляр; 6, 10 — пробки на шлифе; 7 — регулировочный кран для отбора дистиллята; 8, 9 — градуированные приемники для газообразного и жидкого Дистиллята соответственно; 11 — электронагреватель куба; 12 — сосуд Дьюара; 13 — куб.
251
жидкого азота или кислорода. Плотность газа можно определять с помощью весов Штока (рис. 174) [98]. Приборы, измеряющие давление паров (см. разд. 4.4.1), используются для определения содержания н- и изобутана. Подробное описание техники работы с газами дал Миллер [99]. В книге «Чистые газы» [100] Мюллер и Гнаук подробно рассмотрели приборы и аппаратуру для получения и транспортировки газов, а также методы их анализа, разделения и очистки.
Принцип работы установок для низкотемпературной ректификации рассмотрен ниже на примере установки Коха и Гильберата [101 ]. Колонна (рис. 175) выполнена в виде стеклянной трубчатой спирали (подобно колонне Янцена, см. рис. 76). Она обладает низкой удерживающей способностью по жидкости (3—4 мл), поэтому для загрузки колонны достаточно 15—25 г жидкости. Колонна работает так же, как и аппараты для разделения при обычных температурах. Благодаря реализации метода полной конденсации можно устайавливать любое флегмовое число. Дистиллят можно отбирать в газообразном или жидком состоянии [103].
Термометр 3 подвешен на тонкой проволоке. Отверстие капилляра 5 закрыто пицеином. Пробка 6 снабжена штуцером для соединения с небольшой осушительной трубкой.
Перед проведением ректификации проверяют качество герметичности кожуха колонны с помощью высокочастотного течеиска-теля. При герметичности кожуха (в затемненном рабочем помещении) в его полости не должно возникать свечения, возможна зеленая флюоресценция стеклянных стенок кожуха. Если возникает свечение, проводят повторное вакуумирование. Для этого используют трехступенчатый диффузионный ртутный насос. Пары ртути вымораживают в глубокоохлаждаемом адсорбере с активным углем или силикагелем, установленном между насосом и колонной. Для смазки кранов применяют высоковакуумную смазку (см. разд. 9.4). При достижении высокого вакуума, соответствующего остаточному давлению 10~5 мм рт. ст. и ниже, кран закрывают.
Куб 13 охлаждают до требуемой температуры с помощью бани, заполненной смесью метанола с сухим льдом или другим хладо-агентом. Одновременно хладоагент загружают в конденсатор 4. Если по каким-либо причинам нежелательно поддерживать постоянную температуру конденсации с помощью криостата с охлаждающим рассолом, то в качестве хладоагентов можно применять жидкий воздух или азот. Затем в кубе 13 конденсируют высушенную и, при необходимости, освобожденную от СО2 пробу газа. После этого вместо охлаждающей бани используют сосуд Дьюара 12. При правильной установке верхний край сосуда Дьюара должен соприкасаться с держателем штатива, поддерживающим куб. Содержимое куба 13 испаряют, как обычно, с помощью электронагревателя 11. Неперегретые пары поступают в спиральную колонну 1, изолированную посеребренным высоковакууми-рованным кожухом и дополнительно стекловолокном. Преду-252
смотрена возможность измерения температуры в верхней части колонны с помощью термометра <3, помещенного в защитный карман, который исключает переохлаждение термометра стекающим конденсатом. Дистиллят отбирают через регулировочный кран 7, расположенный под конденсатором.
Для сбора жидкого дистиллята предназначен градуированный приемник 9, куда дистиллят поступает в виде капель через припаянный спиральный капилляр. Спиральный капилляр и приемник помещены в сосуд Дьюара. Вследствие конденсации паров дистиллята в спиральном капилляре, расположенном ниже регулировочного крана, возникает постоянное разряжение, которое позволяет отсасывать пары из колонны. Для сбора дистиллята в газообразном состоянии предусмотрен градуированный сборник 8 с запорной жидкостью, также устанавливаемый за регулировочным краном. Требуемое постоянное разряжение в этом слу-
Рис. 176.
Установка Штаге с насадочной колонной для препаративной низкотемпературной ректификации:
/ — колонна; 2 — приемник дистиллята; 3 — сосуд для поддержания постоянного напора.
Изготовитель: фирма Фишера «Лабораторная и исследовательская техника», Бонн—курорт Годесберг.
Рис. 177.
Установка Гроссе—Етриигхауза для низкотемпературной перегонки.
Изготовитель: фирма Нормаг, Хофхайм.
253
Рис. 178.
Автоматизированная установка Подбильияка для низкотемпературной ректификации
Изготовитель: фирма Подбильияка, Департамент обрабатывающей промышленности, Фраиклни парк, Иллинойс, США.
чае получают, изменяя высоту уровня запорной жидкости.
Если температура кипения дистиллята ненамного превышает температуру его затвердевания, то необходимо точно..регулировать температуру в конденсаторе. Штаге [102] разработал для препаративных целей установку низкотемпературной ректификации, в которой температуры куба и конденсатора регулиру
ются автоматически (рис. 176). В качестве хладоагента используется жидкий воздух. В тех случаях, когда температура хладоагента наименьшая, открывается предварительно установленный на контактном термометре магнитный клапан и в поток хладоагента подается дополнительный воздух. Дистиллят отбирается также с помощью магнитного клапана. Низкотемпературный холодильник Гроссе-Етрингхауза (рис. 177) предназначен для перегонки низкокипящих жидкостей или жидкостей с растворенными газами. Пары или газы, несконденсировавшиеся в первом холодильнике, конденсируются в низкотемпературном холодильнике приемника дистиллята, который заполнен подходящей охлаждающей смесью.
Ректификационная установка Подбильияка (рис. 178) модели «Термокон» серии 8700 работает автоматически в интервале температур от —200 до 20° С [104] (см. разд. 8.1).
Видоизмененная ректификационная установка Подбильияка модели «Рургаз» рассчитана на загрузку 3 -4 л газа при атмосфёр,-ном давлении.
Ректификация в установке продолжается примерно' 2 й; столько же времени требуется для предварительной конденсации газа. Встроенный автоматический термограф непрерывно записывает температуру верхней части колонны. ; ‘ .
На рис. 179 показана диаграмма перегонки, проведенной на этой установке [105].
254
Для низкотемпературной ректификации небольших количеств сжиженных газов пригодна колонна Симонса [106], помещаемая в сосуд Дьюара (рис. 180).
Минимальное флегмовое число для процессов низкотемпературной ректификации можно приближенно рассчитать по формуле Подбильияка [107]; эта формула получена в предположении, что концентрация кубовой жидкости составляет 50% (мол.):
( а ( 1 ( хр [ 1
VMHH-^£-а_1 J [*в(1-*в)
где хЕ — концентрация низкокипящего компонента в дистилляте, мол. доли; хв — концентрация никзокипящего компонента в кубе, мол. доли.
Для определения минимального числа теоретических ступеней разделения при низкотемпературной ректификации с бесконечным флегмовым числом служит номограмма Подбильияка, показанная на рис. 181 [107]. Номограмма составлена для концентрации кубовой жидкости 50% (мол.) с учетом относительной летучести компонентов смеси и требуемой концентрации дистиллята. Номограммой пользуются следующим образом. Сначала по давлениям паров определяют относительную летучесть а компонентов разделяемой газовой смеси (см. разд. 4.4). Затем от точки, соответствующей найденному значению а, проводят вертикальную прямую до пересечения с кривой, соответствующей определенной
Рис. 179.
Диаграмма низкотемпературной ректификации, проведенной иа установке модели < Рургаз > .
Рис. 180.
Микрбколоииа Симонса для низкотемпературной ректификации.
255
Рис. 181.
Номограмма Подбильняка для определения минимального числа теоретических ^ступеней разделения пмин при низкотемпературной ректификации с V — ОО . '
концентрации дистиллята [80-99,9% (мол.) ]. От точки пересечения проводят горизонтальную прямую до оси ординат и таким образом находят число теоретических ступеней разделения. В табл. 38 для некоторых смесей низкокипящих углеводородов приведены минимальное число теоретических ступеней разделения при и = оо и минимальное флегмовое число, которые могут служить оценкой для аналогичных процессов разделения [108].
Из данных табл. 38 видно, что разделение низкокипящих смесей является сравнительно простым процессом, который может быть осуществлен в колоннах с небольшой разделяющей способностью. Следует иметь в виду, что приведенные значения чисел теоретических ступеней разделения получены при v — оо, и при конечных флегмовых числах пмии должно соответственно измениться.
256
Таблица 38.
Минимальное число теоретических ступеней разделения и минимальное флегмовое число для процессов разделения некоторых бинарных низкокипящих смесей
Смесь а А в с D
Метан—этилен 61,2 1,32 1,82 0,03 0,164
Метан—этан 107,0 1,14 1,60 0,018 0,093
Этилен—этан 2,3 6,5 9,2 1,60 7,95
Этан—пропилен 8,1 2,52 3,57 0,27 1,4
Этап—пропан 9,3 2,38 3,36 0,23 1,2
Пропилен—пропан 1,3 20,2 28,6 6,65 33,0
Пропан—изобутан 3,3 4,6 6,5 0,89 4,5
Изобутан—«-бутан 1,5 13,0 18,4 3,99 20,0
Изобутан—изобутилен 1,2 28,8 40,8 10,0 50,0
Изобутилен—1 -бутилен 1,03 25,5 36,0 8,98 43,5
н-Бутан—изопентан 2,8 5,3 7,5 1,16 5,8
Изопентан—н-пентан 1,33 18,6 26,3 6,25 30,1
н-Пентан—н-гексан 3,0 4,9 6,95 1,04 5,25
А — необходимое число теоретических ступеней разделения смеси при Xg — 50% (мол.), Xg — 99,5% и v == оо; В — то же при Xg = 10%, Xg = 99,5% и v = оо; С — минимальное флегмовое число для разделения смеси при Xg = 50% и Xg — 100% *; D — то же при Xg ~ 10% и Xg = 100% *.
* Концентрация дистиллята 100% (мол.) указана с соответствующим округлением, поскольку абсолютно чистый дистиллят получить принципиально невозможно. — Прим, ред. ---------------------------------------------—------------
5.3.2. ВЫСОКОТЕМПЕРАТУРНАЯ
И ИЗОТЕРМИЧЕСКАЯ ПЕРЕГОНКА
При температурах выше 250° С органические вещества обычно разлагаются, поэтому процессы перегонки в лаборатории редко проводят в интервале температур от 250 до 400° С. Вместе с тем ( иногда бывает необходимо определить интервал температур кипе-( ния при атмосферном давлении, например у смол, высокомолеку-; лярных парафинов, воска. Горный воск часто подвергают деструк-тивной перегонке, чтобы одновременно посредством крекинга !; выделить низкомолекулярную часть [109]. В других случаях удается избежать разложения и полимеризации с помощью ин-! гибиторов [ПО].
( Для высокотемпературной перегонки высокомолекулярных веществ при атмосферном давлении или вакууме применяют только невысокие колонны, насадка которых служит в основном для предотвращения брызгоуноса. Насадку (обычно из крупных элементов) размещают на опорной решетке из проволочной сетки. Тарельчатые колонны для высокотемпературной перегонки непригодны. Для предотвращения частичной конденсации колонну
9 э. Крель 257
Таблица 39.
Нагревательные бани для высокотемпературной ректификации
Теплоноситель Максимально достижимая температура, °C
Глицерин — 160
Серная кислота —250
Полнгликоли —300
Твердые парафины с температурами плавления 30— 60 °C —300
Минеральное масло —330
Солевая баня, состоящая из 40% (масс.) NaNO2, 53% KNO3 н 7% NaNOg 150—500
снабжают сплошным обогревающим кожухом, простирающимся от куба до конденсатора. Приемник дистиллята также необходимо снабдить обогревающим кожухом или электронагревательной обмоткой. Целесообразно нагревать кран отбора дистиллята инфракрасным или обычным светом во избежание заедания его сердечника. Преимущественно применяют магнитные клапаны.
Для обогрева кубов при высокотемпературной ректификации в лаборатории обычно применяют масляные и солевые бани [111], температуру которых регулируют с помощью контактных термометров. В табл. 39 приведены значения температур, достигаемые в таких банях (см. разд. 7.7).
Большое преимущество солевых бань состоит в том, что они работают без выделения отходящих газов в широком интервале температур. Недостатком этих бань является трудность стабилизации температуры и необходимость их охлаждения. По окончании перегонки необходимо сразу же вынуть куб из бани и смыть водой оставшуюся на нем соль.
При охлаждении дистиллята в условиях высокотемпературной перегонки иногда требуется следить за тем, чтобы не затвердевал дистиллят (сублимированная его часть); в противном случае возможно уменьшение поперечного сечения холодильника вплоть до его закупоривания. Для предотвращения затвердевания дистиллята требуемые температуры конденсации можно устанавливать и регулировать посредством циркуляционных термостатов с водой или гликолем в качестве термостатирующей жидкости. Другой простой метод установки и регулирования температуры конденсации основан на использовании холодильника с кипящим хладо-агентом. Этот холодильник состоит из двух концентричных труб. По внутренней трубе пропускают охлаждающую жидкость, температура кипения которой несколько выше температуры плавления дистиллята, но ниже его температуры кипения. Пары дистиллята, проходящие по кольцевому каналу между трубами, нагревают ох-
258
вакуум-насосу
Шкаф управления
' К форвакуум-насос у
Рис. 182.
Дистилляционная установка Штаге для йерегоики веществ с высокой температурой плавления.
Установка 'снабжена холодильником дистиллята с кипящим хладо-агеитом. Изготовитель: фирма Фишера «Лабораторная н исследовательская техника*, Бонн — курорт Годесберг.
Рис. 183.
Устройства для перегонки металлов с боковым отводом паров (а) и с нижним отводом паров (<>): 1 — печь; 2 — сосуд с расплавом; 3 — приемник; 4 — конденсатор; 5 — конденсат.
лаждающую жидкость и испаряют ее. При этом дистиллят конденсируется на наружной стенке внутренней трубы.
На рис. 182 показана дистилляционная установка Штаге [112], снабженная холодильником дистиллята с кипящим хладоагентом. В работе [112] рассмотрены также другие установки для перегонки сублимирующихся веществ с высокой температурой плавления и подобных веществ в смеси с растворителями.
В последнее время перегонку стали широко применять для очистки металлов, особенно в тех случаях, когда низкокипящую часть сплава можно отделить в виде паров от высококипящей [113]. Это условие соблюдается при перегонке бинарных сплавов А1—Zn, Al—Mg, Pb—Zn, Ag—Zn и Ag—Pb. Значения давлений паров различных металлов можно найти в справочнике Лейбольда [114]. Для реализации процесса разделения металлов в лаборатории, необходимо сначала освоить соответствующую аппаратуру для получения небольших количеств веществ. На рис. 183 показаны два лабораторных устройства для перегонки металлов с боковым и нижним отводами потока паров [115].
Подробный обзор о лабораторной перегонке под вакуумом металлов и сплавов, не содержащих железа, приведен в работе Шпендлеве [116]. Хорслей [117] описал аппаратуру для разгонки щелочных металлов. В соответствии с этими работами металл расплавляют в вакууме, фильтруют и затем перегоняют преимущественно при давлении до 10~4 мм рт. ст. Пары металла конденсируют в конденсаторе, охлаждаемом циркулирующим маслом. Для получения чистого тантала Паркер и Вильсон [118] использовали хлорид тантала ТаС15 (температура кипения 240° С при 760 мм рт. ст.). Безобразов с сотр. [118а] разработал кварцевый аппарат диаметром 40 мм и высотой разделяющей части 1250 мм для аналитической перегонки высококипящих веществ с температурой кипения до 1000° С (сера, селен, теллур, цинк, кадмий, сульфид мышьяка и Др.).
Одним из наиболее часто применяемых в лаборатории процессов перегонки является дистилляция ртути с целью отделения олова, кадмия и благородных металлов. Обычно перед дистилляцией проводят предварительную химическую очистку, при этом в ртути могут оставаться вещества, используемые при очистке.
Прибор непрерывного действия, выполненный из кварца или тугоплавкого стекла, работает на принципе объемного насоса;
260
после предварительного вакуумирования с помощью водоструйного насоса прибор в ходе эксплуатации самостоятельно вакуумируется [119]. Принцип работы подобного прибора наглядно показан на рис. 184. Для работы прибора необходимо, чтобы сливная труба 7 была выполнена в виде капилляра внутренним диаметром 2,0 мм. Перед началом дистилляции целесообразно вакуумировать прибор с помощью ртутного диффузионного насоса и после одно- двухчасовой перегонки закрывать клапан 4. При необходимости повторного вакуумирования клапан снова открывают.
Для получения больших количеств очищенной ртути можно использовать аппаратуру для двукратной дистилляции ртути (рис. 185). После испарения ртути на первой ступени образуется конденсат, поступающий по трубчатой перемычке на вторую ступень, где он снова испаряется. Полученный дистиллят через запорную трубу, работающую на барометрическом принципе, стекает в приемник. С использованием электронагревателей мощностью 300 Вт при остаточном давлении до 1 мм рт. ст., можно получать на каждой ступени около 2 кг/ч дистиллята ртути. По данным спектрального анализа перегнанная ртуть имеет очень высокую степень чистоты. Прибор снабжен сферическими шли-
Рис. 184.
Автоматический прибор Робесона для дистилляции ртути:
/ — сосуд со ртутью; 2 — испаритель; 3, 10 — трубы с внутренним диаметром 20 и 6 мм соответственно; 4 — ртутный поплавковый клапан со штуцером для ртутного диффузионного вакуумного насоса; 5,9— капилляры с внутренним диаметром 2 и 4 мм соответственно; 6 — приемник с переливным устройством; 7 — сливная труба; 8 — промежуточный сосуд; 11 — блок из алюминия; 12 — электронагревательная обмотка. *
Рис. 185.
Аппарат для двукратной дистилляции ртути.
Изготовитель: народное предприятие «Йенский завод стекла Шотт», Иена.
261
фами с прецизионными соединительными поверхностями, не требующими применения вакуумной смазки.
Если перегонку проводят при постоянном давлении, то количество отбираемого дистиллята регулируют, меняя температуру. При изотермической перегонке, наоборот, температуру в кубе стабилизируют с помощью термостата, а давление непрерывно снижают. В этом случае на диаграмме разгонки давление (ось ординат) и количество отбираемого дистиллята (ось абсцисс) указаны при постоянной температуре. Изотермическую перегонку применяют, когда необходимо подобрать степень разрежения, требуемую для испарения определенного количества многокомпонентной смеси, если, например, для обогрева имеется в распоряжении пар определенной температуры. Эхолс и Гелус [120] использовали изотермическую перегонку для того, чтобы определить количества кубовых остатков исходной смеси, которые соответствуют определенному давлению при постоянной температуре. Возможно также построение изотермической кривой равновесия *. Теоретические основы изотермической и изобарической дистилляции обсуждаются Улусоем и Закалоцем [121]. Согласно опытным данным, полученным ими, изотермическая дистилляция пригодна и в других случаях.
5.4. КЛАССИФИКАЦИЯ МЕТОДОВ ПЕРЕГОНКИ ПО РАБОЧЕМУ ДАВЛЕНИЮ
Перед проведением перегонки следует решить, при каком давлении наиболее целесообразно проводить разделение. В тех случаях, когда это возможно, следует работать при атмосферном давлении, поскольку пропускная способность аппаратов, работающих в условиях вакуума, сильно снижается. Кроме того, перегонка под вакуумом или под давлением требует применения дополнительных устройств.
Газовые смеси, содержащие низкокипящие углеводороды, разделяют методом низкотемпературной ректификации при атмосферном давлении или ректификацией под давлением-, а легко разлагающиеся и высококипящие органические вещества перегоняют под вакуумом при остаточном давлении 760—1 мм рт.ст. Высокая производительность ректификационных установок может быть достигнута при использовании расширительной перегонки под вакуумом, соответствующем остаточному давлению 20—1 мм рт.ст. Термически нестойкие вещества нельзя перегонять непосредственно из куба; их разделяют в мягких условиях методом пленочной перегонки при остаточных давлениях 20—10’1 мм рт.ст. Вещества с низкими давлениями паров и большой молекулярной массой в пределах 250—1200 разделяют методом молекулярной
* Изотермические данные по равновесию пар—жидкость более удобны для термодинамического анализа. — Прим. ред.
262
дистилляции под вакуумом соответствующем 10"3—10~6 мм рт. ст., поскольку при этих остаточных давлениях средняя длина свободного пробега молекул соизмерима с размерами аппарата. Техника дистилляции и ректификации термически нестойких веществ под вакуумом подробно рассмотрена Франком и Куче в книге «Перегонка в мягких условиях» [122].
5.4.1. ВАКУУМНАЯ ДИСТИЛЛЯЦИЯ И РЕКТИФИКАЦИЯ
Под вакуумной перегонкой понимают процесс дистилляции и ректификации, который проводят при давлении ниже давления окружающей среды. Обычно остаточное давление в колоннах составляет примерно 0,5 мм рт. ст. Для перегонки при более низких давлениях используют специальную аппаратуру.
Основное преимущество вакуумной перегонки по сравнению с перегонкой при атмосферном давлении заключается в снижении точки кипения, что позволяет проводить процесс в условиях ниже температур разложения разделяемых веществ. Вакуумную перегонку применяют прежде всего для разделения термически нестойких и, особенно, полимеризующихся веществ. В качестве примеров можно назвать промышленную дистилляцию минеральных масел и ректификацию сырой смеси фенолов, которые проводят при 20—60 мм рт. ст., а также перегонку синтетических жирных кислот при давлении 1—20 мм рт.ст. Вакуумную перегонку применяют и в тех случаях, когда нет опасности разложения веществ, однако температуры их кипения при атмосферном давлении настолько высоки, что по теплотехническим соображениям ректификация при пониженном давлении становится предпочтительной. Это имеет место, например, при работе с различными эфирными маслами. Преимущество вакуумной перегонки состоит не только в снижении температуры кипения, но и в увеличении относительной летучести компонентов разделяемой при разрежении смеси.
Если разделяемая смесь образует при атмосферном давлении азеотроп, то с понижением давления азеотропная смесь обогащается низкокипящим компонентом и при некотором остаточном давлении азеотроп может исчезнуть *. Например, смесь этанол— вода при 70 мм рт.ст. азеотропа не образует (см. разд. 6.2.1). Следовательно, вакуумной ректификацией при давлении ниже 70 мм рт. ст. можно получать абсолютно чистый спирт без примесей **. Следует обратить внимание на то, что при таком разрежении температура кипения спирта сравнительно низка (примерно
* Смещение составов азеотропов с изменением давления зависит от соотношения скрытых теплот испарения компонентов. Следует также иметь в виду, что снижением давления не всегда удается разрушить азеотроп, поскольку при этом может раньше произойти кристаллизация разделяемой смеси. — Прим. ред.
** Как уже указывалось в примечаниях выше, путем ректификации получить абсолютно чистый дистиллят невозможно. — Прим. ред.
263
Рис. 186.
Влияние степени предварительного смачивания насадки на разделяющую способность ректификационной колонны по данным Гельбе [124], полученным прн 20 мм рт. ст.
28° С), поэтому при выборе вакуума следует учитывать имеющиеся в распоряжении хладо-агенты для конденсации паров.
Чаще всего в промышленности используется греющий пар
низких параметров, поэтому достаточную разность температур между температурой греющего пара и температурой кубовой жидкости можно обеспечить только при пониженном рабочем давлении. Следует учитывать также и возможность коррозии. Часто повышение температуры выше определенного предела является нежелательным из-за опасности коррозии куба и нижней части колонны. Следовательно, ряд факторов приводит к необходимости применения вакуумной перегонки. Экономические и технические соображения с учетом перечисленных выше факторов позволяют выбрать оптимальный вакуум. Биллет и Райхле [123 ] описали метод расчета рабочего давления ректификации, обеспечивающий минимальный перепад давления потока паров при вакуумной перегонке. В разд. 4.6.2 и 4.10.6 уже обсуждались различные точки зрения о влиянии вакуума на разделительную способность ректификационных колонн. Гельбе [124] указал, что выводы многих работ очень противоречивы. На основе экспериментов, проведенных с использованием колонны диаметром 45,7 и высотой разделяющей части 500 мм с насадкой из пружинных спиралей размером 4 мм, он установил, что при постоянной массовой производительности разделяющая способность колонны не зависит от давления в интервале от 10 до 100 мм рт. ст. При повышении абсолютного давления до 740 мм рт. ст. число единиц переноса постепенно возрастает вследствие увеличения относительной скорости потока паров. Так, при разделении смесей н-декан — транс-декалин число единиц переноса увеличивается примерно на 15% (см. рис. 97). Гельбе предположил, что противоречивость опубликованных экспериментальных данных обусловлена различной степенью эмульгирования газо-жидкостного слоя, имевшую место в опытах разных исследователей. В разд. 4.10.8 отмечался этот факт, там же обсуждались методы работы в режиме эмульгирования.
Гельбе проводил ректификацию при рабочих давлениях ниже 100 мм рт. ст., причем процесс сначала протекал при более высоком давлении, необходимом для увеличения количества флегмы. Это приводило к тому, что в начальный период ректификации барботажный слой, образующийся в нижней части колонны, постепенно поднимался по слою насадки до головки колонны. Слой 264
жидкости, образовавшийся при этом в головке, многократно проходил через насадку. После снижения давления до рабочего колонна продолжала работать в режиме эмульгирования. Степень смачивания насадки, достигнутая таким образом, отчетливо иллюстрирует рис. 186; верхняя прямая на рисунке соответствует оптимальному режиму эмульгирования.
Методика расчета размеров ректификационных колонн, в том числе работающих под вакуумом, описана в разд. 4.11. Следует отметить, что очень важно правильно определять размеры вакуумной коммуникации. Потери давления в трубопроводе диаметром менее 200 мм при умеренном вакууме рассчитывают по формуле Пуазейля [113]:
Др = 8VriZ/(r4n:i;) (178)
где Др — потеря давления; V — объем потока газа; Ц —динамическая вязкость газа; I—длина трубопровода; г—радиус трубопровода; т—время.
Харрис [125] разработал удобную номограмму (рис. 187), с помощью которой можно определить размеры вакуумных линий и производительность насоса. При разработке номограммы предполагалось, что скорость вакуумирования изменяется не более чем на 30 %.
Рис. 187.
Номограмма Харриса для определения размеров вакуумных коммуникаций.
265
Правила пользования номограммой поясняются на примерах 1 и 2.
Пример 1 Пример 2 р — давление в вакуумированном сосуде, мм рт. ст.................................. 1,0 1,0
d — диаметр трубопровода, см .... 1,0 3,0
S — производительность вакуумного насоса, м3/ч................................. 2,0 50,0
I — длина трубопровода, см .............. 350,0 450,0
В примере 2 точка пересечения прямой р — S со шкалой А лежит выше точки А 2 иа 6,5 делений шкалы, поэтому коэффициент полезного действия вакуумного насоса превышает 60%.
а) Определение м-аксимальной производительности вакуумного насоса (см. пример 1). Соединяют прямой линией точки на шкалах d и I, соответствующие значениям диаметра и длины трубопровода, и получают в точке пересечения этой прямой со шкалой А значение гидравлического сопротивления вакуумного трубопровода Аг. Затем проводят прямую через точки А х и р (определяемой на шкале р по значению давления в вакуумированном сосуде), продолжают ее до пересечения' со шкалой S и получают в этой точке пересечения максимальную производительность вакуумного насоса, б) Определение минимально допустимого диаметра трубопровода (см. пример 1).
Соединяют прямой линией точки на шкалах р и S, соответствующие выбранным значениям р и S, и получают в точке пересечения этой прямой со шкалой А величину Аг. Затем проводят прямую через точки и I (определяемой на шкале I по значению длины трубопровода), продолжают ее до пересечения со шкалой d. Точка пересечения показывает значение минимально допустимого диаметра трубопровода.
в) Определение максимальной длины трубопровода (см. пример 1).
Соединяют прямой линией точки на шкалах р и S, соответствующие выбран- ным значениям р и S, и получают в точке пересечения этой прямой со шкалой А величину А^. Затем проводят прямую через точку At и d (определяемой на шкале d по значению диаметра трубопровода), продолжают ее до пересечения со шкалой /и получают в этой точке пересечения значение максимальной длины трубопровода.
Точка пересечения А, обеих прямых для примера 1 соответствует значению коэффициента полезного действия вакуумного насоса (41%), определяемого по формуле:
N = /1 + (Ро/р)2 — Ро/Р (179)
где р0 — нижний предел давления, который можно обеспечить в разрабатываемой вакуумной установке с допустимым коэффициентом полезного действия.
Для проверки правильности выбора вакуумной коммуникации в уже готовой ректификационной установке (см. пример 2) необходимо соединить прямыми линиями точки на шкалах р и S, а также d и I. Точка пересечения прямой d — I со шкалой А дает значение Л2. Если точка пересечения прямой р—S со' шкалой А лежит выше точки Л2, то размеры вакуумной коммуникации достаточны.
Более точный метод расчета параметров вакуумных коммуникаций приведен в оригинальной работе [125]. Для лабораторных и пилотных ректификационных установок пригодны стеклянные трубопроводы диаметром 20—30 мм, собираемые из участков труб длиной 1—2 мм, которые соединяют короткими отрезками вакуумных резиновых шлангов.
Установки для вакуумной дистилляции и ректификации по размерам отличаются от соответствующих установок, работающих при атмосферном давлении (см. разд. 4.11). Необходимы также 266
дополнительные элементы, такие как устройство для присоединения к вакуумной линии, которое обычно совмещают с охлаждаемой ловушкой (рис. 188), и соответствующий вакуумный приемник дистиллята (см. разд. 7.6). Наряду с насадочными колоннами для вакуумной перегонки пригодны полые колонны (см. разд. 7.3.1), а также колонны с неподвижными (см. разд. 7.3.4) и вращающимися (см. разд. 7.3.5) контактными устройствами. Наиболее подходящее отношение диаметров наружной и внутренней труб в охлаждаемых ловушках составляет 1,6. Рекомендуется измерять остаточное давление непосредственно за устройством для присоединения к вакуумной линии, чтобы между точкой измерения давления и точкой измерения температуры не было заметной потери давления. С целью контроля в каждом отдельном случае следует измерять перепад давления между точками измерения давления и температуры (см. разд. 8.3).
Для вакуумных работ предпочтительны стандартные шлифы ряда 1. В тех случаях, когда смазка шлифов легко растворяется, а также для работ при среднем и высоком разрежении применяют высоковакуумные шлифы ряда 0 (см. разд. 3.1). Поскольку обычные краны при работах под вакуумом часто разуплотняются, созданы специальные краны для вакуумных работ. На рис. 189 показан вакуумный кран с закрытой снизу муфтой, а на рис. 190— тот же кран с дополнительным ртутным затвором. Другие специальные краны и клапаны для вакуумных работ описаны в разд. 7.2.1.
Для создания вакуума применяют водоструйные, диффузионные и водокольцевые насосы, а также электрические насосы различных типов. Для получения высокого вакуума необходимы ртутные и масляные диффузионные насосы. Более подробные данные о вакуумных насосах можно найти в соответствующей специальной литературе по вакуумной технике [ИЗ, 114, 119, 122, 126, 127] *.
* По вопросам выбора отечественного высоковакуумного оборудования см. Савинский К., Зав. лаб., № 9, 1111—1127/1955/. — Прим. ред.
Устройства для присоединения вакуумной линии с припаянными охлаждаемыми ловушками к аппаратам без регулирования потерь давления (а) и с регулированием потерь давления (б).
Рис. 189.
Вакуумный кран с закрытой снизу муфтой.
Рис. 190. Вакуумный кран с ртутным затвором.
267
Рис. 191.
Экранированный высокочастотный течеискатель с электродом в виде щетки.
При выборе вакуумного насоса можно 1| также руководствоваться справочником
J Лейбольда по вакуумной технике [114].
В обзоре Арденне [128] рассмотрены области применения и характеристики важнейших типов насосов. После сборки вакуумной установки необходимо испытать ее на герметичность. Сначала проверяют вакуум, создаваемый насосом, путем присоединения его к буферной ёмкости на 5—10 л. Затем проверяют герметичность кранов, шлифовых соединений и мест спаев. Целесообразно размещать краны или клапаны на установке таким образом, чтобы можно было отдельно испытать на герметичность различные ее части. Для проверки герметичности применяют высокочастотный течеискатель типа «Тесла» с электродом в виде щетки (рис. 191). Принцип работы прибора основан на возникновении искры от электрода в месте пропускания воздуха. Можно также проверить герметичность аппарата с помощью стетоскопа или смазать предполагаемые места пропусков мыльным раствором и создать в установке избыточное давление около 0,5 кгс/см2. Изящный метод проверки герметичности состоит в том, что на поверхность вакуумированной установки наносят кисточкой слабощелочной раствор флоуресцина или эозина в метаноле, затем ее облучают в темноте ультрафиолетовым светом, при этом в герметичных местах будет отчетливо наблюдаться флуоресценция. Специальные методы испытаний установок, работающих в условиях высокого вакуума, описаны Лаппорте [119] и Мён-хом [126].
Промышленность освоила выпуск течеискателей модели «Лек-зухэр», предназначенных для проверки герметичности установок, работающих в условиях высокого вакуума и избыточного давления.
Подобный галогенный течеискатель использует в качестве хладоагента фреон 12 (CF2C12). В основу работы прибора положено известное явление, заключающееся в том, что отожженная платина при контакте с галогенами испускает большое число ионов. При проверке герметичности вакуумной аппаратуры трубу тече-искателя присоединяют к местам предполагаемых пропусков. При наличии дефектов уплотнения в испытуемую установку засасывается эталонный газ из течеискателя, что регистрируется указывающим прибором или появляется акустический сигнал. При испытании установок, находящихся под избыточным давлением, газ через неплотности поступает из приборов в трубу течеискателя. Наименьшая течь газов, регистрируемая данным тече-искателем, составляет 10 “ л-мм рт. ст./с.
268
Другие виды течеискателей, использующие в качестве эталонного газа водород или светильный газ, обладают такой же чувствительностью. Для проверки герметичности применяют также прибор, использующий гелий, который подобен масс-спектрометру. Этот прибор может измерять утечки газов до 10“10 л - мм рт. ст./с [129 ].
Если термические нагрузки небольшие, то негерметичные места можно уплотнить пицеином или замазкой. Однако лучше заменить дефектную деталь или запаять негерметичное место.
Герметичность установки Dt характеризуют уменьшением давления Ар в единицу времени, умноженным на суммарный объем ректификационной установки:
D[ = V p/t (л-мм рт. ст./с) (180)
Значения D{ при удовлетворительной герметичности должны лежать в интервале от 10 3 до 10 6 л-мм рт. ст./с.
5.4.2. РАСШИРИТЕЛЬНАЯ ПЕРЕГОНКА
Расширительная перегонка относится к непрерывным промышленным методам перегонки. Несмотря на большие преимущества, этот метод еще не нашел широкого применения в лабораторной и промышленной практике.
Стеклянная установка автора [130], разработанная для реализации подобного процесса, показана на рис. 192. Основной частью этой установки является хорошо теплоизолированная расширительная колба 8 объемом 250—500 мл с термометром на стандартном шлифе. Температуру обогрева колбы регулируют с помощью контактного термометра. К центру колбы подведен сменный патрубок 2, служащий для впрыскивания перегоняемой жидкости. За операцией впрыскивания можно постоянно наблюдать через небольшое смотровое окно. Расширительная колба освещается небольшой лампой накаливания. Исходную смесь из бутыли 1 передавливают в напорный бак 4, работающий по принципу сосуда Мариотта. В баке 4 с помощью термостатирую-щего кожуха поддерживают определенную температуру. Дальнейший подогрев происходит в теплообменнике 3, в который подают термостатированную жидкость или пар. Для регулирования температуры теплоносителя предназначен контактный термометр 6. Мерной бюреткой5 контролируют, а краном 7 регулируют расход исходной смеси, поступающей в колбу 8.
Подогретую исходную смесь непрерывно с определенной скоростью впрыскивают в вакуумированную колбу 8. При этом жидкость распределяется по стенкам колбы в виде тонкой пленки. В результате дополнительного разрежения при впрыскивании низкокипящая фракция сразу же испаряется. Пленка, образованная на нагретых стенках колбы, имеет большую поверхность испарения, поэтому в течение небольшого периода времени испа-
269
— Рис. 192.
Установка Креля для расширительной перегонки:
1, 13, 17 —бутыли для исходной смеси, дистиллята и кубового продукта соответственно; 2 — патрубок для впрыскивания исходной смеси; 3 — подогреватель исходной смеси; 4 — напорный сосур, Мариотта; 5, 16 — мерные бюретки для исходной смеси и кубового продукта соответственно; 6, 14 — контактные термо-
метры; 7 — регулировочный край для подачи исходной смеси; 8 — расширительная колба; 9 — трубчатая перемычка;
10 — конденсатор; И — штуцер для присоединения вакуумной линии; 12 — приемник Аншюца — Тиле &ля дистиллята; 15 — колонна.
Изготовитель: народное предприятие «Комбинат технического стекла», Ильменау.
Рис. 193.
Диаграмма .пробной расширительной разгонки головной (?) и кубовой (2) фракций сырой смеси жирных кислот.
Рабочее давление перегонки для головного продукта 15 мм рт. ст., для кубового продукта 6 мм рт. ст.
Выход дистиллята , % (масс.)
ряется и оставшаяся часть низкокипящей фракции. Пары дистиллята проходят по изолированной трубчатой перемычке 9 с термометром к конденсатору 10, где конденсируются. Конденсат через' приемник Аншюца—Тиле 12 поступает в бутыль 13. Вакуумную линию присоединяют к штуцеру 11. Вакуум стабилизируют с помощью регулятора давления (см. разд. 8.3). Высококипящая фракция стекает из расширительной колбы вниз и с помощью перфорированной воронки распределяется по поперечному сечению короткой колонны 15, снабженной обогревающим кожухом. Если высококипящая фракция содержит низокипящие компоненты, то их отделяют в неадиабатически работающей колонне, к стенкам которой дополнительно подводится тепло. Температуру обогревающего кожуха колонны 15 регулируют с помощью контактного термометра 14. Кубовый продукт непрерывно отводят в бутыль 17 через мерную бюретку 16.
270
260
2<Ю
КО
120
80
Концентрация} °/о(о6.)
800—1500 г/ч.
Рис. 194.
Диаграмма пробной расширительной разгонки головной и кубовой фракций фенольного масла.
/—5 _ кривые трех разгонок дистиллята; j 4 — 6 — то же кубового продукта.
Непрерывная расширительная перегонка особенно пригодна для отделения низкокипящей фракции при давлении 1 — 20 мм рт. ст. Производительность колонны состав-получении низкокипящей
ляет фракции кислот С4—С10 из сырой смеси жирных кислот методом расширительной перегонки под вакуумом, соответствующем 15 мм рт. ст., дистиллят отбирают со скоростью 1200 г/ч.
На рис. 193 показаны результаты анализа дистиллята и кубовой жидкости, полученные при пробной перегонке. Граница разделения соответствует компоненту С10- Предварительное фракционирование дистиллята фенольного масла также осуществляли путем расширительной перегонки. При атмосферном давлении смесь разделяется в интервале температур 210—230 °C. Полученные результаты представлены на рис. 194. Кривые, соответствующие трем пробным перегонкам дистиллята и кубовой жидкости при атмосферном давлении, накладываются друг на друга (точность около 15 °C). Особенно высокой производительности до 3 л/ч можно достигнуть при большой разности температур кипения компонентов смеси, например при перегонке смеси глицерина с растворителем, кипящим при 150 °C [130].
Смесь фенолов, содержащую 9% воды, обезвоживали путем расширительной перегонки, проводимой на аналогичной установке с расширительной колбой, не имеющей колонны. При абсолютном давлении 23 мм рт. ст. и температуре исходной смеси 80 °C оптимальная производительность установки составляла 4 л/ч. Такую высокую производительность вряд ли могут иметь обычные лабораторные установки периодического действия. Серьезным препятствием для использования метода расширительной перегонки является возможное вспенивание смесей.
Данный метод наиболее пригоден для препаративного разделения многокомпонентных смесей на отдельные фракции. Этим методом можно быстро и в мягких условиях фракционировать смеси объемом до 100 л с последующей аналитической ректификацией отдельных фракций на установках периодического действия.
271
5.4.3. ПЛЕНОЧНАЯ ПЕРЕГОНКА
(ПЛЕНОЧНОЕ ИСПАРЕНИЕ)
Пленочная перегонка осуществляется из пленки жидкости в мягких температурных условиях. Этот метод соответствует одноступенчатой дистилляции, для которой разделяющее действие установки максимально приближается к одной теоретической ступени разделения. Степень разделения можно повысить путем последовательного соединения большого числа пленочных аппаратов. Опыты автора показали, что как при атмосферном давлении, так и при остаточных давлениях 5—0,1 мм рт. ст. пленочная дистилляция имеет существенные преимущества [130], заключающиеся в малом времени пребывания жидкой фазы в нагретой зоне и в отсутствии необходимости нагревания больших слоев жидкости, как это имеет место при перегонке из куба.
Поясним данные преимущества на примере. Предположим, что требуется разделить при абсолютном давлении 1 мм рт. ст. смесь насыщенных жирных спиртов С1о—С18 нормального ряда. Если в качестве куба использовать круглодонную колбу объемом 1 л, то высота слоя жидкости составит 54 мм. При этом в колбе получается следующее соотношение давлений (рис. 195п): на поверхности жидкости давление равно 1 мм рт. ст.; вследствие гидростатического давления слоя жидкости (54 : 13,5 = 4 мм рт. ст.) давление на дне куба повышается до 5 мм рт. ст. В начале перегонки фракция С1о отгоняется при давлении 1 мм рт. ст. и температуре 76 °C, при этом у дна куба температура составляет 142 °C.
При пленочной же перегонке очень тонкая пленка жидкости, находящаяся в нагретой зоне очень малое время, практически не вызывает увеличения давления. Поэтому для получения первой фракции достаточна температура пленконесущей поверхности, равная 78 °C (рис. 1956). Мягкие температурные условия пленочной перегонки наиболее пригодны для переработки термически нестойких веществ. Пленочную перегонку целесообразно применять для предварительного разделения исходной смеси в узком температурном интервале.
Если высококипящая смесь содержит незначительное количество низкокипящих компонентов, то даже при высокой температуре часто бывает невозможно целиком выделить низкокипящую фракцию перегонкой из куба. При перегонке под вакуумом (для предотвращения разложения высококипящей фракции) возникают трудности при конденсации паров низкокипящих компонентов. Этих затруднений можно избежать, если проводить пленочную перегонку при атмосферном давлении.
Различают следующие области применения непрерывной пленочной перегонки (см. также [122] и гл. 5 и 6): перегонка термически нестойких веществ в мягких температурных условиях; перегонка смеси высококипящих веществ, которую нельзя разделить обычной перегонкой из куба; разделение смесей с неболь-272
шим содержанием низкокипящей фракции и высоким содержанием высококипящей фракции; дегазация жидкостей; выпаривание жидкостей; перегонка легко вспенивающихся веществ.
При пленочной перегонке, или точнее, при пленочном испарении, применяют испарители следующих типов.
1) Испарители с падающей (гравитационно стекающей) пленкой. В состав этих испарителей входят неподвижные обогреваемые вертикальные трубы или трубчатые змеевики, по наружной поверхности которых стекает пленка жидкости (см. рис. 196, 198, 199, 212), вращающиеся контактные устройства для обеспечения циркуляции пленки жидкости, выполненные в форме щеток (см. рис. 201), стеклянных спиралей (см. рис. 210) или скребковых роторов со щетками, лопастями или роликами (см. рис. 201, 202, 211). 2) Проточные испарители, расположенные горизонтально или наклонно. Эти испарители применяют обычно для молекулярной дистилляции (см. рис. 205, 209). 3) Испарители с диспергированием жидкости. Эти испарители применяют для расширительной перегонки (см. рис. 192). 4) Роторные испарители, имеющие вращающийся куб (см. рис. 203), барабан для перемешивания пленки жидкости (см. рис. 200) и испарительные диски, обеспечивающие распределение жидкости под действием центробежных сил (см. рис. 213).
Геммекер и Штаге [132] дали подробный обзор пленочных испарителей и испарителей мгновенного действия с разбрызгиванием
Рис. 195.
Соотношение давлений в обычном перегонном кубе (а) н в пленочном аппарате (б).
Рис. 196.
Пленочный дистиллятор Креля:
/ — электронагреватель внутренней трубы;
2 — обогреваемая внутренняя труба; 3 — устройство для точной установки расхода исходной смеси с поплавком 6 4 — напорный <5ак; 5 — фотоэлемент; 7 — стеклянная воронка для подачи исходной смеси; 8 — передвижное металлическое кольцо; 9 — бутыль для „ исходной смеси.
/ Изготовитель и разработчик: народное предприятие «Комбинат технического стекла», Ильменау.
273
жидкости, а также пленочных ректификационных колонн. Сведения о пленочных аппаратах можно найти также в монографии [122].
На рис. 196 показан испаритель с падающей пленкой, в котором жидкость стекает по неподвижной вертикальной трубе. В аппарате имеются две концентричные трубы. Внутренняя сменная труба с нагревателем является поверхностью испарения, а наружная труба служит поверхностью конденсации.
Утцингер [133] указал на трудность обеспечения равномерного распределения пленки жидкости (см. разд. 4.2). Крель предложил для уменьшения образования струй на обогреваемом цилиндре исходную смесь подавать в углубление, находящееся в его верхней части; при этом жидкость равномерно стекает через края цилиндра. Кроме того, штуцер для ввода исходной смеси целесообразно погружать в это углубление, что уменьшает капле-образование. Для дополнительного улучшения распределения жидкости поверхность обогреваемого цилиндра выполняли шероховатой с помощью пескоструйного аппарата. Передвижные металлические кольца 8 (см. рис. 196), насаженные на обогреваемый цилиндр, обеспечивали циркуляцию пленки, а расположенная над углублением для подачи исходной смеси стеклянная воронка 7 предотвращала попадание исходной жидкости на поверхность конденсации.
Аппарат снабжен прибором для автоматической установки расхода исходной смеси с помощью фотоэлемента и автоматическим регулятором температуры нагревателя. Дистиллятор заключен в охлаждающий кожух, что позволяет разделять методом пленочной перегонки низкокипящие вещества.
Регулирование работы дистиллятора и всех вспомогательных аппаратов проводится с центрального пульта управления. Исходная смесь заливается в бутыль 9, вмещающую около 10 л. Из бутыли смесь передавливается воздухом по трубопроводу в напорный бак 4, расположенный над дистиллятором. Расход исходной смеси регулируется с помощью поплавка 6, который посредством фотоэлемента 5 включает или выключает воздушный насос. Таким образом гарантируется непрерывная подача подогретой исходной смеси в напорный бак. Устройство 3 для точной установки расхода обеспечивает точное и надежное дозирование разделяемой смеси из напорного бака на наружную поверхность внутренней обогреваемой трубы 2 дистиллятора. Дистиллят и кубовый остаток могут отводиться в отдельные приемники без нарушения технологического режима процесса дистилляции. Дистиллятор предназначен для работы при атмосферном давлении и под вакуумом.
Как показали испытания дистиллятора, на результаты разделения при постоянных условиях (вакуум и температура) значительное влияние оказывает скорость подачи исходной смеси. При повышении скорости подачи увеличивается количество кубового продукта, а при ее уменьшении возрастает количество отби-274
раемого дистиллята при одновременном повышении четкости разделения. Изменяя рабочую температуру при постоянном вакууме, можно определить оптимальные условия процесса.
Если известен состав исходной смеси, то устанавливают такую скорость ее подачи, чтобы количества головной и кубовой фракций соответствовали необходимой степени разделения. При этом всегда целесообразно при первой пленочной разгонке отобрать возможно большее количество дистиллята. Такое «превышение» позволит получить кубовый продукт с минимальным содержанием низкокипящей фракции. Высококипящие продукты, попавшие в первичный дистиллят, путем повторной пленочной дистилляции переводят к кубовый остаток, причем после второй перегонки обычно получают очень чистый дистиллят. С помощью градуированных приемников можно непрерывно контролировать необходимое соотношение скоростей отбора дистиллята и кубового продукта (рис. 197).
Методом непрерывной пленочной дистилляции можно выделять фракции жирных кислот до кислотного числа 90, так что в дистиллят попадают кислоты с числом атомов углерода, меньшим 30. Пленочная перегонка предпочтительна также для разделения воска и силиконовых масел, которые особенно хорошо смачивают греющую поверхность. Гутвассер и Мюллер [22] разработали установку с пленочным испарителем, предназначенную для разделения смеси спермацета и жирных кислот. На рис. 198 показана схема этой установки и пленочного испарителя.
Аппараты фирмы Лейбольд—Хэраеуз (Кёльн), предназначенные для работы при 1 —10~3 мм рт. ст., имеют особенно развитую поверхность испарения и обеспечивают постоянное обновление «активной поверхности» жидкости путем интенсивного перемешивания пленки. Эта фирма выпускает ректификационные аппараты следующих типов.
1) Колонны с перемешивающейся пленкой (рис. 199), служащие для дистилляции жидких смесей, кубовый остаток которых при рабочей температуре может отбираться только в виде капель. Такими смесями могут быть высокомолекулярные фракции нефти и сложные эфиры, ароматические вещества, моноглицериды и растительные экстракты. В этих колоннах восходящий поток паров движется перпендикулярно к потоку жидкости. Пленочный испаритель
Рис. 197.
Диаграмма разгонки кубовой (/) и головной (2) фракций смеси жирных кислот С8—С25 методом непрерывной пленочной дистилляции. Выход дистиллята отнесен к суммарному количеству обеих фракций.
275
Рис. 198.
Установка Гутвассера—Мюллера [22], предназначенная для пленочного испарения жидкостей под вакуумом.
а — Схема установки:
1 — дистилляционный куб; 2 — предохранительный холодильник; 3 — пленочный испаритель; 4 — обогревающий кожух; 5 — термостат; 6,7 — штуцеры для присоединения термостата; 8, 10 — термометры; И — распределитель пара и жидкости; 12 — конденсатор головки испарителя; 13, 14 — охлаждаемые ловушки; 15 — ионизационный вакуумметр; 16 — ртутный диффузионный насос; 17 — масляный манометр.
б — Пленочный испаритель со стандартными шлифами NS70: /<
13 — запорные карманы; 2 — сферический шлиф для присоединения приемника дистиллята; 3,6 — штуцеры для установки термометров; 4 — конденсатор паров дистиллята; 5 — штуцер для присоединения вакуумной линии; 7, 8 — штуцеры для присоединения термостата; 9 — распределитель конденсата нз головки испарителя; 10 — конденсаторы, совмещенные со вторичными испарителями; И — стеклянная пластина распределителя; 12 — основная испарительная труба.
Изготовитель: народное предприятие «Комбинат медицинской и лабораторной техники», завод V, Лейпциг.
• - обогрев
& - конденсации вещество
Рис. 199.
Схема колонн с перемешивающейся пленкой жидкости.
Рис. 200.
Барабан для перемешивания пленки жидкости.
Обозначения см. на рис. 199.
276
модели 11 пригоден для получения клейких продуктов.
2) Барабаны с перемешивающейся пленкой жидкости (рис. 200), предназначенные для перегонки смесей веществ, имеющих вязкие или твердые кубовые остатки. К таким смесям относятся каменноугольный пек, натуральный или синтетический воск и битуминозный (горючий) сланец.
3) Колонны с вращающимися щетками (рис. 201), служащие для дистилляции смесей веществ, дистиллят и кубовый остаток которых при рабочей температуре в колонне имеют вязкость до 800 сП, например талловое масло, ди- и трихлорфенилы, ланолин [146].
Для этих аппаратов выпускаются стандартные взаимозаменяемые детали, с помощью которых можно собирать многоступенчатые пленочные дистилляторы любых типов, предназначенные для дегазации и перегонки жидкостей при различных температурах. Дальнейшим развитием данных аппаратов являются дистилляционные устройства для перегонки «на коротком пути» со скребковыми роторами и внутренним охлаждающим устройством, из которых также могут собираться многоступенчатые дистилляторы (см. разд. 5.4.4).
Испарители с падающей пленкой, снабженные вращающимися контактными устройствами, аналогичны по конструктивному выполнению роторным ректификационным колоннам (см. разд. 7.3.5).
рвогрев о —
- дещестдо
& —конденсация
Колонна с вращающимися щетками.
Рис. 202.
Дистилляционная установка со стеклянным испарителем модели Самбай:
1 — воронка для подачи исходной смеси;
2 — игольчатый клапан; 3 — регулируемый приводной механизм ротора; 4 — магнитная муфта; 5 — змеевик для подогрева исходной смеси; 6 — калиброванная стеклянная труба; 7 — скребковый ротор с лопастями; 8 — конденсатор; 9, 12 — промежуточные вставки со штуцером для вакуумметра; 10 — дезодорирующая промежуточная вставка; 11, 13 — приемники для кубового продукта и дистиллята соответственно; 14 — термостат.
Изготовитель: фирма «Стеклянная техника», Висбаден --Ширштайи.
277
Основное отличие состоит в том, что в роторных колоннах жидкая смесь испаряется в кубе и массообмен происходит при противоточном движении паровой и жидкой фаз, за счет чего обеспечивается высокая степень разделения. Однако как в роторных испарителях, так и в роторных ректификационных колоннах с помощью ротора обеспечивается циркуляция пленки жидкости и тем самым предотвращается обеднение ее поверхности низкокипящим компонентом. Проблемы механической турбулизации тонких слоев жидкости обсуждены в работе Яносфиа [134].
Испарители с падающей пленкой могут работать как при прямо-или противотоке паровой и жидкой фаз, так и на принципе скоростной дистилляции (см. разд. 5.4.4). Подвод тепла обычно осуществляется снаружи (см. рис. 198, 200—203, 211), однако есть аппараты и с внутренним подводом тепла (см. рис. 196, 199, 210, 212).
Пленочные испарители хорошо зарекомендовали себя в промышленности [135]. Производительность и массообменные характеристики различных испарителей сравнил Дитер [136]. Математическое моделирование и методы расчета роторных пленочных испарителей разработал Биллет [137].
На рис. 202 показана дистилляционная установка с пленочным испарителем модели Самбай. Испаритель снабжен скребковым ротором. Эта установка выпускается фирмой QVF «Стеклянная техника».
Исходная смесь поступает в испаритель из воронки 1 с капельницей, снабженной обогревающим кожухом. Точное дозирование жидкости обеспечивается с помощью игольчатого клапана 2 воронки, которая работает как сосуд Мариотта. Перед входом в испаритель смесь нагревается в обогреваемом змеевике 5 до температуры кипения, вследствие чего рабочая зона испарителя полностью используется только для испарения жидкости. Одновременно в змеевике происходит дегазация исходной смеси, что исключает образование брызг в рабочей зоне испарителя.
Пленка образуется на внутренней поверхности калиброванной стеклянной трубы 6 при вращении скребкового ротора 7 с лопастями (для уменьшения коррозии ротор выполняется из тантала или нержавеющей стали марки VA). Регулируемый приводной механизм 3 со ступенчато изменяющейся скоростью вращения соединяется с ротором магнитной муфтой 4, которая лишена всех недостатков сальникового уплотнения. Нижний конец ротора снабжен самоустанавливающимся шарикоподшипником в виде тефлонового шара, размещенного в стеклянной опоре. Смазкой подшипника служит стекающий кубовый продукт. Источником тепла является циркуляционный термостат 14 мощностью электрообогрева 1,5 или 2 кВт. При температурах до 200° С в качестве теплоносителя используют парафиновое масло, а при более высоких температурах — силиконовое масло. Эти масла полностью прозрачны.
278
В комплект установки входят также приемник кубового продукта 11, служащий для отбора высококипящей фракции, и промежуточная вставка 9 со штуцером для вакуумметра. При дезодорации жидкостей или дистилляции смесей с небольшим содержанием низкокипящей фракции промежуточную вставку 9 заменяют на дезодорирующую вставку 10. Через боковой штуцер этих вставок 9, 10 можно вводить паровой поток низкокипящей фракции для увеличения скорости движения паров в направлении конденсатора и уменьшения возможного испарения дистиллята. Наличие сифона способствует предотвращению конденсации паров в приемнике кубового продукта.
Низкокипящая фракция конденсируется в конденсаторе 8 и собирается в приемнике дистиллята 13. Штуцер промежуточной вставки 12 служит для присоединения установки к вакуумной линии.
Обогревающая поверхность испарителя составляет 0,016 м2, а охлаждающая поверхность конденсатора — 0,2 м2. Максимальная рабочая температура равна 220° С. При этой температуре и при давлении 25 мм рт. ст. наибольшая производительность установки составляет 480 г/ч.
Лабораторные и промышленные испарители с вращающимся кубом, применяющиеся в различных ректификационных установках, стандартизированы. Их применяют как в пилотных дистилляционных установках, так и в лабораторных приборах, предназначенных для микроперегонки. Данные испарители имеют вращающийся куб в виде трубы с шаровым расширением (см. разд. 5.1.1) или круглодонной колбы, емкость которых может изменяться в интервале от 1 мл до 100 л. Наряду с дегазацией масел и смол испарители с вращающимся кубом используют для отделения растворителей и пенящихся веществ в мягких температурных условиях. На рис. 203 показана принципиальная схема данного испарителя. Конструкции таких испарителей и области их применения подробно рассмотрены Эгли [138]. Частота вращения колбы 4 может ступенчато изменяться и регулироваться в интервале от 10 до 220 об/мин. Для удобства эксплуатации установка снабжена механическими и автоматическими
Рис. 203.
Испаритель «Ротавапор» с вращающейся колбой модели R-10:
/ — приемник дистиллята; 2 — конденсаторы; 3 — штуцер для присоединения вакуумной линии; 4 — вращающаяся перегонная колба; 5 — обогревающая баня; 6 — пульт управления.
Изготовитель: фирма В. Бюхи, Флавиль, Швейцария.
279
подъемными приспособлениями для куба. Система взаимозаменяемых деталей для сборки испарителей с вращающимся кубом и ее возможности рассмотрены в работе Крамера [138а].
5.4.4. МОЛЕКУЛЯРНАЯ дистилляция
Методом молекулярной дистилляции перегоняют высококипя-щие, часто термически нестойкие вещества при абсолютном давлении, не превышающем 10“3 мм рт. ст. При таких давлениях расстояние, проходимое молекулами между испаряющей и охлаждающей поверхностями, меньше средней длины свободного пробега молекул. Таким образом достигают того, что большинство молекул испаряющейся жидкости попадает на поверхность конденсации без столкновения с молекулами другого газа. Под средней длиной свободного пробега молекулы понимают теоретически рассчитанное расстояние, которое молекула может пройти, не столкнувшись с другими молекулами. Например, для триглицеридных жиров нормального ряда с молекулярной массой 800 эта величина имеет следующие значения (в мм) [141]:
При остаточном давлении 8-Ю-3 мм рт. ст...................... 7
» » » 3-10'3 мм рт. ст................... 25
» » » 1 • 10'3 мм рт. ст................. 50
При молекулярной дистилляции не происходит кипения жидкости. Этот процесс можно определить как «молекулярное испарение». Равновесное состояние между жидкостью и ее паром постоянно нарушается из-за конденсации. При этом должно устанавливаться новое равновесие, т. е. новые молекулы должны испариться с поверхности жидкости. Таким образом, молекулярная дистилляция является ярким примером прямоточной дистилляции, которую также можно назвать «однократным испарением» [141].
Области применения молекулярной дистилляции рассмотрены в монографиях Барроуза и Холо с сотр. [139]. Ридгвэй-Уатт [140] дал подробный обзор установок молекулярной дистилляции различной производительности (от микроаппаратов до промышленных установок). В разд. 1.5 и в работе [1221 можно найти ссылки на ряд других обзорных статей по данному методу перегонки [108, 131, 145, 156, 157].
Скорость испарения зависит от давления паров вещества р° при температуре поверхности испарения, а также от молекулярной массы перегоняемого продукта. Формальное математическое описание этой зависимости предложил Лэнгмюр [142]:
D = 0,0583/?° К М/Т (181)
где D— максимальная удельная скорость испарения, г/(с-см2); /?“—давление паров вещества при температуре Г, мм рт. ст.; М — молекулярная масса вещества; Т — температура дистилляции, К.
280
Эта формула получена при допущении, что испарению не препятствуют молекулы другого вещества. Поскольку в реальных процессах испарившиеся молекулы при движении к поверхности конденсации сталкиваются с молекулами другого газа, значение D в большинстве случаев не достигается. С целью его коррекции вводят коэффициент испарения а, который учитывает остаточное давление другого газа. Для процессов молекулярной дистилляции, осуществляемых в современных промышленных установках, а равен 0,9. Очевидно, что значение а будет тем ближе к 1, чем ниже остаточное давление в дистилляторе. Барроуз [143] исследовал применимость формулы (181) и получил ряд полуэмпирических уравнений, которые лучше соответствуют различным условиям процесса молекулярной дистилляции.
Теоретически достижимые скорости молекулярной дистилляции очень малы, что наглядно видно из данных, приведенных в табл. 40 [108 ].
Если принять коэффициент испарения равным 0,8, то из данных таблицы можно определить количества дистиллята, получаемые за 1 ч с испаряющей поверхности 100 см2. Эти количества лежат в интервале от 15 до 22 г/ч.
Для достижения высокой производительности установок молекулярной дистилляции следует работать при абсолютных давлениях выше 10 :| мм рт. ст. по методам Утцингера [133] и Яекеля [144], последний из которых предпочтительно применять в полупромышленных и промышленных установках. В большинстве случаев бывает достаточно, если остаточное давление приближается к 10 мм рт. ст. При этом давление паров перегоняемой жидкости может быть во много раз больше и достигать порядка 1 мм рт. ст. Молекулы оставшегося газа при столкновении с поверхностью конденсации отражаются от нее, а молекулы паров жидкости, напротив, задерживаются на этой поверхности [145].
Области применения молекулярной дистилляции весьма разнообразны. Прежде всего этим методом перегоняют термически не-
Таблица 40.
Теоретически достижимые скорости молекулярной дистилляции при остаточном давлении 10-3 мм рт. ст.
Вещество Молекулярная масса D . 104 по формуле (181) р° при 120 °C, мм рт. ст. К м/т при 10-3 мм рт. ст. ро/1/~М При 120 °C
г/(с. см2) МОЛЬ
(с см2)
Стеариновая кис- 284 0,52 0,0021 35,0 0,90 2,07
лота Холестерин 387 0,56 0,0014 0,5 0,97 0,025
Тристеарин 891 0,76 0,00С9 10~4 1,32 —
281
стойкие вещества с молекулярной массой от 250 до 1200. В качестве примера укажем следующие области применения молекулярной дистилляции (см. [122, 158] и гл. 5, 6): получение масел для вакуумных насосов, а также высоковязких смазочных масел, вязкость которых мало зависит от температуры; исследование триглицеридных масел и жиров, а также восков, смол и высокомолекулярных жирных кислот и спиртов; получение витаминов и концентратов гормонов; очистка пластификаторов и других веществ с низким давлением паров; дезодорация высокомолекулярных веществ; очистка эфирных масел и ароматических веществ.
В последнее время молекулярную дистилляцию применяют для исследования высокомолекулярных веществ, содержащихся в нефтяных и других кубовых остатках, которые получают при обычной перегонке. Методом многократной дистилляции получают вещества максимально возможной степени чистоты. Обзор аппаратуры и методов молекулярной дистилляции жирных кислот и липидов представил Перри [145]. Франк [146] рассмотрел специальные вопросы, касающиеся данного способа, и описал конструкцию многоступенчатых испарителей.
Важным преимуществом молекулярной дистилляции является возможность разделения смеси веществ с одинаковыми давлениями паров, но разными молекулярными массами.
Из формулы Лэнгмюра
D =-p/]^2nMRT (182)
где D—максимальная удельная скорость испарения, моль/(с-см2); р—давление пара, дин/см2; М — молекулярная масса; R—универсальная газовая постоянная, равная 8,3-10’ эрг/(К-моль); Т — абсолютная температура, К-следует, что при постоянной температуре количество образующихся паров зависит от отношения р/1/ М. Относительную испаряемость компонентов бинарной смеси при молекулярной дистилляции по аналогии с относительной летучестью можно представить в виде [1081:
О!,/О2= (Л КОМКАЛ) (183)
где pj и р2 — парциальные давления паров компонентов.
Из этой формулы следует, что два вещества с разными молекулярными массами можно разделить путем молекулярной дистилляции даже при равных парциальных давлениях их паров.
. Конструкции аппаратов для проведения молекулярной дистилляции должны удовлетворять следующимтребованиям. Они должны иметь устройство для присоединения к вакуумной линии с большим диаметром условного прохода, чтобы потери давления были минимальными (см. разд. 5.4.1). В этих аппаратах должно обеспечиваться равномерное распределение пленки жидкости по поверхности испарения и малое время ее пребывания на этой поверх-282
кости (см. разд. 5.4.3). Расстояние между поверхностями испарения и конденсации должно быть примерно равно средней длине Свободного пробега молекул (1—2 см), разность температур между Йтими поверхностями должна составлять 50—100 °C. Установку Следует оборудовать устройствами для предварительной дегазации перегоняемых веществ с целью удаления растворенных инертных газов.
Имеющиеся колонны для молекулярной дистилляции по принципу действия можно разделить на следующие группы: аппараты с тонким слоем жидкости в плоскодонном кубе; дистилляторы с поверхностью испарения в виде горизонтальных или наклонных лотков; колонны с вертикально стекающей (падающей) пленкой жидкости; аппараты центрифужного типа.
Чтобы более полно отделить низкокипящий компонент, в колоннах этих групп можно проводить многократную перегонку путем циркуляции разделяемой смеси. Отметим также, что в данных дистилляторах испарение происходит только с поверхности пленки жидкости и молекулы селективно покидают эту поверхность без какого-либо механического воздействия [147].
Наиболее простыми по конструкции являются аппараты с тонким слоем жидкости в плоскодонном кубе. На рис. 204 показан современный дистиллятор подобного типа. С помощью таких аппаратов можно успешно разделять вещества с молекулярными массами до 300. Дистилляцию из куба с плоским дном и тонким слоем жидкости обычно применяют для получения предваритель-
ных данных о температурных пределах перегонки и способности веществ к разложению.
Микромолекулярная дистилляция, осуществляемая по методам «холодного пальца» и падающей пленки жидкости и предназначенная для перегонки 0,5—5 г исходной смеси, описана в разд. 5.1.1 (см. рис. 131—133). Наиболее распространенным аппаратом с испаряющей поверхностью в виде лотка является дистиллятор Ут-цингера [133, 147]. Утцингер
Рис. 204.
Высоковакуумный молекулярный дистиллятор. Условия работы: температура 20 — 200 °C, загрузка исходной смеси 2,5 или 10 мл. Снабжен регуляторами остаточного давления до Ю“* мм рт. ст. и регуляторами мощности электрообогрева.
Изготовитель: фирма Фишера «Лабораторная и исследовательская техника», Бонн —курорт Годесберг.
283
Рис. 205.
Аппарат Утцингера для получения трех фракций методом дистилляции на «коротком пути» .
Изготовитель: фирма «Завод стекла Шотт», Майнц.
ввел понятие дистилляции «на коротком пути» *, обозначающее однократную перегонку при остаточных давлениях более 10'3 мм рт. ст. Аппарат, созданный им еще в 1943 г., в дальнейшем был видоизменен применительно к процессам непрерывного фракционирования. На рис. 205 показана новейшая модель этого дистиллятора. Установка работает следующим образом.
Исходная смесь из колбы для дегазации поступает по капилляру в дистиллятор, в котором она стекает тонким слоем по наклонному лотку, обогреваемому циркулирующим высококипя-щим минеральным маслом. Вдоль лотка возникает градиент температур, поскольку температура повышается в направлении течения дистиллируемой жидкости. Пары, окружающие лоток, конденсируются в холодильнике, наклоненном под тем же углом, что и лоток. Установка имеет три зоны конденсации и четыре штуцера для отбора трех фракций дистиллята и кубового остатка.
Незначительный перепад давления между колбой для дегазации и дистиллятором (оба сосуда перед проведением перегонки подключаются к одному и тому же вакуумному насосу) облегчает ввод исходной смеси в лоток дистиллятора и позволяет измерять скорость ее подачи. Предусмотрена возможность регулирования скорости течения перегоняемой жидкости по обогреваемому лотку путем соответствующего изменения наклона дистиллятора с помощью сферического шлифа, а также возможность изменять скорость течения во время дистилляции. Регулирование этих параметров необходимо для установления оптимального соотношения между количеством отбираемых фракций.
* В отечественной технической литературе этот термин не применяют. — Прим. ред.
284
Циркулирующее обогревающее масло нагревается с помощью горелки Бунзена или электронагревательной обмотки, размещенной на входном штуцере циркуляционного контура. Продольный температурный градиент в обогреваемом лотке зависит от скорости подачи исходной смеси, скорости течения ее по лотку, состава перегоняемой жидкости, а также от типа вакуумного насоса. В описанном стеклянном дистилляторе температурный градиент между началом и концом лотка составляет 10—20° С. Рабочую температуру дистилляции определяют косвенным путем, измеряя температуру кипения небольшой пробы дистиллята при стабильном вакууме. Термометр, погруженный в циркулирующее обогревающее масло, обычно показывает температуру на 60—80° С выше измеренной температуры кипения. Это означает, что пленочное испарение приводит к сильному охлаждению испаряющей поверхности лотка. Средняя пропускная способность установки составляет около 100 мл/ч, что соответствует количеству отбираемого дистиллята 50—70 мл/ч.
Дистиллятор Утцингера был усовершенствован народным предприятием «Йенский завод стекла Шотт», на котором были созданы макро- и микрокаскадные аппараты (рис. 206). На этих аппаратах, собранных из стандартных взаимозаменяемых деталей,
^Рис. 206.
I Мнкрокаскадный аппарат для молекулярной дистилляции н дистилляции на «коротком пути» .
№: Изготовитель: народное предприятие «Йенский завод стекла Шотт», Иена. ,
ЕрРис. 207.
К Дегазатор Утцингера.
285
0 20 ОО 60 80 100
Выход дистиллята,У а
Рис. 208.
Разделяющая способность одноступенчатого (/) и многоступенчатого (2) испарителей.
Молекулярная дистилляция смеси диизооктилсебацииат (ДИОС), молекулярная масса 426, — днизооктилфталат (ДИОФ), молекулярная масса 391 ‘[146].
Рис. 209.
Установка Малюсова—Жаворонкова [149] для многоступенчатой моле-кулярной дистилляции:
1 — штуцеры для присоединения вакуумной линии; 2 — устройство для отбора дистиллята; 3 — штуцеры для подачи охлаждающей воды; 4 — труба для подачи исходной смеси; 5 — электронагреватели.
можно получать любое необходимое число фракций. В каждом элементе каскада путем циркуляционного способа нагревания можно устанавливать любую температуру. Предварительную дегазацию исходной смеси проводят отдельно в устройстве, показанном на рис. 207. При этом учитывают то обстоятельство, что процессы дегазации и дистилляций протекают с существенно различными скоростями.
Мельпольдер с сотр. [148] разработал двадцатиступенчатый аппарат для молекулярной ректификации, предназначенный для разделения высококипящих фракций нефти. В работе [140] предложен десятиступенчатый испаритель, который медленно вращается в наклонном положении. Разделение исходной смеси в этом случае происходит на всей поверхности испарителя. Образовавшийся на первой ступени охлаждения конденсат поступает во вторую ступень испарения и таким образом продвигается далее до места отбора дистиллята. Сравнение разделяющих способностей одноступенчатого испарителя (см. рис. 204) и десятиступенчатого испарителя [140] показано на рис. 208 [146]. Методы расчета чисел теоретических ступеней разделения, достигаемых в подобных многоступенчатых аппаратах, представлены в работах Жаворонкова и Малюсова с сотр. [149, 150]. На рис. 209 показана установка для молекулярной дистилляции, использованная этими исследователями.
Ректификация по принципу «падающей пленки» нашла наиболее широкое применение. Большую часть работающих на этом прин-286
ципе аппаратов, описанных в разд. 5.4.3, можно использовать также и для молекулярной дистилляции. Благодаря малому времени пребывания гравитационно стекающей пленки жидкости на поверхности испарения, составляющему для лабораторных приборов 3—4 с и для промышленных аппаратов до 0,001 с [92], перегонка веществ протекает в исключительно мягких температурных условиях. В лабораторных приборах расчетная толщина слоя жидкости лежит в пределах 0,1—0,2 мм, что соответствует слою из 50 000 молекул, а в промышленных аппаратах толщина слоя может составлять 0,001—0,005 мм, что соответствует слою из 400 молекул. Теоретически идеальным условием перегонки являлся бы мономолекулярный слой.
Очень важными факторами для аппаратов, работающих по методу молекулярной дистилляции, являются форма выполнения поверхности испарения и, конечно, расстояние между поверхностями испарения и конденсации. Хорошее смачивание поверхности испарения и оптимальная циркуляция пленки жидкости обеспечиваются в аппаратах «с падающей пленкой» жидкости, описанных в разд. 5.4.3 [122].
На рис. 210 показана установка для молекулярной дистилляции с испарителем, снабженным спиральным ротором. Стеклянный спиральный ротор 8 испарителя вращается вокруг испарительной свечи 10. Такое конструктивное решение обеспечивает получение тонкослойной жидкой пленки толщиной около 0,1 мм и хорошую циркуляцию жидкости. Время пребывания жидкости в аппарате составляет всего несколько секунд. Установка имеет следующие технические данные: условная производительность — 1000 г/ч; производительность* — 250—2000 г/ч; максимальная скорость испарения * — 18 000 г/ч; частота вращения ротора — около 40—90 об/мин; площадь поверхности испарения — около 600 см2; максимальная температура дистилляции — 300 °C; рабочее напряжение электросети 380—220 В; потребляемая мощность — 2 кВт; расход охлаждающей воды — около 350 л/ч. В качестве вымораживающих хладоагентов рекомендуется использовать жидкий воздух или азот, а в подходящих случаях смесь СО2 — ацетон.
При разработке лабораторной установки модели КДБ4, ;в основу работы которой положен метод дистилляции на «коротком пути», фирма Лейбольд—Хэраеуз (Кёльн) исходила из того, чго первой стадией молекулярной дистилляции является дегаза-?ция исходной смеси, за которой следует большое число стадий дистилляции, проводимых при различных температурах. Уста-сновка снабжена испарителями с роликовым скребковым ротором, j/изготовленным из тефлона или тефлоновой керамики. Ротор размещается между внутренним охлаждаемым цилиндром и наружным обогреваемым корпусом. Для обеспечения многоступенчатой
* Зависит от разделяемой смеси.
287
Рис. 210.
Установка для молекулярной дистилляции модели MDRR600.
1 — пульт управления; 2 — охлаждаемая ловушка для дегазатора; 3 — клапан для точной дозировки исходной смеси; 4 — привод ротора; 5 — дегазатор; 6 — охлаждаемая ловушка дистиллятора; 7 — сосуд для исходной смеси; 8 — спиральный ротор; 9 — дистиллятор с конденсационным кожухом; 10 — испарительная свеча; 11 — термопара; 12 — высоковакуумный масляный диффузионный насос; 13 — электромагнитные клапаны сменных сборников дистиллята и кубового продукта; 14 — сборник дистиллята; 15 — сборник кубового продукта; 16 — двухступенчатый шиберный вакуумный насос; 17 — одноступенчатый шиберный вакуумный насос; 18 — вакуумный запорный клапан; 19 — универсальный лабораторный термостат.
Изготовитель: народное предприятие «Йенский завод стекла Шотт», Иена.
дистилляции испарители, выполненные из стекла или нержавеющей стали, устанавливают друг за другом. Схема лабораторной установки KDL4 непрерывного действия показана на рис. 211. Давление на различных ступенях дистилляции может достигать 1(Г3 мм рт. ст. Минимальная производительность установки составляет 100 г/ч. Хабендорфф [140а] представил рекомендации по монтажу и вводу в эксплуатацию промышленных установок для дистилляции «на коротком пути», а также технологические параметры процесса.
288
; Ю Э. Крель
Некоторые аппараты «с падающей пленкой» снабжают устройством для циркуляции перегоняемой жидкости (рис. 212). Циркуляцию осуществляют с помощью электромагнитного насоса. J Обычно в месте ввода жидкости размещают кольцо из проволочной сетки для обеспечения равномерного распределения исходной смеси [152].
Основной проблемой при разработке аппаратов, работающих ; по принципу «падающей пленки», является предотвращение воз-• никновения струйного течения на поверхности испарения. Ее решают одним из следующих способов: поверхность испарения выполняют шероховатой путем обработки ее пескоструйным аппа-
Рис. 211.
Лабораторная установка модели KDL4, работающая по методу дистилляции «на коротком пути», с роторными испарителями.
Ротор испарителя снабжен скребками в виде роликов. 1 — распредели-ельные электрощиты со штепсельными розетками; 2 — дозирующий со-£УД‘, 3 — привод ротора; 4 — штуцер для присоединения вакуумного на-са 1 ступени; 5 — охлаждаемые ловушкн; 6 — стеклянный дистиллятор; __ — металлический дистиллятор; 8 — циркуляционные термостаты; 9 — ^Мерный сосуд (в разрезе); 10 — штуцер для присоединения вакуумного асоса 2 ступени, зготовитель: фирма Лейбольд—Хэраеуз, Кельн.
Рис. 212.
ппарат Хикмана для молекулярной дистилляции с циркуляционным уст-йством.
289
ющую поверхность подают
Рис. 213.
Аппарат Хикмана для молекулярной дистилляции с центробежным распределением жидкости: 1 — насос для подачи исходной смеси; 2 — устройство для отвода кубового продукта; 3 — поверхность конденсации; 4 — вращающийся обогреваемый диск; 5 — сборник кубового продукта; 6 — емкость для исходной смеси.
ратом, травления или наплавлива-ния стеклянного порошка (см. рис. 196, 205, 206); в месте ввода исходной смеси устанавливают кольцо из проволочной сетки либо плотно прилегающий цилиндр из проволочной сетки или стеклоткани (см. рис. 196, 212); поверхность испарения выполняют волнистой или в виде спирали (см. рис. 210); жидкость на испаря-с помощью ротора со скребковыми
устройствами или при вращении всей поверхности испарения (см. рис. 200—203, 211).
Центробежный принцип получения наиболее тонких слоев жидкости развит Хикманом 1153), он применил его для разработки промышленных аппаратов большой производительности [151 ]. На рис. 213 показан лабораторный аппарат с вращающимся диском. Разделяемая смесь из емкости 6 подается насосом 1 на вращающийся обогреваемый диск 4, с которого под действием центробежной силы жидкость распределяется в виде тонкой пленки. Поверхность конденсации 3 расположена на небольшом расстоянии от центрифуги. Дистиллят можно либо отбирать с поверхности конденсации, либо возвращать в емкость 6. Кубовый остаток с помощью устройства 2 отводится в сборник 5, откуда он может быть отправлен в емкость 6 для дальнейшей переработки.
Хикман [152] и Эмбре [154] ввели для молекулярной дистилляции понятие «дистилляционная способность», под которой понимают отношение числа молекул вещества, покидающих в единицу времени поверхность испарения, к числу молекул того же вещества, остающихся при данных условиях в пленке жидкости. Многократной циклической перегонкой можно полностью получить вещество в виде дистиллята. При этом продолжительность времени дистилляции удается сократить путем повышения температуры испарения. «Кривую выделения» находят следующим образом. Смесь перегоняют при стабилизированном вакууме и постоянной скорости повышения температуры (например, последовательно повышая температуру на 10 °C) и определяют концентрацию низкокипящего компонента в дистилляте. Типичные «кривые выделения» показаны на рис. 214. Как видно из рисунка, концентрация вначале растет до максимума, а затем снижается
290
г до нулевого значения. Форма кривой зависит, конечно, от физико- химических свойств веществ и прежде всего от удельной энталь-5 пии испарения, а также от времени пребывания жидкости в испа-рителе (которое для возможности сравнения экспериментальных г данных должно быть постоянным). Максимум на «кривой выделения» соответствует температуре кипения при обычной дистилляции. При большем времени пребывания продукта в горячей зоне «кривая выделения» смещается таким образом, что ее максимум соответствует более низкой температуре (см. кривую 2, рис. 214) [159]. Для достижения оптимальной производительности установок молекулярной дистилляции необходимо предварительно тщательно исследовать условия ее проведения путем определения «кривых выделения». Такое теоретическое исследование проведено Горрицом с сотр. [155].
Данный метод получил дальнейшее развитие благодаря применению стандартных веществ, в большинстве случаев красителей, » которые удобно анализировать колориметрическим методом [108].
Если стандартного вещества мало, то для получения хорошей • пленки жидкости добавляют другое вещество — носитель.
Хикман и Тревой [141 ] при исследовании процессов высоковакуумного испарения и молекулярной дистилляции получили интересные данные. Оказалось, что коэффициент испарения чистых жидкостей может иметь значение 1. Отклонения чаще всего обусловлены незначительными загрязнениями. При определении • относительной летучести смеси ди-2-этилгексилфталат (ЭГФ)— ' ди-2-этилгексилсебацинат (ЭГС) было установлено, что она в значительной степени зависит от условий определения (рис. 215).
Малюсов с сотр. [160] при исследовании процессов перегонки под вакуумом, соответствующем 10~4 мм рт. ст., определял раз-
Рис. 214.
«Кривые выделения» , полученные при кратковременном (/) и длительном (2) пребывании продукта на поверхности испарения.
Рис. 215.
Зависимость относительной летучести для смеси дн-2-этнлгекснлфталат (ЭГФ) — ди«2-этилгекснлсебацннат (ЭГС) от температуры.
Температура кипения ЭГФ 183—181 °C н ЭГС 199 — 202 °C при 0,5 мм рт. ст. Кривые получены при работе на приборе для исследования фазового равновесия, снабженном кубом (/), н на приборе <с падающей пленкой» (2).
Г— 10*
291
ницу между неравновесным анравн и равновесным аравн факторами разделения. С повышением температуры отношение аирави/арави приближается к 1, что соответствует графику на рис. 215.
Уеха и Хагихара [163] исследовали эталонную смесь ди-2-этилгексилфталат—ж-трикрезилфосфат при остаточном давлении менее 1,5-10”® мм рт. ст. (2-10-3 мбар) в пределах температур 130—160 °C. Оказалось, что в области низких температур эта смесь в интервале концентраций ди-2-этилгексилфталата от 20 до 80% (мол.) ведет себя как идеальная смесь.
5.4.5. РЕКТИФИКАЦИЯ ПОД ДАВЛЕНИЕМ
Под давлением выше атмосферного можно перегонять смеси, которые при комнатной и более высокой температурах находятся в газообразном состоянии. В данном случае в качестве хладоагента можно применять воду, что делает процесс перегонки более экономичным. Однако для получения избыточного давления необходима дополнительная аппаратура.
Ректификацию под давлением целесообразно использовать в тех случаях, когда относительная летучесть компонентов смеси увеличивается с повышением температуры. Например, для смеси 2,4-диметилпентан — 2,2,3-триметилпентан разность температур кипения при атмосферном давлении составляет лишь 0,2 °C, а значение а возрастает с 1,006 при 80 °C до 1,055 при 200 °C (см. разд. 4.6.2).
Известно лишь небольшое число лабораторных установок для ректификации под давлением. Для давлений выше 5 кгс/см1 2 в качестве конструкционного материала для колонн стекло не-
Рис. 216.
Установка Симонса для ректификации под давлением до 5 кгс/см2:
1, 16 — клапаны для автоматической - стабилизации давления; 2 — коиус поплавка клапана; 3 — седло клапана; 4, 5 — штуцеры для подачи сжатого воздуха и вывода его в атмосферу соответственно; 6 — поплавок клапана; 7, 15 — груши клапанов; 8,9 — конденсаторы дистиллята; 10 — капилляр с электронагревательной обмоткой; 11 — воронка для подачи флегмы; 12 — колонна; 13 — куб; 14 — электронагреватель куба; 17 — приемник дистиллята; 18, 19 — трубы для подачи дистиллята; 20 — край.
Рис. 217.
Кран для работы под давлением с предохранительным устройством.
292
►применимо. Шнайдер описал металлический ректификационный (аппарат, предназначенный для работы под давлением до 45 кгс/см2, "который состоит из колонны диаметром 25 мм и куба емкостью 3 л [161 ].
Ректификационную установку Симонса [162], полностью изготовленную из стекла, можно применять до давлений, не превышающих 5 кгс/см2 (рис. 216). Автоматическая стабилизация давления сжатого воздуха, подаваемого по штуцеру 4, осуществляется (с помощью клапана 1, заполненного ртутью. Клапан имеет поплавок 6, оканчивающийся конусом 2. Этот конус запирает отверстие в седле клапана 3. Уровень ртути в клапане устанавливают ’С помощью груши 7, таким образом поплавок 6 поддерживает в системе необходимое давление. Если давление превысит заданное, то поплавок 6 опустится вниз и избыток воздуха через штуцер 5 будет сбрасываться в атмосферу до тех пор, пока снова не (установится равновесие сил давления и выталкивания, и клапан (опять закроется. Таким образом в системе поддерживается постоянное давление.
Разделяемую смесь загружают в куб 13, обогреваемый электронагревателем 14. Колонну 12 заполняют необходимой насадкой. Пары, выходящие из колонны, конденсируются в холодильнике 9. (Конденсат в виде капель поступает в воронку 11, снабженную небольшим отверстием, через которое часть конденсата — фле-Гма — стекает назад в колонну. Дистиллят, протекая по капилляру 10 с электронагревательной обмоткой, вновь испаряется, Тйатем вторично конденсируется в холодильнике 8 и по трубе 19 втекает в клапан 16, идентичный клапану 1. Поднятием и опусканием груши 15 клапан 16 открывают или закрывают. При этом часть дистиллята собирается в слое, расположенном над )тутью в клапане 16, а остальная его часть постоянно перетекает . Цо трубе 18. Если закрыть клапан 16 и открыть кран 20, то давле-йе под клапаном 16 уменьшится и отобранная низкокипящая фракция испарится. Ее конденсируют в приемнике дистиллята 17, Охлаждаемом жидким воздухом. Таким образом отбирают пробы,. ie изменяя давления в установке.
При соответствующих мерах предосторожности обычные стек-|1янные аппараты можно применять в ректификационных уста-овках, работающих под давлением до 2 кгс/см2 *. В этих случаях все шлифовые соединения должны быть укреплены прочими стягивающими пружинами или, еще лучше, кольцевыми айбами. После внесения соответствующих изменений обычные >аны могут быть использованы для работы под давлением ЙИс. 217).
* В этих случаях необходимо выполнять все требования техники безопас-' ОсТи, предъявляемые при работе со стеклянной аппаратурой под вакуумом едохранитедьные ксжухи, защитные очки и пр.). — Прим-, ред........
6
Селективные методы перегонки
К селективным методам перегонки относятся такие методы дистилляции и ректификации, разделяющая способность которых обеспечивается или дополнительно повышается при добавлении в исходную смесь какого-либо вещества в газообразном, жидком или твердом состоянии. Сюда же относят и методы, связанные с образованием новых веществ в результате каких-либо реакций, протекающих в процессе перегонки. К селективным методам можно также причислить комбинированные методы, дополняющие перегонку другими методами разделения, например газовой хроматографией.
Согласно правилу фаз Гиббса, двухфазная система, состоящая из двух компонентов с неограниченной взаимной растворимостью, может быть обогащена ректификацией в отличие от трехфазной системы, содержащей два взаимно нерастворимых компонента (см. рис. 29 а—г). С другой стороны, известно, что трехкомпонентная система с ограниченной взаимной растворимостью компонентов, т. е. система с двумя жидкими фазами и одной паровой фазой, может быть разделена ректификацией. Типичный пример такого процесса разделения — получение абсолютного спирта азеотропной ректификацией с бензолом.
6.1. ПЕРЕГОНКА В ПАРАХ НОСИТЕЛЯ
Наиболее известным примером метода перегонки в парах носителя является перегонка высококипящих веществе водяным паром. Носитель вводят в паровую фазу, во-первых, чтобы извлечь из нелетучего кубового остатка более летучие компоненты смеси, и, во-вторых, чтобы снизить температуру кипения и смягчить термический режим перегонки. При этом достигаются условия, близкие к тем, которые имеют место при вакуумной перегонке. Давление паров летучего вещества рх меньше, чем общее давление системы р. Разность р — рг соответствует парциальному давлению водяного пара р2. При вакуумной перегонке по мере уменьшения содержания легколетучего компонента в кубе необходимо либо увеличить температуру, либо повышать вакуум. При перегонке с водяным паром требуется лишь постепенно увеличивать расход водяного пара [2].
Перегонку с водяным паром применяют, главным образом, для очистки и разделения нераствормых или мало растворимых в воде 294
J веществ, например, эфирных масел, жирных кислот, спиртов жирного ряда, анилина, таллового масла и восков, а также бензинов и фракций смол различного состава. Водяной пар вытесняет из установки воздух и предохраняет тем самым загруженный материал от окислительных процессов. Перегонка с водяным паром
дает хорошие результаты при разделении смазочных масел и при отгонке веществ из сильных оснований в препаративных целях.
Как показано в разд. 4.3 и 4.5, при перегонке с водяным паром оба компонента и их пары ведут себя так, как если бы при той же температуре присутствовал только один компонент. Общее давление над кипящей смесью складывается из парциальных давлений ’t обоих компонентов, один из которых вода. Парциальные давления и температуру кипения смеси можно определить графически с по-; мощью суммирующей формулы (39), как описано в разд. 4.5. ‘ Целесообразно использовать диаграмму Бэджера—Кэба [2], на
которую нанесены кривые давления паров перегоняемых веществ (рис. 218). Кривые, соответствующие давлению паров воды для общего давления 760, 300 и 70 мм рт. ст., показывают разность между общим давлением над смесью и парциальным давлением водяного пара. Например, давление паров воды при 60,1 °C равно 150 мм рт. ст., тогда ордината точки кривой А с абсциссой 60,1 °C
800
100
600
В 500 Cj
в ^600
, 300
200
100
О 20 60 60 80 100 120 1<Ю 160 180 200
t,°C
'Рис. 218.
(^Диаграмма Бэджера—Мак-Кэба для определения температуры кнпення н Парциальных давлений при перегонке с водяным паром;
f —• простой эфир; 2 — сероуглерод; 3 — ацетон; 4 — метанол; 5 — че-Ыреххлористый углерод; 6 — бензол; 7 — этанол; 8 — муравьиная кислота; 9 — толуол; 10 — уксусная кислота; 11 — терпентин; 12 — фенол; !8 — аннлнн; 14 — крезол; 15 — нитробензол; 16 ~ нитротолуол. А — •Ода прн 760 мм рт. ст.; Б — вода при 300 мм рт, ст.; В — вода при >0 мм рт. ст.
295
Рис. 219.
Взаимосвязь температуры кипения и давления перегонки для насыщенных жирных кислот ряда с не-разветвленной углеродной цепью и четным числом атомов углерода в присутствии водяного пара и без него.
будет равна разности 760—150 = 610 мм рт. ст. Точки пересечения кривой 80 КО 160 200 2W 280 320 360 для ВОдЫ с крИВОЙ ДЭВле-’ ния паров любого компо-
нента соответствуют температуре кипения смеси и парциальному давлению этого компонента. Диаграмму можно построить также и в логарифмических координатах.
Исходя из парциальных давлений, по формуле (35) рассчитывают состав паров над смесью. Соотношение массовых количеств отгоняемого компонента и носителя — водяного пара определяют по формуле
Gx/G2 = рМ(р2Мг) (184)
Например, для смеси толуол — вода получаем:
Gi/Ga =337-92/(423.18)= 4,1 (185)
Это означает, что для выделения 4,1 кг толуола потребуется 1 кг водяного пара. По формуле (35) содержание толуола в парах составит 80,3% (масс.), температура кипения смеси в соответствии с диаграммой на рис. 218 равна 84,4 °C.
Перегонку легко разлагающихся веществ с водяным паром можно проводить также и под вакуумом. Согласно диаграмме, представленной на рис. 218, температура кипения смеси, содержащей анилин (98 °C при 760 мм рт. ст.), снижается до 75 °C при остаточном давлении 300 мм рт. ст. При этом предполагается, что водяного пара подводят ровно столько, сколько нужно для взаимного насыщения компонентов.
Перегонку с водяным паром выгоднее проводить под вакуумом также для выделения веществ с очень низким давлением паров, например, жирных кислот с длинной углеродной цепью, или при разделении смесей, один из компонентов которых содержится в незначительных количествах, например следы растворителя в маслах.
Вопросы применения перегретого водяного пара при перегонке обстоятельно рассмотрены Штаге. Перегретый пар широко используют в промышленности при перегонке смол, минерального масла и жирных кислот, а также при очистке глицерина. На примере гомологического ряда насыщенных жирных кислот С4—С20 с прямой углеродной цепью показано, что при перегонке с насыщенным паром температуры кипения снижаются приблизительно 296
на 160—240 °C, однако при этом разность температур кипения кислот с различным числом углеродных атомов настолько уменьшается, что разделить компоненты уже невозможно (рис. 219). Если же работать при остаточном давлении 10 мм рт. ст. с 10%-ной добавкой перегретого водяного пара, то температура процесса снизится только на 20 °C. При этом сохранятся различия в температурах кипения, а также такие важные преимущества перегонки с водяным паром, как хорошее перемешивание кубовой жидкости и уменьшение опасности ее перегревания [31. Следует указать, что подобные смеси жирных кислот и спиртов сравнительно хорошо разделяются в присутствии водяного пара также методом многоступенчатой парциальной конденсации [4 ].
Водяной пар вследствие низкой молекулярной массы, большой теплоте парообразования и хорошей конденсируемости широко применяют в качестве носителя. Для снижения парциального давления перегоняемой смеси в промышленности используют также и другие газы. Например, при перегонке каменноугольных смол для получения твердого пека в качестве газа—носителя применяют отходящие газы, которые содержат СО2, N2 и водяной пар [6 ].
Не следует опасаться значительного снижения эффективности колонны в результате введения в нее инертного газа 171. Алгер-миссен (81 разработал графический мётод расчета процесса конденсации паров из смеси с газами.
Аппарат для перегонки с водяным паром можно собрать из обычных стандартных деталей. На рис. 220 представлена схема установки для перегонки с насыщенным водяным паром при атмосферном давлении и под вакуумом. Перегонную колбу 1 тщательно изолируют стекловатой или шлаковатой. Колбу рекомендуется обогревать, чтобы исключить конденсацию в ней водяного пара.
Патрубок для ввода водяного пара 2 снабжен 'краном для слива водного конденсата, и он может служить также для подачи другого газа—носителя. На рис. 221 показана установка для ректификации с перегретым водяным паром. Для получения водяного пара применяют металлический куб 1 с во-
Рис. 220.
Установка для перегонки с насыщенным водяным паром:
1 — перегонная колба; 2 — патрубок для ввода водяного пара;
3 — предохранительный клапан;
4 — капельница.
297
Рис. 221.
Установка для ректификации с перегретым водяным паром:
1 — металлический куб для" получения водяного пара; 2 — металлический змеевик для перегрева водяного пара; 3 — предохранительный клапан.
Рис. 222.
Прибор Поццн—Эскота для дистилляции с водяным паром малых количеств вещества.
домерной стеклянной трубкой. Пар перегревается в металлическом змеевике 2, размещенном в коническом корпусе, за которым установлен брызгоуловитель с термометром. Целесообразно в обеих описанных установках предусмотреть предохранительный клапан 3.
Для получения перегретого пара очень удобен пароперегреватель конструкции Тропша [7]. Для получения воспроизводимых результатов при перегонке в колонну необходимо подавать постоянные количества пара. Достаточно равномерного дозирования пара можно достигнуть, если над колбой с водой установить градуированный цилиндр с капельницей 4 (см. рис. 220), с помощью которой регулируют подачу воды при ее постоянном уровне в колбе. Более надежным является приспособление, описанное Меркелем [9 ], в котором количество пара регулируется по перепаду давления, контролируемого с помощью манометра. Штаге с сотр. [10] разработал устройство, обеспечивающее точную дозировку пара за счет того, что вода из измерительной бюретки непрерывно подается в генератор водяного пара, который почти наполовину засыпан крупным песком для интенсификации теплопередачи.
Для перегонки малых количеств вещества удобен прибор Поцци—Эскота [11], в котором колба для получения водяного пара служит одновременно баней для перегонной колбы (рис. 222). Для микродистилляции пригоден прибор Парнасса и Вагнера [12]. 298
| Оборы методов перегонки с водяным паром можно найти I у Бернхауэра и Торманна [14]. Вебер [15] подробно рассматри-I вает теорию и практику процессов перегонки с водяным паром на I примере ректификации эфирных мас1ел. Ригамонти [16] разра-Е ботал номограмму для расчета расхода водяного пара при задан-I ной степени обогащения.
I 6.2. АЗЕОТРОПНАЯ И ЭКСТРАКТИВНАЯ
I ПЕРЕГОНКА
I Методы азеотропной и экстрактивной перегонки широко приме-I няют в промышленности в тех случаях, когда обычное ректифика-I ционное разделение связано со значительными трудностями. В ла-I бораториях же для разделения близкокипящих компонентов не-L идеальных смесей без азеотропа, а также азеотропных смесей I обычно используют другие методы, например химические методы, | экстракцию или хроматографию. Ниже показано, что для разделе-I ния указанных выше смесей селективные методы перегонки, та-г кие как азеотропная и экстрактивная ректификация, имеют су-I щественные преимущества. Общий отличительный признак этих I обоих методов перегонки заключается в том, что они основаны на I влиянии специально подобранного вещества на отношение коэф-I фициентов активности разделяемых компонентов [17]. Кюммерле К [18] показал, что возможна также комбинация обоих методов — I азеотропноэкстрактивная ректификация. Герстер [19] сравнил | эти методы и обычную ректификацию с экономической точки В зрения.
t Мюллер [19а] сопоставил между собой экстракцию и экстрак-| тивную перегонку применительно к выделению ароматических к соединений из продуктов пиролиза бензина и риформинга. Он fc показал, в каких случаях экстракция и экстрактивная ректи- фикация имеют определенные преимущества одна перед другой, К и привел экономические расчеты. Вопросы азеотропной и экстрак-Ц тивной перегонки подробно рассмотрены в монографии Хофф-К мана, которая содержит многочисленные примеры расчетов для В бинарных, тройных и многокомпонентных систем.
Ц Неидеальные смеси, не образующие азеотропа, характери-Ц зуются тем, что их кривая равновесия асимптотически прибли-Ц жается к диагонали в верхнем или нижнем углу диаграммы Ц (например, см. рис. 29, е и з). Разделение подобных смесей требует К значительного числа теоретических ступеней разделения даже при К сравнительно большой разнице в температурах кипения компо-Е центов. Неидеальные смеси с одной азеотропной точкой (их кратко Е называют азеотропными) при ректификации образуют (см. разд. Е 4.6.2), либо азеотроп с минимальной температурой кипения Е (положительный азеотроп), либо азеотроп с максимальной темпе-к ратурой кипения (отрицательный азеотроп).
299
Если азеотроп положительный, то в головном продукте (дистилляте) получают смесь, обоих компонентов, а в кубовом остатке перегонки содержится чистый компонент. Если азеотроп отрицательный, то дистиллят представляет собой чистый компонент, а кубовый остаток — смесь обоих компонентов. Какой из компонентов получится в чистом виде, зависит от состава исходной смеси. После получения азеотропного состава дальнейшее обогащение смеси легколетучим компонентом не может быть достигнуто методом обычной ректификации. Положительные азеотропы (см. пример на рис. 43, тип 3, ряд III) встречаются значительно чаще, чем отрицательные азеотропы (см. рис. 43, тип 5, ряд III). По таблицам Леката [20], которые содержат сведения о 6287 азеотропных и 7003 неазеотропных смесях, количественное соотношение между положительными и отрицательными азеотропами примерно составляет 9:1.
Хорслей [51] приводит данные по азеотропам, опубликованные до 1962 г. В справочнике по азеотропным смесям Когана с сотр. [21 ] содержатся сведения о 21069 смесях, в том числе о 19735 бинарных, 1274 тройных и 60 многокомпонентных смесях. В справочнике наряду с таблицами также дано введение в теоретические основы и технику экспериментального исследования явления азеотропии. Здесь же приводятся кроме того сведения о влиянии температуры на состав азеотропных смесей, о методах расчета азеотропного состава и о методах исследования свойств азеотропных смесей.
Вопросы теории азеотропных смесей рассмотрены с термодинамической и молекулярно-кинетической точек зрения в ряде работ [22], [23], [27] и [28]. Эти работы содержат сведения о тройных и четверных азеотропах. В работе Отмера [28а] приводится подробный обзор достижений в области теории и практического использования азеотропной ректификации.
В первом томе своей фундаментальной монографии Светослав-ский [27] излагает теорию азеотропии, обсуждает методику экспериментального исследования этого явления, приводит классификацию разновидностей азеотропов, а также анализирует возможности использования явления азеотропии и полиазеотро-пии в промышленности. Во втором томе монографии явление азеотропии рассмотрено с термодинамической точки зрения.
Превосходное введение в азеотропную и экстрактивную ректификацию в лабораторных условиях как с теоретической, так и с практической точки зрения дает Рёк [17]. Азеотропии бинарных систем обсуждается в первом томе книги Шуберта [29] по термодинамике растворов.
При перегонке смесей близкокипящих компонентов обычно следует считаться с возможностью образования азеотропа. Азеотропии проявляется не только при наличии специфического взаимодействия между молекулами, но также наблюдается у смесей неполярных веществ, например бензол—циклогексан или бен-300
зол—четыреххлористый углерод. В. Кун и Г. Дж. Кун [24] объясняют распространенность азеотропии у смесей близкокипящих компонентом тем, что в таких смесях эффекты, основанные на различии в давлениях паров чистых компонентов, компенсируются, а тепловые эффекты смешения компонентов суммируются. Циборак [30] показал, что при добавлении в смесь этанол—вода углеводородов в процессе для обезвоживания образуется ряд четырехкомпонентных гетероазеотропов и трех компонентных гомоазеотропов. Орзаг [31 ] исследовал различные полиазеотропные жидкие смеси и влияние образовавшихся азеотропов на процесс перегонки. Малесинский [32] и Штеки [33] дают классификацию гомо- и гетероазеотропных систем. В обзоре, посвященном выбору добавок для азеотропной и экстрактивной перегонки, Берг [34 ] рассматривает классификацию жидкостей по водородным связям их молекул.
Разумеется, для разделения близкокипящих компонентов и неидеальных смесей, не образующих азеотропа, можно подобрать ректификационные колонны эффективностью в 100 и более теоретических ступеней разделения, поскольку насадка с ВЭТС, равной 1—2 см, сейчас не является уже редкостью. Однако вместо применения колонн с 200 или даже 300 теоретическими ступенями разделения (относительная летучесть а = 1,03 — 1,02) такие смеси можно разделить, если воздействовать на фазовое равновесие в направлении повышения значений а и достижения более благоприятных условий разделения. В качестве примера рассмотрим экстрактивную ректификацию смеси близкокипящих компонентов w-гептан — метилциклогексан, для которых разность температур кипения составляет 2,7 °C (а = 1,075). При обычной ректификации с бесконечным флегмовым числом требуется 48 теоретических ступеней, чтобы сконцентрировать смесь от 15,3 до 95,4% (мол.). Если же в смесь добавить 70% (масс.) анилина, то такого же обогащения можно достигнуть при числе теоретических ступеней 12,4 и флегмовом числе v = 35. При этом относительная летучесть возрастает с 1,07 до 1,30 [35]. Если смесь является азеотропной, то чистые компоненты можно получить только с помощью селективного метода разделения.
С одной стороны, явление азеотропии крайне нежелательно, например при концентрировании смеси этанол—вода и других азеотропных смесей. С другой стороны, это явление дает возможность разрушить уже имеющийся азеотроп и разделить смесь весьма близкокипящих компонентов. Введением дополнительного компонента * сознательно вызывают образование азеотропа между этим компонентом и одним из компонентов бинарной смеси * ** и легко отделяют новый азеотроп от второго компонента благо-
* В советской литературе добавочный компонент называют разделяющик
агентом. — Прим. ред.
** При азеотропной перегонке. — Прим. ред.
301
Рис. 223.
Схема установки для азеотропной ректификации а периодическом режиме (а) и в непрерывном режиме (<Г):
1 — гетероазеотроп; 2 — разделяющий агент (возврат на орошение); 3 — компонент А; 4 — в начале перегонки исходная смесь разделяющий агент; в конце — компонент В; 5 — исходная смесь -|- разделяющий агент; 6 — компонент В.
даря увеличившейся разнице в температурах кипения. Конечно, необходимо условие, чтобы азеотропный конденсат легко расслаивался на составные части. Это достигается охлаждением, высаливанием, химическим выделением разделяющего агента, экстракцией выделяемого компонента или также повторной азеотропной перегонкой.
302
Если после полной конденсации паров азеотропного состава образующийся дистиллят не расслаивается, то говорят о гомогенном азеотропе (например, смесь этанол—вода), в то время как азеотроп, расслаивающийся в условиях конденсации на две фазы, называют гетерогенным (например, смесь бензол—вода).
При экстрактивной перегонке благодаря введению в исходную жидкость смесь высококипящего разделяющего агента с более высокой (на 50—100 °C) температурой кипения добиваются того, чтобы разделение вообще стало возможным (если в исходной
Рис. 224.
Схема установки для экстрактивной ректификации в периодическом режиме (а) и в непрерывном режиме (£):
1 — дистиллят А н В; 2 — разделяющий агент (ввод); 3 — экстракционная зона; 4 — в начале перегонки исходная смесь, в конце — разделяющий агент; 5 — дистиллят А; 6 — исходная смесь (ввод); 7 — дистиллят В; 8 — разделяющий агент (вывод).
303
смеси разность равновесных концентраций Ах между паром и жидкостью равна 0) или стало более легким за счет увеличения Ах [17].
Разделяющий агент не должен образовывать азеотропа с компонентами разделяемой смеси, должен легко отделяться от разделяемой смеси и специфически воздействовать на один из компонентов, вызывая тем самым увеличение относительной летучести компонентов. Обычно используют те же разделяющие агенты, которые применяют при жидкостной экстракции, либо подобные им вещества. Принцип азеотропной и экстрактивной ректификации в периодическом и непрерывном режимах пояснен на рис. 223 и 224.
6.2.1. АЗЕОТРОПНАЯ ПЕРЕГОНКА
Азеотропную перегонку применяют для разделения смесей близкокипящих компонентов, которые в большинстве случаев уже сконцентрированы обычной ректификацией, и для разделения азеотропных смесей, которые близки по составу к азеотропу.
Как правило, с помощью подобранного разделяющего агента добиваются образования с одним из разделяемых компонентов положительного азеотропа. Однако возможно также, что разделяющий агент образует двойные или даже тройные положительные азеотропы с обоими разделяемыми компонентами. Конечно, при этом необходимо, чтобы состав новых азеотропов отличался от состава исходной смеси.
Характерным примером разделения смесей близкокипящих компонентов азеотропной ректификацией является разделение смеси индол—дифенил с применением диэтиленгликоля в качестве разделяющего агента. При атмосферном давлении разность температур кипения указанных веществ составляет всего 0,6 °C. Благодаря добавке диэтиленгликоля разница в температурах кипения образовавшихся азеотропов достигает уже 12,2 °C. Кипящие соответственно при 230,4 и 242,6 °C азеотропные смеси дифенил—диэтиленгликоль и индол—диэтиленгликоль, которые содержат почти по 60% диэтиленгликоля, можно легко разделить, даже используя малоэффективные колонны, при небольшом флегмовом числе. Так как дифенил в отличие от индола мало растворим в диэтиленгликоле, то больших количеств гликоля не требуется. Отгоняемый в первую очередь азеотроп дифенил—диэтиленгликоль расслаивается в приемнике дистиллята, и гликоль непрерывным потоком возвращают на стадию ректификации. Из полностью отогнанной смеси индол—диэтиленгликоль индол осаждают, разбавляя смесь водой [361.
Имеющийся в продаже Р-пиколин представляет собой смесь а-пиколина, Р-пиколина и 2,6-лутидина. Разница в температурах кипения Р-пиколина и 2,6-лутидина при атмосферном давлении составляет 0,15 °C. Методом азеотропной ректификации с до-304
бавлением уксусной или пропионовой кислоты удается сконцег трировать отдельные основания до 95—98% [37]. Другие при ложения азеотропной ректификации в промышленности описан] Думметом [38]. Следует коротко остановиться на некоторы: примерах, представляющих интерес для промышленности.
Согласно Хунсманну и Суммроку [39] при разделении трои ной смеси вода—муравьиная кислота—уксусная кислота следуе ожидать образования бинарного (В) высококипящего азеотроп. 4ип = 107,65 °C, состоящего из 56,7% (мол.) муравьиной кислоте и 43,3% воды и тройного (Т) азеотропа (107,1 °C) состоящей из 39,3% (мол.) воды, 48,2% муравьиной кислоты и 12,5% уксус ной кислоты. Весь интервал концентраций трехкомпонентно! смеси можно разделить на четыре отдельных области перегонк! (рис. 225). Смесь обезвоживают азеотропной перегонкой с однш из высших эфиров.
Закономерности ректификации трехкомпонентных смесей с азео тропными точками были изучены Петликом и Аветьяном [39а ] Лино с сотр. [40] методом азеотропной ректификации с добавкоЁ бромистого этилена удалось удалить из ацетона следы воды вплоп до концентрации менее 1400 рргп.
Обзор всех известных приемов азеотропной перегонки был бь слишком громоздким. Техническая литература, в том числе е патентная, по данному вопросу исключительно обширна. Уже приведенные примеры показывают, насколько велики возможности этого метода перегонки. Поэтому целесообразно указать лишь классы веществ, которые особенно выгодно разделять азеотропной перегонкой. Азеотропную перегонку широко применяют для обезвоживания органических веществ, таких как муравьиная
кислота, уксусная кислота и пиридин, а также для выделения угле-
водородов из спиртов, очистки ароматических углеводородов, разделения моно- и диолефинов и т. д. Мэйр, Глазгов и Россини [41, 42], как и Берг [34], провели систематическое исследование процесса разделения углеводородов азеотропной ректификацией. Разделение изомеров метилнаф-
талина в присутствии ундеканола показывает, что при азеотропной перегонке имеет значение также правильный выбор рабочего давления. Разделение происходит наилучшим образом при 200— 300 мм рт. ст. В кубовом остатке
Муравьиная кислота
Рис. 225.
Схематическое изображение четырех областей перегонки трехкомпонентной смеси вода—муравьиная кислота—уксусная кислота с,б (парным (В) и тройным <Т> азеотропом 139].
R — состав исходной смеси.
305
Рис. 226.
Зависимость состава азеотропа от давления для смесей этанол—вода (/) и вода—фенол (2).
Рис. 227.
Графический метод Наттинга—Хорсли для определения интервала давлений, для которых существует азеотроп.
/ — положительный азеотроп; II — отрицательный азеотроп; Л, В — чистые вещества; С — бинарный азеотроп.
получают чистый а-метилнафталин, а в дистилляте — смесь, содержащую 80% р-метилнафталина [43].
Если азеотроп не стабилен, то его можно исключить, изменяя внешние термические условия (температура или давление) путем проведения перегонки под вакуумом или при избыточном давлении, не прибегая к азеотропной или экстрактивной перегонке. Для гомоазеотропных двухкомпонентных систем Шуберт [44] приводит приближенные зависимости, по которым можно ориентировочно рассчитать условия, необходимые для устранения азео-тропии за счет изменения давления, температуры или путем подбора определенной минимальной концентрации разделяющего агента (применительно к азеотропной или экстрактивной перегонке).
В общем случае понижение давления сказывается таким образом, что азеотропная смесь обогащается легкокипящим компонентом. Во многих случаях в конце концов при некотором вакууме, азеотропный состав исчезает. В качестве примера можно указать на разделение смесей этанол—вода и вода—фенол (рис. 226). Вакуумной перегонкой при 70 мм рт. ст. получают абсолютный спирт и без разделяющего агента. Азеотропная точка на кривой равновесия смеси вода—фенол исчезает при остаточном давлении 32 мм рт. ст. Шнайнкер и Пересслени [45] установили, что азеотропная точка смещается следующим образом: азеотроп смеси муравьиная кислота—вода при 55 мм рт. ст. содержит 66% (масс.) муравьиной кислоты, а при 200 мм рт. ст. — 72% (масс.) кислоты; азеотроп смеси бутанол—бутилацетат при 50 мм рт. ст. содержит 37% (мол.) бутанола, а при 760 мм рт. ст. — 79% (мол.) спирта. 306 **
Рютер и Лю [46] разработали методы расчета азеотропного состава, которые были экспериментально проверены на системах этилацетат—этанол, этанол—вода, метанол—бензол.
Можно привести примеры смесей, для которых применение избыточного давления перегонки также оказывает такое влияние на фазовое равновесие, что азеотропная точка на кривой равновесия исчезает.
В соответствии с графическим методом Нуттинга и Хорслея [47 ] можно очень просто определить интервал давлений, в котором еще существует азеотроп. На рис. 227 показаны кривые давления насыщенных паров чистых исходных компонентов и азеотропа, нанесенные на диаграмму Кокса в координатах 1g р — 1/(Т + 230). Вследствие прямолинейности зависимости такого рода для ее построения нужны только две точки. При давлениях вне интервала, ограниченного ординатами точек пересечения Р и Р' прямой для азеотропа с прямыми для чистых веществ, азеотроп уже не существует. Если указанные прямые не пересекаются, то это означает, что азеотроп сохраняется при всех давлениях. Согласно методике Иоффе [48 ] достаточно знать состав азеотропа при какой-либо одной температуре (давлении), чтобы вычислить состав азеотропа при других температурах (давлениях) методами экстраполяции или интерполяции. Малесинский [49] предлагает зависимости, по которым можно рассчитывать температуры кипения тройных азеотропов.
Если нельзя достигнуть эффективного разделения исходной смеси за счет изменения рабочего давления перегонки, то подбирают подходящее вещество, которое образует с одним из компонентов смеси гетеро- или гомоазеотроп, легко разделяемый на составные части [34].
Рассмотрим следующий пример. Смесь вода—пиридин образует при 92 °C положительный азеотроп, содержащий 54% (масс.) пиридина. Воду можно удалить в виде азеотропа, подобрав подходящий разделяющий агент (выноситель). К такому разделяющему агенту предъявляются следующие требования [34, 50]. Он должен образовывать с водой положительный азеотроп с температурой кипения ниже 92 °C и не должен образовывать азеотропа с пиридином, а также тройного азеотропа. При этом он должен связывать в виде азеотропа возможно большее количество воды и не должен практически растворяться в воде (чтобы обеспечить разделение гетероазеотропа при охлаждении).
Замечено, что положительный азеотроп образуется, когда силы притяжения между неодинаковыми молекулами меньше, чем между одинаковыми. В противном случае образуется отрицательный азеотроп. Обработав имеющиеся данные по азеотропам, собранные Легатом [20] и Хорсли [51], Эвелл, Харрисон и Берг [52 ] разработали теорию, согласно которой под силами притяжения, обуславливающими образование азеотропа, следует иметь в виду водородные связи, которые значительно превосходят
307
Таблица 41.
Классификация жидкостей по водородным связям
А. Классы по Бергу [34, 52]
Классы Тип молекул Примеры
I Молекулы, способные образовывать трехмерную структуру Н2О, многоатомные спирты, аминоспирты, оксикислоты, полифенолы, гидроксиламин
II Молекулы, содержащие как активные атомы Н, так и электроотрицательные атомы со свободными парами электронов (О, N, F) Спирты, кислоты, фенолы, первичные и вторичные амины, оксимы, нитросоединения и нитрилы с атомами Н ва-положении, аммиак, гидразин, фтористый и цианистый водо-
III Молекулы с электроотрица- род Простые и сложные эфиры, кетоны,
тельными атомами, но без активных атомов Н альдегиды, третичные амины, пиридин, нитросоединения и нитрилы, не имеющие атомов Н ва-положении
IV Молекулы с активными атомами Н, но без электроотрицательных атомов СНС13, СН2С12, СН3СНС12, СН2С1СН2С1, СН2С1СНС12 и т. д.
V Молекулы, не способные образовывать водородные связи Углеводороды, CS2> СС14, сульфиды, меркаптаны, металлоиды
Б. Смеси жидкостей с ожидаемыми отклонениями от закона Рауля
Классы Характер отклонения Водородные связи
1+ V | и+V/ Всегда положительное; смеси I + + V часто характеризуются ограниченной взаимной растворимостью компонентов Ослабляются
III + IV 1+ IV 11+ IV Всегда отрицательное Всегда положительное; смеси I + IV характеризуются ограниченной взаимной растворимостью компонентов Образуются Частично ослабляются, частично образуются вновь; первый эффект преобладает
1+ I I + II I + III 11+ II II + III Обычно положительное, но возможно и отрицательное; иногда образуется отрицательный азеотроп Частично ослабляются, частично образуются вновь
III + III III + V IV + IV IV + V v+ V Квазиидеальные системы; отсутствие отклонения или слабое положительное отклонение; изредка образуется положительный азеотроп Отсутствуют
308
другие межмолекулярные силы, например дипольные или индукционные. На основе этого представления все жидкости можно разделить на пять классов, различающихся по числу и прочности водородных связей (табл. 41).
Если оба компонента данной разделяемой смеси отнести по признаку водородной связи к соответствующим классам, то по данным табл. 41Б можно оценить предполагаемые отклонения каждого компонента от представителей другого класса и таким образом подобрать подходящий разделяющий агент. В табл. 42 указаны компоненты, которые образуют отрицательный азеотроп (см. [29]).
В табл. 43 указаны возможные разделяющие агенты для смеси вода—пиридин. Вода (класс I) проявляет сильные отклонения от закона Рауля в смесях с жидкостями классов III, IV и V, в то время как для пиридина (класс III) в смесях с теми же веществами характерны слабые положительные отклонения или вообще их отсутствие.
Из данных табл. 43 видно, что точка кипения вновь образовавшегося азеотропа тем ниже, чем меньше температура кипения чистого разделяющего агента. С учетом растворимости и содержания воды в азеотропном конденсате подбирают оптимальный разделяющий агент, который образует азеотроп с минимальной температурой кипения, которая не должна превышать 92 °C.
Берг и сотр. [34, 52] изучили ряд смесей близкокипящих углеводородов и подобрали для их азеотропной перегонки опти-Таблица 42.
Вещества, образующие отрицательные азеотропы
Компоненты
Примеры
Вода I
Сильные кислоты J
Вода |
Ассоциированные жидкости J
Жидкости класса III )
Неассоциированные жидкости J
Органические кислоты 1
Амины /
Фенолы 1
Амины J
Органические кислоты
Кислородсодержащие соединения
класса III
Фенолы
Кислородсодержащие соединения
класса III
Фенолы
Спирты
Вода с HCI, НВг, HI, HNO3
Вода с муравьиногГкислотой, с этилендиамином
Ацетон—хлороформ, циклогексанон —-бромоформ, бутилацетат — 1,2,2-три-хлорпропан
Уксусная кислота — триэтиламин, пропионовая кислота — пиридин
Фенол — анилин, о-крезол —диметил-анилин
Муравьиная кислота — диэтилкетон, масляная кислота — циклогексанон
Фенол — метилгексилкетон, о-крезол— этилоксалат
Фенол — н-октиловый спирт, о-крезол — гликоль
309
Таблица 43.
Разделяющие агенты для обезвоживания пиридина азеотропной ректификацией
Класс Разделяющий агент Темп, кип. при 760 мм рт. ст.» °C Темп, кип. азеотропа с водой, °C Количество разделяющего агента на 1 масс. ч. удаляемой воды, масс ч. Растворимость в 100 масс. ч. воды, масс. ч.
III Изобутилформиат 98,2 80,4 11,8 1,0
Этилпропионат 99,0 81,2 9,0 2,4
Метилбутират 102,0 82,7 7,7 1,5
Этилизобутират 111,7 85,2 5,6 Незначительная
Диизобутиловый эфир 122,2 88,6 3,4 То же
Амилформиат 132,0 91,6 2,5 То же
IV • Дихлорэтан 83,0 72,0 11,0 0,9
1,2-Дихлорпропан 96,8 78,0 7,4 0,3
V Бензол 80,2 69,3 10,2 0,1
Толуол 110,7 84,1 6,4 Не растворяется
Таблица 44.
Оптимальные разделяющие агенты для азеотропной перегонки углеводородов
Разделяемая смесь углеводородов Относительная летучесть без азеотропа а Разделяющий агент Изменения относительной летучести
Неогексан—циклопентан 1,006 н-Пропиламин 0,987
Метилциклогексан—изооктан 1,046 Этанол или 1,4-диоксан 1,084
З-Метилпентан—гексен-1 1,009 Метиленхлорид 1,159
З-Метилпентан-2-этил бутен-1 1,037 Этилформиат 1,156
2-Этилбутен-1-н-гексан 1,056 Хлороформ 1,094
2,2,4-Триметилпентан-2,2,4- 1,040 Изопропилацетат 1,129
тр иметил пентен-1
н-Гептан-2,4,4-триметилпентен-1 1,045 Изопропилацетат 1,129
Этилбензол-п-ксилол 1,035 2-Метилбутанол 1,079
Ксилол-.и-ксилол 1,020 2-Метилбутанол 1,029
Ксилол-о-ксилол 1,105 Метилизобутилкарбинол 1,150
мальные разделяющие агенты (табл. 44). Как видно из данных табл. 44, при введении разделяющего агента относительная летучесть компонентов а резко увеличивается.
Для экстрактивной перегонки также весьма важен вопрос о том, образует ли разделяющий агент азеотроп с компонентами или нет. При экстрактивной перегонке разделяющий агент не должен давать азеотропа ни с одним из компонентов разделяемой
310
смеси. Поэтому рассмотрим еще несколько эмпирических методов подбора разделяющих агентов, важных как для азеотропной, так и для экстрактивной перегонки.
Мэйр, Глазгов и Россини [41 ] предлагают простой графический метод определения параметров азеотропа. Рис. 228 представляет температуры кипения азеотропных смесей бензола с различными углеводородами как функцию состава смеси [53]. Если соединить точку кипения каждого углеводорода (на оси ординат) с точкой, взятой на кривой и соответствующей температуре кипе
ния азеотропа, то можно получить прямые линии с различным наклоном. На основе диаграммы можно предсказать, что углеводороды с точками кипения ниже 68 °C и выше 100 °C не дают азеотропа с бензолом. Далее, для любого углеводорода ‘можно определить температуру кипения и состав его азеотропа с бензолом, если от значения температуры кипения чистого вещества провести линию, параллельную ближайшей уже построенной наклонной прямой. Наклон прямой зависит от температуры кипе-
ния чистого углеводорода и степени разветвленности его молекулы. Если, например, на диаграмме отметить точку кипения 3-этилпентана (93,5 °C), то на кривой линии найдем в хорошем
соответствии с опытными данными точку кипения азеотропа при 80 °C, а на оси абсцисс — состав азеотропа, равный 96% (мол.) бензола. Подобные диаграммы можно строить по методу Хорсли [54], а также Мейсснера и Гринфельда [55]. Графический метод, предложенный Новиковой и Наградзе [56], основан на исполь
зовании трехмерной координатной системы, изменение параметров в которой выражается пространственной кривой. На оси абсцисс
откладывают значения состава азеотропа в % (мол.), на оси орди-
нат — значение, обратное
температуре кипения, а на аппликате (третьей оси) — логарифм давления. Если известна характеристика азеотропа при двух различных давлениях, то для бинарных азеотропных смесей можно также точно определить: характеристику азеотропа при любом давлении, давление и концентрации, при котором может появиться азеотропная смесь.
Литвинов [26] разработал графический метод для вычисления
Рис. 228.
Графический метод Мэйра, Глазгова и Россини определения параметров азеотропных смесей бензола с углеводородами:
1 — 2,2,4-триметилпентаном; 2 — н-гептаном;
3 — 2,3-диметилпеитаиом; 4 — 2,2,3-триме-тилбутаном; 5 — 2,4-диметилпентаиом; 6 — циклогексаном; 7 — метилциклопентаном; 8 —
н-гексаном.
311
Рис. 229.
Графический гметод Смольника для определения параметров азеотропных смесей бензола с углеводородами:
1 — 2,2,4’Триметилпентаиом; 2 — 2,3-диметил-пентаном; 3 — 2,4-диметилпеитаиом; 4 — 2,2,3-триметилбутаиом; кривая I — соответствует формуле (186), кривая II — формуле (187).
вероятности появления азеотропа в тройных смесях. Показано, что тройной азеотроп может также обра-
зоваться в системе, в которой отсутствуют бинарные азеотропы. По ^методу Скольника [57 ] возможно также чисто расчетное определение параметров азеотропа. Для членов одного и того же гомологического ряда (например, углеводородов) и какого-либо второго компонента (например, бензола) имеют силу следующие линейные зависимости:
1g хаз = А (273,1 + /аз) + В (186)
где хаз — содержание второго компонента в азеотропной смеси, % (мол), ta3 — температура кипения азеотропа, °C, А и В — константы, и
lg(P-fa3) =£-Л273,1 +/) (187)
где D, Е и F — константы; t — температура кипения рассматриваемого члена гомологического ряда.
На основе зависимостей (186)"и?( 187) можно получить кривые, которые представлены на рис. 229.
Г Непосредственно по кривым могут быть определены параметры азеотропов для углеводородов одного гомологического ряда. Например, 2,2-диметилпентан кипит при 79,2 °C. По кривой 2 находим, что состав азеотропа составляет 50% (мол.), а по кривой 1 определяем температуру кипения' азеотропа с бензолом (75,8 °C). Кривая 2 служит также для определения интервала температур кипения, в котором азеотроп с бензолом еще возможен. Получающийся^ при этом интервал 65—98 °C хорошо согласуется с результатами определения по графическому методу Мэйра, Глазгова и Россини. Эти температурные интервалы кипения для парафинов, дающих с бензолом гомоазеотропы, называют «азеотропным эффектом» бензола [57 ]. По данным инфракрасной спектроскопии, Наканиси и сотр. [58] разработали диаграмму, с помощью которой для бинарных смесей с метанолом можно выяснить, образуется или нет азеотроп с определенным компонентом данной смеси при заданной температуре в интервале 30—150 °C.
Брандани [58а ] сообщает о применении предельных коэффициентов активности для определения состава азеотропа при постоянной температуре и частичной взаимной растворимости компонентов в бинарных жидких смесях.
312
Расчет условий азеотропной перегонки, необходимых для разделения, например, числа теоретических ступеней разделения и флегмового числа проводят в обычном порядке, как описано в гл. 4.
Для расчета числа теоретических ступеней разделения при ректификации правильных систем с азеотропным составом, в которых абсолютная теплота смешения мала, Кун [59] приводит следующее уравнение, которое соответствует формуле Фенске (108в) для непрерывной ректификации при v = оо:
ЛМИИ —
1 2хаз -1 бо 2(1 — хаз)
ХЕ -хЕ
(188)
где 60 = а — 1; хаз — концентрация азеотропа.
Это уравнение справедливо при условии, что хЕ < хаз, т. е. когда требуемая концентрация легколетучего компонента в дистилляте меньше, чем в азеотропе. Для хЕ = хаз необходимое число теоретических тарелок составляет пмин = оо.
Шапиро [60] привел формулы для расчета процесса разделения бинарных смесей с малым содержанием менее летучего компонента, а также для расчета почти азеотропных смесей. В качестве примера проведен расчет процессов разделения смесей вода — этиловый эфир уксусной кислоты (1,75%), бензол —толуол (4%) и этанол —вода (11%). На примере разделения смеси 1,3-бута-диена — 1-бутен азеотропной ректификацией с метиламином Хунсманн [61а] указывает последовательность расчета азеотропной колонны. Сначала исследуют фазовое равновесие двух- или трехкомпонентных систем в технически важной области давлений, результат представляют рядом уравнений и проводят последовательный расчет от нижней тарелки к верхней тарелке. Влияние температуры, а также избытка амина подтверждается вычислениями. Результаты расчетов подтверждены опытами для условий ректификации на маленькой колонне; получилось весьма удовлетворительное согласие.
Принцип работы и особенности гетероазеотропной перегонки раскрыты Вольфом и сотр. [61] на примере разделения смеси вода — уксусная кислота.
В заключение рассмотрим процесс абсолютирования спирта методом ректификации с бензолом в качестве разделяющего агента, как пример азеотропной перегонки, при которой образуется тройной азеотроп. К этанолу концентрацией 95% (масс.), находящемуся в кубе добавляют рассчитанное количество бензола. При перегонке в периодическом режиме при 64,9 °C получается тройной азеотроп, который содержит 74,1% (масс.) бензола,
313
18,5% этанола и 7,4% воды. При охлаждении до 28 °C азеотроп расслаивается на две жидкие фазы. Верхний слой, составляющий 84,7% (масс.) от всего количества тройного азеотропа, содержит 85,6% (масс.) бензола, 11,6% этанола и 2,8% воды. Нижний.слой, составляющий 15,3% (масс.) от всего количества азеотропа, содержит 8,1% (масс.) бензола, 53,1% этанола и 40,6% воды.
После удаления воды при 68,3 °C отгоняют бинарный азеотроп, содержащий 32,4% этанола и 67,6% бензола. Наконец, при 78,4 °C начинает отгоняться безводный спирт концентрацией 99,8%. Конечно, можно работать непрерывно, как обычно принято в промышленности, однако дополнительное усложнение аппаратуры в лабораторных условиях не оправдано. Целесообразнее оба слоя образовавшегося тройного азеотропа, а также бинарный азеотроп разделять перегонкой в периодическом режиме.
6.2.2. ЭКСТРАКТИВНАЯ ПЕРЕГОНКА
Экстрактивную перегонку, также как и азеотропную, применяют, если смесь необходимо обогатить до концентрации выше азеотропной или разделить близкокипящие компоненты и неидеальные смеси с относительной летучестью компонентов, близкой к 1. Однако, экстрактивная перегонка благодаря почти неограниченному числу возможных разделяющих агентов имеет намного большее распространение, чем азеотропная.
Количество добавляемого разделяющего агента следует устанавливать по возможности точнее, так как оно оказывает заметное влияние на эффективность разделения.
В качестве примера экстрактивной ректификации смеси близкокипящих компонентов уже было рассмотрено разделение смеси w-гептан-метилциклогексан с применением анилина. Система метилциклогексан—толуол имеет кривую равновесия, которая приближается к диагонали диаграммы равновесия пар—жидкость асимптотически. Чтобы получить чистый метилциклогексан, необходимо практически бесконечно большое число теоретических тарелок. Благодаря добавке 55% (мол.) такого полярного растворителя как анилин разделение сильно упрощается.
Экстрактивная перегонка удобна также и для разделения азеотропных смесей. На азеотропную смесь циклогексан—бензол можно воздействовать тем же разделяющим агентом (анилином) таким образом, что в дистилляте получится чистый циклогексан. Кортюм и Биттель [61 ] сообщили о разделении первичных, вторичных и третичных ароматических аминов с помощью таких разделяющих агентов^ как глицерин и парафиновое масло. Достойна внимания полностью автоматизированная установка этих исследователей для периодической и непрерывной экстрактивной ректификации. Экстрактивная перегонка оправдала себя и при разделении многокомпонентных смесей, содержащих вещества различных классов. Так, экстрактивной перегонкой можно извлечь
314
ароматические вещества из их смесей с неароматическими углеводородами с помощью одного из бутандиолов (например, 2,3-бу-тандиола) с добавкой октилового спирта в качестве растворителя [62].
Представляет также интерес выделение НО из азеотропа соляной кислоты экстрактивной перегонкой в присутствии H2SO4 по методу Гревера [63]. В этом случае для ректификации применяли колонну, изготовленную из графита. На рис. 230 приведена диаграмма равновесного кипения системы НО—H2SO4—Н2О при постоянном давлении.
В качестве разделяющего агента применяют также твердое вещество. Ино и сотр. [94] успешно использовали для удаления из ацетона малых количеств хлористого метилена соль ZnBr2, растворимость которой в ацетоне превышает 50% (масс.). Содержание хлористого метилена в ацетоне удалось снизить до 0,09%. Соль растворяли в ацетоне, применяя куб с мешалкой.
Принцип экстрактивной перегонки состоит в том, что благодаря разделяющему агенту увеличивается разность концентраций Ах между парами и жидкостью при равновесном испарении. Путем добавления третьего вещества можно даже преобразовать положительный азеотроп в отрицательный [63]. Обычно высококипящий разделяющий агент подбирают таким образом, чтобы относительная летучесть компонентов заметно повышалась. Агет вводят непрерывно в верхнюю часть колонны, чтобы его концентрация оставалась постоянной в жидкой фазе по всей высоте колонны.
Рис. 231 иллюстрирует, как изменяется форма кривой равно веси я азеотропной смеси ацетон—хлорофор в зависимости от ко личества добавляемого разделяющего агента (метилизобутилен кетона). Система имеет максимум температуры кипения при 34,5% (мол.) ацетона; этот максимум уже при добавке 30 % (мол.) разде ляющего агента исчезает; дальнейшее добавление агента вызывает
значительное увеличение от носительной летучести ком понентов смеси [50].
Павлов с сотр. [63а] ис следовал экстрактивную пе регонку углеводородов с 4 i 5 атомами углерода с по мощью 19 различных вспо
Рис. 230.
Диаграмма равновесного кипенн; смеси НС1 —H3SO4—Н2О прн 750 mi рт. ст.
— мольная доля H2SO< в жидко фазе. —— линии постоям
ных составов пара; — — —- — изс термы.
31
Рис. 231.
Зависимость формы кривой равновесия для смеси ацетон—хлороформ от количества введенного метнлнзооутил• кетона.
могательных веществ; найденные значения относитель-ной летучести были представлены в виде диаграмм, отражающих влияние добавки высококипящих углеводородов на относительную летучесть.
Разделяющий агент, применяемый для проведения экстрактивной перегонки,
должен отвечать следующим требованиям. Он не должен образовывать азеотропа с компонентами разделяемой смеси, должен легко отделяться от компонентов разделяемой смеси, давать с одним из разделяемых компонентов возможно большее положительное отклонение от закона Рауля, а с другим компонентом — возможно меньшее или даже противоположное (отрицательное) отклонение, причем разделяющий агент должен обеспечивать максимальное увеличение относительной летучести исходных компонентов [64].
При рассмотрении азеотропной перегонки мы уже познакомились с приемами, позволяющими установить для какой-либо смеси вероятность образования азеотропа (см. разд. 6.2.1). Дополнительные возможности в этом направлении представляет методика Шайбеля [65], основанная на использовании коэффициентов активности компонентов разделяемой смеси. Выделение экстрагированного компонента относительно высококипящего разделяющего агента перегонкой не представляет трудностей. Согласно Бергу [34, 52] в соответствии с данными табл. 41 (см. разд. 6.2.1) наиболее подходящими разделяющими агентами для экстрактивной ректификации являются вещества, принадлежащие классам I и II. Это, главным образом, вещества, которые имеют склонность к образованию прочных водородных связей и могут взаимодействовать и как доноры протонсв, и как доноры электронов. Сюда относят фенолы, ароматическе амины (анилин и его производные), высшие спирты, гликоль и т. д.
При исследовании процессов экстрактивной перегонки смесей хлористый аллил хлористый пропил и «-бутан-трц«с-бутен-2 Гарбер и Мироненко [65а ] разработали универсальный метод подбора наиболее подходящего разделяющего агента. Хакенберг и Шмидт предложили метод парсфазной газовой хроматографии для оценки избирательной способности разделяющих агентов. Согласно этому 316
методу определяют отношение площадей хроматографических пиков веществ 1 и 2, распределенных в паровой фазе над некипящими смесями в присутствии и в отсутствие разделяющего агента.
Оценка избирательной способности разделяющего агента требует проведения многочисленных опытов. Целесообразно определять кривые равновесия для образцов разделяемой смеси, в которые добавлены различные количества разделяющего агента. Роуз 135 ] описывает результаты таких опытов для смеси «-гептан—метилциклогексан, в которую добавляли различные разделяющие агенты. Относительную летучесть компонентов в присутствии растворителя обозначают аэ. Чтобы можно было сравнить действие различных разделяющих агентов, относительную летучесть компонентов, равную 1,07 в отсутствие агента, условно приравняем к 1. Тогда полезный эффект, вызванный введением разделяющего агента, будет равен N = аэ/а. Очевидно, введение 92% (мол.) анилина в жидкую фазу оказывает наибольшее влияние на фазовое равновесие. Влияние разделяющего агента на относительную летучесть разделяемых компонентов можно также оценить, исходя из интегральных теплот смешения [50].
Коган [66] показал, что при добавлении третьего компонента к бинарной смеси увеличивается относительная летучесть того компонента смеси, в котором менее всего растворяется третий компонент. Другие приемы подбора разделяющего агента рассмотрены Кафаровым и Гордиевским [67], а также Коганом [68]. Рёк [69 ], а также Портер и Джонсон [70 ] показали, что с помощью распределительной газовой хроматографии при простом аппаратурном оформлении можно подобрать разделяющие агенты, подходящие для экстрактивной перегонки.
Определение разности между температурами кипения, специально приготовленных эталонных смесей позволяет быстро оценить эффективность разделяющего агента. Для этого смешивают в равных объемах каждый из разделяемых компонентов с предполагаемым разделяющим агентом и опытным путем определяют температуры кипения обеих полученных смесей. Затем эти температуры вычисляют, исходя из мольных концентраций компонентов, при этом предполагают линейную зависимость температуры от состава. Наконец, вычисляют разность температур кипения полученных смесей, при этом используют как экспериментальные, так и расчетные значения температур кипения. Если найденная из опыта разность температур кипения смесей значительно превышает расчетную, то разделяющий агент считают пригодным [35].
Другим решающим фактором, влияющим на выбор разделяющего агента, действующего избирательно на легкокипящий компонент, является относительная полярность компонентов смеси. Жидкости с одинаковой полярностью образуют идеальные растворы; при смешении же компонентов с различной полярностью следует считаться с отклонениями от идеального поведе-
317
ния в соответствии с разностью полярностей компонентов. Например, введением неполярного высококипящего масла в сильно полярную смесь уксусная кислота—вода можно увеличить летучесть воды (по отношению к кислоте). Добавление к неполярной смеси циклогексан—бензол такого полярного разделяющего агента как анилин повышает летучесть циклогексана по отношению к бензолу. Методы подбора разделяющих агентов по относительной полярности компонентов смеси были развиты Бергом [52], а также Гильденбрандом [71].
Экстрактивную перегонку используют не только для разделения бинарных смесей, ее применяют также для выделения отдельных компонентов из многокомпонентных смесей, например бензола из нефтяных фракций [72]. Смеси насыщенных и ненасыщенных углеводородов с почти одинаковыми температурами кипения разделяются экстрактивной ректификацией в присутствии эфиров кетокислоты [73]. В последнее время большое значение приобретает разделение низших углеводородов Ci—С2 [74]. Кар-нер с сотр. [75] исследовал эффективность разделения смеси метилциклогексан—толуол в насадочных колоннах при экстрактивной ректификации с добавлением фурфурола; на основании полученных данных были выведены уравнения для расчета процесса ректификации.
При экстрактивной перегонке, как и при азеотропной, после подбора оптимального разделяющего агента необходимо построить по возможности более точные кривые паро-жидкостного равновесия для тройных или многокомпонентных смесей. Нулл и Пальмер [76 ] дают рекомендации, как можно получить необходимые данные по равновесию при минимуме экспериментального материала.
Необходимое число теоретических ступеней разделения определяют расчетным путем по формуле Фенске, подставляя вместо аэ значение относительной летучести, полученное в присутствии разделяющего агента (см. разд. 4.7.5.3). Точно также можно выполнить расчет графически по методу Мак-Кэба, учитывая количество исходной смеси и применяя кривую равновесия, которая соответствует возросшей величине аэ (см. разд. 4.7). Как показал Гельбин [7], число теоретических ступеней разделения при экстрактивной ректификации можно рассчитать с помощью метода полюсных точек Бошняковича.
Нагель и Зинн [78] предлагают модифицированный метод Мак-Кэба для расчета экстрактивной ректификации. Кортюм и Фальтуш [79 ] обсуждают проблемы, возникающие при реализации подобных селективных методов разделения. К ним относятся конструирование автоматизированной аппаратуры для непрерывной экстрактивной перегонки (из стали V4A) с подбором избирательно действующего агента, а также расчет минимального флегмового числа и необходимого количества разделяющего агента.
Требуемое количество разделяющего агента рассчитывают с учетом количества флегмы. Принимая 83% (мол.) за содержание 318
разделяющего агента (анилина) в его смеси с низкокипящим компонентом А («-гептаном), получим, что объем анилина относится к объему «-гептана как 3:1. Если согласно вычислениям оптимальными условиями ректификации являются нагрузка (расход паров) 1100 мл/ч «-гептана и флегмовое число 10, то при таком режиме разделения в головную часть колонны следует непрерывно подавать 3000 мл/ч анилина. При этом отбирают 100 мл/ч чистого «-гептана в качестве дистиллята, а 1000 мл/ч «-гептана и 3000 мл/ч анилина возвращают в колонну на орошение. Разделяющий агент (анилин) можно накапливать в кубе, пока низкокипящий компонент А не перейдет полностью в дистиллят. Только затем периодической ректификацией отделяют высококипящий компонент В, после чего в кубовом продукте остается разделяющий агент, который имеет более высокую температуру кипения, чем компоненты исходной смеси. Можно также одновременно получать второй компонент (В) в качестве головного продукта (дистиллята) на параллельно включенной колонне. При этом кубовый остаток параллельной колонны (разделяющий агент) непрерывно выводят и снова направляют в верхнюю часть первой колонны (см. рис. 224).
Таблица 45.
Примеры разделения идеальных, неидеальных и азеотропных смесей экстрактивной ректификацией
Разделяющий агент н его полярность, выраженная дипольным моментом (при 20 °C) Разделяемая смесь Разность температур кипения компонентов при 760 мм рт. ст., °C Примечания
Анилин, 1,51 н-Гептан—метилциклогексан Бензол—ци клогексан н-Гептан—толуол 2,7 0,7 12,8 Идеальная смесь (а = = 1,07) Образует азеотроп Неидеальная смесь
Фенол, 1,70 н-Гептан—толуол Изооктан—толуол Метилциклогексан— толуол 12,8 11,4 9,5 с асимптотическим приближением кривой равновесия к диагонали диаграммы
Монобутиловый эфир этиленгликоля Мети лэти л кетон — вода 20,4 Образует азеотроп
Диэтиловый эфир Этанол—вода 21,6 То же
Высшие кетоны или высшие спирты (для октанола 1,68) Ацетон—метанол 8,5 Образует азеотроп
Высшие сложные эфиры или высшие спирты Этил ацетат—этанол 1,3 То же
Высшие кетоны или внешне хлорпроиз-водные Ацетон—хлороформ 5,0
319
Рис. 232.
Головка с электромагнитным флегмоделителем и декантатором для колонн азеотропной ректификации с отбором легкой (а) и тяжелой (б) фаз.
Изготовитель: фирмы Отто Фриц, фирма Нормаг, Хофхайм (Таунус).
В табл. 45 дано несколько примеров [64 ] успешного применения экстрактивной ректификации, которые иллюстрируют приведенные выше теоретические положения.
Как для азеотропной, так и для экстрактивной перегонки в периодическом и непрерывном режимах работы можно применять традиционные лабораторные аппараты. Пригодны как насадочные, так и тарельчатые колонны. Лишь в особых случаях требуются специальные дополнительные устройства (см. рис. 223 и 224).
Рассмотрим сначала некоторые вопросы аппаратурного оформления процесса азеотропной перегонки. Если азеотроп гомогенный, то в ректификационную установку не требуется вносить каких-либо принципиальных изменений. Если азеотроп гетерогенный, то на орошение в колонну возвращают не азеотроп, а только разделяющий агент. Для этого применяют специальную «азеотропную» головку (рис. 232), которая позволяет выборочно применять в качестве орошения либо легкую, либо тяжелую фазу. Орошающую жидкость (флегму) целесообразнее подавать не в самую верхнюю часть колонны, а несколько пониже — во вторую сверху царгу. При работе в непрерывном режиме с образованием гомогенного азеотропа разделяющий агент примешивают к исходной смеси. При перегонке с образованием гетероазеотропа определенное количество разделяющего агента непрерывно возвращают в цикл с помощью азеотропной головки (см. рис. 223 и 232). Другие азеотропные головки описаны в разд. 7.5.3.
320
При экстрактивной ректификации принципиально необходима экстракционная зона, куда непрерывно подают разделяющий агент. Чтобы получить в головке колонны дистиллят, не содержащий следов разделяющего агента, последний вводят на одну царгу ниже верха колонны. При непрерывной ректификации исходную смесь подают в верхнюю треть колонны (см. рис. 224). Можно усовершенствовать метод, если кубовый продукт непрерывно выводить, а затем разделять в параллельно включенной колонне на высококипящий компонент и разделяющий агент, который затем возвращают в цикл.
Следует отметить, что в принципе не исключено применение разделяющего агента, который кипит при более низкой температуре, чем разделяемая смесь. Однако в этом случае, например, при экстрактивной ректификации смеси этанол—вода в присутствии диэтилового эфира разделяющий агент вводят в колонну несколько выше куба.
6.3. СОЛЕВАЯ ПЕРЕГОНКА
Описанные выше селективные методы перегонки основаны на смещении фазового равновесия при введении в смесь разделяющего агента в виде пара (перегонка с водяным паром) или в виде жидкости (азеотропная и экстрактивная перегонка). Смещение фазового равновесия можно вызвать также путем растворения в разделяемой смеси соли, что приводит к изменению соотношения парциальных давлений исходных компонентов.
Метод солевой перегонки до сих пор применяли для разделения смесей, один из компонентов которых вода. Кривая равновесия смеси вода—уксусная кислота (для атмосферного давления) при концентрации уксусной кислоты выше 96% (масс.) подходит очень близко к диагонали диаграммы, следовательно, полное удаление воды методом обычной перегонки затруднено. Кроме того, с практической точки зрения недостатком этого способа является (особенно при высоком содержании воды в исходной смеси) то, что энтальпия испарения для воды почти в пять раз больше, чем для уксусной кислоты. Поэтому было бы выгоднее при непрерывной ректификации в дистилляте получать уксусную кислоту, а в кубе — воду. Этого можно достичь добавкой к смеси более 8% (масс.) хлорида кальция [80]. Например, при 760 мм рт. ст. относительная летучесть компонентов смеси вода—уксусная кислота с содержанием 30% (масс.) уксусной кислоты в результате добавления 20% (масс.) хлорида кальция изменяется от 1,36 до 0,525. Таким образом, относительная летучесть воды и уксусной кислоты становится меньше 1, а это означает, что в головке колонны конденсируется уксусная кислота, а в кубе накапливается вода. Разумеется, при этом предполагается, что разделяющий агент (хлорид кальция) равномерно распределен по всей высоте колонны. В соответствии с систематическими измерениями Даубаха [81 ],
11 Э. Крель 321
Рис. 233.
Кривые равновесия смеси этанол—вода без добавки (/) и’с добавкой 10 г СаС12 на 100 мл смеси (2).
Рис, 234.
Данные для разделения смеси метанол—ацетон:
а — кривая равновесия без добавки (/) и с добавкой 2,3 М раствора CaClg (2); б — схема установки.
На осях диаграммы указаны значения концентраций ацетона в жидкости и в парах.
добавление 10 г хлорида кальция к 100 мл азеотропной смеси этанол—вода вызывает исчезновение азеотропной точки (рис. 233). Таким образом, благодаря введению солевого раствора в непрерывнодействующую колонну получают дистиллят высокой степени чистоты [82]. Аналогичные исследования с NaNO3 и K2SO4 провели Гурси и Томпсон [83]. Новелла и Тарассо [84] использовали соли, растворимые в обоих компонентах разделяемой смеси, например СиС12 и КС2Н3О2. Они установили, что благодаря этому при ректификации исключается образование азеотропа. О влиянии солей металлов на равновесие в системе жидкость— пар сообщает Шир [85].
Азеотропную смесь метанол—ацетон можно обогатить до концентрации, значительно превышающей азеотропную, если к 1 объему смеси добавить 3,5 объема водного раствора хлорида кальция плотностью 1,2 г/см3 (при 20 °C это соответствует 2,3/И раствору) [86]. На рис. 234 приведены кривые равновесия смеси метанол—ацетон с добавкой соли и без нее, дана также упрощенная схема установки.
При перегонке смеси вода—фенол путем добавления 17% NaCl достигают смещения азеотропного состава с 91% (масс.) до 84% воды; поэтому можно использовать область ограниченной растворимости системы [87]. Насыщая нитратом калия смесь этанол—вода в интервале концентраций этанола от 15 до 70%, также достигают большего обогащения, чем без добавления соли [88]. Гайер с сотр. [89] исследовал влияние солевых добавок на парожидкостное равновесие смеси муравьиная кислота—вода. При содержании в системе 35,5% соли азеотроп больше не обра-322
зуется, благодаря чему можно получить высокую концентрацию муравьиной кислоты. Систематические исследования солевого эффекта в системе этанол—вода провел также Миро с сотр. [90].
В своем обзоре Фуртер [91 ] анализирует степень влияния солевого эффекта на перегонку с точки зрения возможности использования подобных методов в промышленности, особенно при экстрактивной ректификации. Ряд работ Шуберта с сотр. [92] посвящен исследованию влияния различных хлоридов металлов и других простых солей на изотермическое фазовое равновесие систем пропанол—вода, н-бутанол—вода, а также метанол—вода.
Уравнение Жака и Фуртера [95 ] для оценки влияния соли на паро-жидкостное равновесие бинарных смесей описывает взаимосвязь температуры и концентрации жидкости для трехкомпонентной смеси этанол—вода—соль. Это уравнение имеет шесть констант. Фуртер и Меранда [96] обсуждают теорию солевого эффекта. Допустив упрощения, Сада с сотр. [97] вывел соотношение для расчета паро-жидкостного равновесия для неводных бинарных систем, в которых соль растворяется только в одном компоненте (например, бензол—этанол с добавлением хлорида лития или кальция).
Уже эти немногие примеры показывают, что для решения трудных проблем разделения все возрастающее значение приобретают селективные методы перегонки; систематические исследования позволяют открыть новые области их применения. Сочетание процессов перегонки с адсорбционными эффектами привело к разработке «адсорбционной перегонки». Фукс и Рот [93] исследовали влияние материала насадки на процесс разделения смеси вода— уксусная кислота.
11
Материалы и аппаратура
Специальные устройства, применяемые в технике лабораторной перегонки, были рассмотрены в гл. 1—6. В настоящей главе описываются новейшие детали, узлы и приборы, предназначенные для различных процессов перегонки. Путем сравнительного анализа большого числа принципиально тождественных элементов ректификационных установок автором отобраны те элементы, которые можно стандартизировать. Преимущества стандартизации проявляются как при изготовлении, так и при использовании ректификационной аппаратуры. Очевидно, что условием стандартизации отдельных элементов ректификационных установок является возможность использования .их в любых процессах перегонки, применяемых в промышленности при оптимальных условиях.
7.1. МАТЕРИАЛЫ ДЛЯ ИЗГОТОВЛЕНИЯ РЕКТИФИКАЦИОННОЙ АППАРАТУРЫ
В качестве материалов для изготовления различных аппаратов ректификационных установок применяют в основном стекло, а также металлы, фарфор и кварц. Металлы используют для изготовления установок высокотемпературной перегонки или ректификационных установок, работающих под давлением, когда прочность стекла становится недостаточной. Наиболее широкое применение находят аппараты из нержавеющей стали V2A *, которые могут быть использованы для высокотемпературной перегонки и перегонки под давлением, а также для ректификации агрессивных веществ. Фарфор применяют в тех случаях, когда металлы и стекло не могут использоваться вследствие их коррозионной неустойчивости. Из кварца изготовляют в основном аппараты для перегонки воды и высокотемпературной ректификации. Отдельные детали и узлы выполняют также из пластмасс, например тефлона.
Стекло, применявшееся уже около 3000 лет назад в древнем Египте, для изготовления косметических сосудов, амулетов и позднее приборов для алхимии, не потеряло своего основного значения в лаборатории и промышленности до настоящего времени. Широкое использование стекла обусловлено его ценными свой
* По составу близка к стали марки 12Х18Н10Т, выпускаемой в СССР. — Прим. ред.
324
ствами: этот материал успешно применяется как для изготовление лабораторной аппаратуры, предназначенной для проведения химических и физико-химических процессов, так и для производстве промышленных колонных аппаратов.
Прозрачность стекла позволяет наблюдать за ходом процесса. В адиабатических процессах, протекающих при температурах примерно до 120 °C, кожух из стекла, вакуумированный дс остаточного давления 10-в мм рт. ст., обеспечивает достаточнук термоизоляцию аппарата. При более высоких температурах, а также при использовании крупногабаритных аппаратов в качестве термоизоляционного материала применяют стекловолокно: в слое изоляции оставляют смотровую щель, предназначенную для визуального наблюдения за ходом процесса (см. разд. 7.7). Важным преимуществом стекла является его высокая коррозионная стойкость. Поэтому многие химические реакции и процессы разделения проводят в аппаратах и установках, изготовленных из стекле или других керамических материалов. Широкому примененик стекла в химической промышленности способствует высокая твердость и незначительная шероховатость поверхности стеклянных изделий. Стенки стеклянных аппаратов во время работы незначительно загрязняются и легко поддаются очистке. Ценным свойством стекла является также сравнительно небольшой коэффициент линейного расширения. Использование стеклянных аппаратов при переработке фармацевтических продуктов и однократной или двойной перегонке воды дает возможность получать продукть: без запаха, вкуса и, главное, без примесей металлов.
Основным недостатком стекла, по сравнению с металлами, является его хрупкость, а также недостаточная устойчивость к резким изменениям температуры. Прочность стекла можно повысить, если его расплав охлаждать по специальному режиму, способствующему возникновению в глубине материала определенных внутренних напряжений. Перспективным направлением развития стекольной промышленности является разработка и получение различных марок стекла, свойства которых были бы наиболее оптимальными для изготовления аппаратуры применительно к конкретным физико-химическим процессам. Стекло с универсальными свойствами, приближающееся к металлическим сплавам по возможностям его применения и обработки, по-видимому, никогда не будет получено. Однако при правильном выборе марки стекла можно в значительной мере удовлетворить необходимые требования [1, 5].
В настоящей работе автор не счел целесообразным привести физико-химические свойства стекла различных марок, так каг испытания последних проводили, как правило, в несопоставимых условиях. Все необходимые характеристики стекол можно найти в каталогах промышленности стекольных изделий [1].
Подробные данные о свойствах стекол приведены Мёнхом [2 и Айтелем с сотр. [3]. В работе центральной лаборатории сте
32J
кольной промышленности ГДР в Ильменау [4] представлена сравнительная характеристика физико-химических свойств стекла, фарфора, металлов и пластмасс. Кроме того, в данной работе подробно описаны возможные области применения стекла, а также характеристики стекловолокна различных видов и изделий из них.
Сведения об использовании полупромышленных стеклянных аппаратов в лабораторных и пилотных ректификационных установках приведены Ригером [5]. Применение аппаратов из специальных марок стекла в ректификационных установках, работающих под избыточным давлением до 4,3 кгс/см2, описано в работе [6]. Камель [7] рассмотрел вопросы, связанные с использованием фарфора в лаборатории.
7.2. СТАНДАРТНЫЕ И СБОРНЫЕ ПРИБОРЫ
Стандартизованные методы перегонки описаны в разд. 3.1. На рис. 235 показан дистилляционный прибор Энглера со стандартными шлифовыми соединениями. В этом приборе глубина погружения термометра и расположение капельницы в трубке для отвода дистиллята точно фиксированы. Автоматические дистилляционные установки, в состав которых входит прибор Энглера, подробно описаны в разд. 8.1.
Прибор Энглера целесообразно применять для скоростной
перегонки в промышленных условиях, однако он не пригоден для проведения более точных работ. Этот прибор не пригоден также и для вакуумной дистилляции, при перегонке на нем высококипящих веществ возможно частичное разложение исходной смеси,
искажающее полученные результаты.
Для определения температурных пределов выкипания при остаточных давлениях около 10 мм рт. ст. применяют дистилляционный прибор с колбой Кляйзена (рис. 236). Следует отметить,
что измерение давления за приемником дистиллята может привести к заметным погрешностям. Для получения точных результатов необходимо измерять давление и температуру по возможности в одном месте. Место установки вакуумметра при работе в интервале остаточных давлений ниже 10 мм рт. ст. показано на рис. 236 штриховой линией.
Недостаток прибора Энглера и прибора с колбой Кляйзена
в том, что они не позволяют точно определить температуру начала
перегонки. При перегонке с использованием прибора Энглера в соответствии с указанием стандарта фиксируют температуру
Рис. 235.
Дистилляционный прибор Энглера со стандартными шлифовыми соединениями.
326
Рис. 236.
Дистилляционный прибор с колбой Кляйзеиа для перегонки под вакуумом.
Рис. 237.
Дистилляционный прибор с насадочной колонной, собранной из узлов и де* талей, выполненных по нормалям «Дестинорм» .
начала перегонки в момент отбора первой капли дистиллята. В приборе с колбой Кляйзена температура начала перегонки также завышается, поскольку в первую порцию дистиллята попадает некоторая часть высококипящей фракции. Искажения температуры начала перегонки особенно сильно проявляются при загрузке небольших количеств исходной смеси (до 100 г), т. е. именно тех количеств, которые обычно используют при аналитическом контроле в производственных условиях. В последнее время широко применяются дистилляционные приборы для пробной дистилляции, снабженные небольшой насадочной ректификационной колонной (рис. 237), в куб которой можно загружать от 200 до 300 мл исходной смеси. В таких приборах можно точно определять температуру начала отгонки при работе с бесконечным флегмовым числом. Воспроизводимость результатов при вакуумной перегонке можно обеспечить, если снабдить данный прибор точными регуляторами давления, нагрузки колонны по пару и флегмового числа. Для полной отгонки исходной смеси к ней добавляют вещество-вытеснитель, температура кипения которого примерно на 50° С выше конечной температуры дистилляции. После окончания дистилляции целевого продукта в кубе колонны остается только вещество-вытеснитель [8].
В лабораторной практике обычную дистилляцию применяют в основном для регенерации растворителей и других жидкостей, а также для предварительного отделения большей части основного
327
Рис. 238.
Детали и аппараты дистилляционных установок:
1 — капилляр, применяемый для устранения пульсаций давления при кипении; 2 — приставка; 3,9 — воздушные холодильники; 4 — перемычка;
5, 11 — холодильники Либиха и Димрота соответственно; 6, 12 — форштосы; 7 — холодильник с винтовой охлаждаемой поверхностью; 8 — шариковый холодильник; 10 — змеевиковый холодильник; 13, 14, 16 — сборники дистиллята, разработанные Аншюцем—Тнле, Бредтом и Брюлем соответственно; 15 — градуированная цилиндрическая бюретка.
вещества от исходной смеси. Для проведения дистилляции используют колбу с приставкой, снабженной термометром на шлифе, устройства для конденсации пара и сбора дистиллята, а также приспособление для присоединения системы к вакуумной линии. Во многих случаях для предотвращения уноса брызг полезно
328
снабдить колбу короткой насадочной колонной высотой 5—10 см. На рис. 238 показаны стандартные детали и аппараты дистилляционных установок [9].
Приставку 2 к кубу в виде колбы применяют для дистилляции низко- и высококипящих веществ, а перемычку 4 используют в основном при работе с высококипящими и легкозатвердевающими веществами. При дистилляции высококипящих веществ обычно достаточно воздушного охлаждения паров дистиллята, поэтому в этих случаях пригодны детали 3 и 9. Если полученный дистиллят затвердевает, то его можно легко расплавить с помощью горелки Бунзена или электроспирали. Однако в целях безопасности лучше применять холодильники 7 и 8, которые охлаждаются жидкостью из термостата и легко очищаются после окончания процесса дистилляции. Особенно удобен винтовой холодильник 7, в котором деталь с винтовой поверхностью при необходимости очистки может быть вынута через шлиф.
При дистилляции низкокипящих веществ, требующих интенсивного охлаждения, часто бывает недостаточно холодильника Либиха 5, так же, как и трубки с узкой охлаждаемой рубашкой Веста. Более эффективное охлаждение обеспечивают змеевиковый холодильник 10 и холодильник Димрота 11, позволяющие, кроме того, собирать установки, более компактные по высоте.
Для вакуумной дистилляции наиболее подходящим является сборник дистиллята Аншюца—Тиле 13 (TGL 13839), предназначенный для отбора любого количества фракций без нарушения технологического процесса. Это устройство хорошо работает и при атмосферном давлении в тех случаях, когда процесс пере-
Рис. 239.
Приставки к кубам в виде колб, используемые при дистилляции вспенивающихся жидкостей:
а — приставка Райтмайра; б — приставка Фридрихса.
Рис. 240.
УстройствоХФридрихса для дистилляции легковоспламеняющихся жидкостей.
325
Рис. 241.
Детали для сборки соединительных коммуникаций:
/ — труба; 2, 3 — колена с углом изгиба 135 и 90° соответственно; 4, 5 — Т- и Y-образные тройники; 6,7 — краны.
Рис. 242.
Подвижная соединительная коммуникация из колеи со шлифами, предложенная Фридрнхсом.
гонки сопровождается выделением газов. В качестве сборников дистиллята удобно использовать градуированные цилиндрические бюретки 15, с помощью которых можно непрерывно контролировать количество отбираемого дистиллята. Следует отметить также форштосы 6 (TGL 9972) и 12 (TGL 9971), применяемые в тех случаях, когда дистиллят не разделяют на отдельные фракции. Сборник дистиллята Бредта 14 (TGL 13841), выполненный в виде «коровьего вымени», рассчитан на четыре фракции; аналогичное устройство Брюля 16 имеет семь сосудов размером 120 X х 30 мм. Для устранения пульсаций давления при кипении нередко в дистилляционный прибор через капилляр подают воздух или азот, при этом в качестве куба удобно применять колбу Кляйзена (см. рис. 236) или трехгорлую колбу, в которую на шлифе вводят капилляр 1. Для перегонки сильно вспенивающихся веществ используют специальные приставки Райтмайра (рис. 239, а) или Фридрихса (рис. 239, б). При дистилляции низкокипящих и легковоспламеняющихся жидкостей лучше использовать другое устройство Фридрихса (рис. 240), обеспечивающее безопасность работ. К трубе А можно присоединить шланг для отвода выделяющихся газов в атмосферу.
Ректификационные установки, также как и дистилляционные приборы, можно собирать из отдельных деталей. Без особых 330
трудностей можно собрать даже подобные промышленным сложны установки непрерывного действия с двумя ректификационным колоннами (см. рис. 134 и 162) [10, 11]. Комбинируя различны детали, выполненные по нормали «Дестинорм», можно собрат не только разнообразные установки для перегонки, определени давления паров и данных по равновесию систем пар—жидкость но и более сложные установки для непрерывной ректификаци при атмосферном давлении и под вакуумом (см. рис. 223 и 224J Серия деталей для установок типа «Лабодест» разработана Штаг (изготовитель: фирма Фишера «Лабораторная и исследовательска техника», Бонн—курорт Годесберг). Фирма Нормаг (Хофхайы и предприятие «Стеклянные приборы с нормализованными шли фами» (Вертхайм) также изготавливают отдельные детали, пред назначенные для различных ректификационных установок.
На рис. 241 показаны детали для сборки соединительных коы муникаций. Длинные трубопроводы целесообразно составлять и прямых труб 1 длиной 1 — 1,5 м, снабженных стандартными шли фами. С помощью колен с углом изгиба 135° (2) или 90° (3), а такж Т- или Y-образных тройников 4, 5 можно собирать любые комму никации, включающие при необходимости краны 6 и 7. Дл: обеспечения некоторой подвижности коммуникаций можно ис пользовать соединительные детали со сферическими шлифам) (см. рис. 19) или к штуцеру аппарата подсоединять коммуникации из колен со шлифами, предложенную Фридрихсом (рис. 242) Шлифы различных размеров можно соединять с помощью пере ходов (рис. 243). Ниже приведены диаметры (в мм) муфты и ко нуса переходов, выпускаемых промышленностью:
Муфта WS 12,5 14,5 14,5 24 29 29 29 29 29 29 29 29 29
Конус NS 14,5 14,5 29 29 24 29 34,5 45 55 60 70 85 100
Переход 6 имеет два стандартных конуса, а переход 7 имеет ко нус NS 14,5 и наконечник для шланга.
В табл. 46 указаны некоторые стандартные детали для ректи фикационных установок.
Трубопроводные коммуникации можно собирать также и: деталей с плоскими и сферическими шлифами. При этом макси мальная длина труб для коммуникаций промышленных установоь не превышает 3 м. Для полупромышленных и пилотных установо! стекольная промышленность изготавливает детали, подобньк
Рис. 243.
Переходы:
1—4 — переходы с диаметром конуса, равным или пре вышающим диаметр муфты; 5 — переходы с диаметроь конуса меньшим, чем диаметр муфты; 6 — переходь с двумя конусами; 7 — переходы с конусом NS 14,5 i наконечником для шланга.
Таблица 46.
Стандартные детали для ректификационных установок
Стандарт Наименование
TGL 13828 13831 13834 13835 13836 13837 13838 9966 9973 9978 Соединительные перемычки, прямые * Соединительные перемычки, изогнутые под углом 90° * Соединительные перемычки S-образной формы * Соединительная перемычка с одноходовым краном * Соединительная перемычка с пароотводной трубкой * Соединительная перемычка с каплеотбойником * Соединительная перемычка с воронкой и краном * Переходы * U-образные переходы с двумя шлифовыми муфтами Капилляры, применяемые для устранения пульсаций дав-
DIN 12404/63 ления при кипении * Капилляры со стандартными шлифами, применяемые для
12565/70 устранения пульсаций давления при кипении Капельная воронка
* Все вышеуказанные детали снабжены стандартными шлифами.
деталям, показанным на рис. 241. Типы выпускаемых деталей можно найти в каталогах [1, 5, 71. Кроме того, к настоящему времени выпущен ряд стандартов на подобные детали (TGL 10192— 10200, 11641—11644, 12023—12032, 20277 и проекты стандартов DIN 28800—28808).
7.2.1. КРАНЫ И КЛАПАНЫ
Стандартные стеклянные краны, предназначенные для лабораторных ректификационных установок, приведены в табл. 47.
На рис. 189 и 190 показаны стеклянные краны для работы под вакуумом. Многоходовой кран Кинца [12], предназначенный для работы под вакуумом, устроен так, что его сердечник вращается в одном направлении, и поэтому ошибки в его установке исключены (рис. 244).
В обзоре Вольфа [13] рассмотрены краны специальных конструкций. Один из подобных кранов — плоский кран «Вестале» — показан на рис. 245. Для полупромышленных установок этот кран может выполняться в виде проходного, выпускного, а также трех-и четырехходового крана.
Обычные стеклянные краны с жировой смазкой в качестве уплотнительного материала хорошо заменяют краны со сменными сердечниками из тефлона. Предприятие Отто Фриц Нормаг (Хоф-хайм/Таунус) выпускает различные стеклянные краны с ограничительными кольцами. Миниатюрные краны французской фирмы Гафлон выполняются полностью из тефлона. Стеклянные трубы 332
Таблица 47.
Стандартные стеклянные краны для ректификационных установок
Стандарт Наименование
TGL 21734 группа 1 группы 2—7 13843 группа 1 группа 2 группа 3 DIN 12541/67 группа 1 /70 группа 1 Краны (описание) Специальные краны Высоковакуумные краны Одноходовые краны (размеры) Двухходовые краны (размеры) Одноходовые конусные краны (масса) Одноходовые конусные краны (испытание гер-
12551/67 группа 1 метичности) Одноходовые конусные краны с иезаменяе-
12553/61 мым массивным сердечником Двухходовые краны с незаменяемым сердечником
12554/67 Трехходовые конусные краны с незаменяемым
12563/67 сердечником Трехходовые конусные краны с незаменяемым массивным сердечником, снабженные отверстиями, смещенными по окружности на 120°
присоединяют к этим кранам с помощью тефлоновых болтовых соединений с прокладками также из тефлона.
Обычно стеклянные краны снабжают устройствами, предотвращающими выход сердечника крана из его гнезда при вращении (см. рис. 217). В качестве таких устройств можно использовать резиновые шайбы или кольца, либо, еще лучше, предохранители из пружинной стали (рис. 246).
Рис. 245.
Плоский край «Вестале» (патент ФРГ 1112846):
а — детали крана; б — кран в сборке.
Изготовитель: фирма Эдмунд Бюлер. Тюбинген.
333
Рис. 247.
Схема клапана для точной регулировки расхода жидкостей и газов с седлом из стекла и шпинделем из тефлона (диаметр седла или диаметр подводящей трубы 4 илн 7 мм):
а — угловой клапан; б — проходной клапан.
Изготовитель: народное предприятие «Йенский завод стекла Шотт», Иена.
В полупромышленных и пилотных установках не рекомендуется использовать стеклянные краны с жировой смазкой, особенно при диаметрах условного прохода трубопроводов NW выше 40 мм. В этих случаях целесообразно применять клапаны современных конструкций, выполненные из стеклянных и тефлоновых деталей [1]. Вместо кранов в лабораторных установках часто используют клапаны с тефлоновыми шпинделями (рис. 247). Прецизионные клапаны Совиреля системы «Торион» предназначены для установки на трубопроводах диаметром 2,5—10 мм. Клапаны должны надежно работать при остаточных давлениях до 10-4 мм рт. ст.
В клапанах для промышленных установок в качестве запорного устройства применяется тефлоновый сильфон с запорной пластиной или запорным конусом из тефлона (рис. 248). Установка
положения клапанов может осуществляться вручную с помощью электромагнитов, электромотора или пневматического привода, что позволяет применять эти клапаны в автоматизированных ректификационных установках.
Промышленность стекольных изделий выпускает клапаны различных моделей с плоскими или сферическими шлифами, имеющими диаметр условного прохода NW до 150 мм.
Рис. 248.
Пневматический регулируемый клапан (без сервопривода).
Диаметр условного прохода NW25, 32, 40 и 50 мм.
Изготовитель; народное предприятие «Йенский завод стекла Шотт», Иена.
334
Различают клапаны следующих типов (см. разд. 5.1.3.2): проходные, выпускные, угловые, дроссельные, обратные с заслонкой или шаром, предохранительные для максимальных скачков давления, не превышающих 5 кгс/см2.
7.3. РЕКТИФИКАЦИОННЫЕ КОЛОННЫ
Физические процессы массо- и теплообмена протекают в ректификационной колонне на поверхности раздела жидкой и паровой фаз (см. разд. 4.1). Следовательно, конструкция колонны должна обеспечить протекание этих процессов в условиях противоточного движения пара и жидкости при наличии развитой поверхности контакта фаз, причем удерживающая способность колонны не должна превышать допустимую (см. разд. 4.10.5). Оптимальная конструкция колонны характеризуется минимальным значением ВЭТС и минимальной удерживающей способностью по жидкости.
Хекманн [14] предложил критерии выбора колонн с целью их систематизации. Число разработанных конструкций колонн чрезвычайно велико, однако можно выделить следующие основные типы колонн: безнасадочные, насадочные, тарельчатые, с неподвижными контактными устройствами и с вращающимися контактными устройствами.
Райхельт [14а] описал тарельчато-насадочные колонны. В этих колоннах на колосниковых решетках или тарелках размещены слои насадки, состоящие обычно из легких шариков. При работе колонны в условиях противотока фаз на тарелках образуются кипящие слои насадки, обеспечивающие повышение пропускной способности колонны.
7.3.1. БЕЗНАСАДОЧНЫЕ КОЛОННЫ
Особым преимуществом безнасадочных колонн являются малая удерживающая способность по жидкости и небольшой перепад давления. Это обусловливает их применение, главным образом, для микроперегонки (см. разд. 5.1.1), высокотемпературной ректификации (см. разд. 5.3.2) и вакуумной дистилляции (см. разд. 5.4.1). В колоннах этого типа процессы массо- и теплообмена протекают между паром, поднимающимся в свободном пространстве, и жидкостью, стекающей в виде пленки по стенкам. Области применения пленочных колонн, а также проблемы, возникающие при их эксплуатации, подробно рассмотрены Малевским [15]. Он предложил некоторые конструкции подобных аппаратов, а также указал на нерешенные проблемы.
Ректификационные колонны могут быть выполнены в форме прямой или спиральной трубы, трубы с развитой поверхностью, а также в виде канала кольцевого сечения. Подобные колонны благодаря малому гидравлическому сопротивлению имеют сравнительно высокую пропускную способность по пару, однако их
335
эффективность, особенно при больших нагрузках, невелика вследствие ограниченной поверхности контакта фаз. Исключение составляют лишь колонны, выполненные в виде канала кольцевого сечения. Вертикальные гладкостенные трубы диаметром 20—50 мм в настоящее время редко применяют в качестве ректификационных колонн, обычно их используют для отделения брызг при перегонке. Только при нагрузках ниже 30 мл/ч удается достичь ВЭТС менее 10 см. Разделяющую способность этих колонн можно повысить путем увеличения поверхности ее стенок. Например, сферическая колонна Вюрца (см. рис. 15) и грушевидная колонна Юнга имеют более высокую эффективность, однако обладают при этом большей удерживающей способностью. Характеристики безнасадочных колонн приведены в табл. 48.
Для увеличения поверхности контакта фаз на внутреннюю стенку трубы наплавляют стеклянный порошок или припаивают горизонтально и наклонно небольшие стеклянные стержни, как в известной колонне с елочным дефлегматором. Наклонные стержни служат также для подачи флегмы, стекающей по стенкам, к оси колонны (рис. 249). В модифицированной модели, предложенной Рэем [17], стержни расположены перпендикулярно к стенкам. Достигаемая при этом эффективность разделения указана в табл. 48. Колонну с елочным дефлегматором используют в несложных процессах перегонки, требующих минимального времени пребывания жидкости в колонне. Например, в таких колоннах проводят
Таблица 48.
Характеристики безнасадочных колонн
Тип колонны Диаметр, мм Рабочая длина, мм Нагрузка, мл/ч ВЭТС, см Объем удерживаемой жидкости, приходящийся на 1 теор. ступень разделения, мл Литература
Полая вертикальная труба 30 1000 25 [16]
Труба с наплавленным на внутреннюю поверхность стеклянным порошком 7 500 12,5 [16]
Колонна с елочным дефлег- 12 460 54 5,4 [18]
матором. 96 7,1 0,46
294 7,7 0,62
540 7,7 0,68
24 460 120 10,2 1,3
240 12,1
510 11,5 1,8
Модифицированная колонна с елочным дефлегматором, предложенная Рэем 6 1200 60 2,0 <0,02 [17]
Колонна Янцена 4—6 50—200 4—6 0,2—0,6 [19]
336
Рис. 249.
Колонна с елочным дефлегматором и изоляционным кожухом, заполненным воздухом.
Рис. 250.
Трубчатые змеевиковые колонны Янцена с вакуумированным изоляционным кожухом со змеевиком длиной 3 м (а) и 6 м (д’).
микродистилляцию или высокотемпературную ректификацию смол К' и других высококипящих веществ, которые необходимо разделить г под вакуумом с отгонкой до 98% от общего количества исходной смеси [18]. Для аналогичных целей применяют и змеевиковую трубчатую колонну Янцена (рис. 250), которая выполняется со змеевиками длиной 3 и 6 м. Она обладает удерживающей способов ностью, равной 0,2—0,6 мл на одну теоретическую ступень раз-91 деления [19]. При испытании подобной колонны на смеси че-тыреххлористый углерод—бензол автор получил следующие дан-ные:
К Предел захлебывания....................... 900 мл/ч
| Объем удерживаемой жидкости, приходя-щийся на одну теоретическую ступень
г разделения................................ 0,4 мл (при нагрузке
| 50 мл/ч)
I ВЭТС..................................... 4,3 см (при нагрузке
I 50 мл/ч)
I 5,4 см (при нагрузке
| 90 мл/ч)
К. 5,6 см (при нагрузке
к 190 мл/ч)
337
Рис. 251.
Схема, поясняющая принцип повышения разделяющей способности ректификационной колонии, разработанный Куном.
Рис. 252.
Миоготрубчатая колонна Куна:
1 — трубы; 2 — термостат; 3 — капилляры, обогреваемые снаружи.
Рис. 253.
Трубчатая щелевая колонна Куна:
1 — дефлегматор; 2 — штуцер для присоединения к вакуумному насосу;
3 — охлаждаемый сборник дистиллята; 4,5 — концентрические трубы;
6 — вакуумированный кожух; 7 — куб; 8 — электронагреватель.
Испытанная колонна имела змеевик с диаметром 50 мм, шагом 12 мм и длиной 6 м; змеевик был изготовлен из трубы диаметром 6 мм.
Колонну с елочным дефлегматором обычно изготавливают со съемным теплоизоляционным кожухом, а колонну Янцена — с вакуумированным кожухом. Для всех безнасадочных колонн важно поддерживать в ходе ректификации температуру стенки ниже температуры стекающей флегмы, так как в противном случае нарушается равномерное образование пленки на поверхности стенки. Следует отметить, что эффективность безнасадочных колонн возрастает при уменьшении нагрузки. Используя многотрубчатые колонны, состоящие из параллельно соединенных труб небольшого диаметра, можно получить любую необходимую производительность. Подобную многотрубчатую колонну еще в 1936 г. запатентовал Фенске [20].
Экспериментально установлено, что при нагрузке трубчатой ректификационной колонны диаметром 0,6 см, равной 10 мл/ч, ВЭТС составляет около 1,73 см [21]. Колонна указанного диа-338
метра высотой 1 м обладает при этом гидравлическим сопротивлением 2,7 мм рт, ст. и удерживающей способностью по жидкости около 0,4 мл. На основе этих данных Кун [22 ] разработал новый принцип повышения разделяющей способности ректификационной колонны (рис. 251). Этот принцип основан на многократном увеличении сравнительно небольшого эффекта разделения, имеющего место в поперечном сечении колонны (в направлении вектора а) при стационарном режиме путем создания продольного потока, изменяющего свое направление на концах трубчатой ректификационной колонны (векторы ui и и2).
Для достижения высокой эффективности разделения необходимо иметь в колонне достаточно низкую скорость потока паров при значительном флегмовом числе. Чтобы получить при этом удовлетворительную производительность колонны, нужно, как уже указывалось выше, использовать многотрубчатую колонну (рис. 252). Целесообразным оказалось образование флегмы непосредственно в трубах 1, помещенных в общий термостат 2; при этом дистиллят отбирают из верхних концов труб 1 с помощью обогреваемых снаружи капилляров 3 (см. также рис. 157).
Данные о производительности подобной колонны, содержащей 100 параллельных труб диаметром 0,4 см и длиной 2 м, полученные Куном с сотр. [22] при различных исходных и конечных концентрациях и разностях температур кипения компонентов, приведены в табл. 49.
При расчетах производительности принимали, что молекулярная масса компонентов составляет 100, а температура кипения — около 90 °C. Другая колонна Куна, содержащая 61 трубу диаметром 0,2 см с рабочей высотой 1,5 м, была использована для разделения изотопов 12С и 13С с разностью температур кипения всего 0,03 °C. В этом случае число теоретических ступеней разделения определяли по данным анализа продуктов перегонки на масс-спектрометре. Удерживающая способность одной трубы диаметром 0,2 см и высотой 2 м составила около 0,5 г [22].
Таблица 49.
Производительность колонны Куна, содержащей 100 труб диаметром 0,4 см и длиной 2 м
Разность температур кипения компонентов, °C Производительность колонны (в г/сут) при исходной и конечной концентрациях ннзкокипящего компонента (xQ -> *кон» % мол.)
40 -> 60 10 -> 90 10 -> 99 50 -> 90 1 -> 99
0,1 30
0,31 300 5 2,9 22 0,2
1,0 3 000 50 29 220 2
3,1 30 000 500 290 2 200 20
10,0 300 000 5000 2900 22 000 200
339
7
100 200 300 ЧОО 500-
Нагрузка, мл/ч
Рис. 254.
Диаграмма разделения изомеров ксилола с помощью колонны Куна высотой 0,8 м.
Рис. 255.
Зависимость числа теоретических ступеней разделения, приходящихся на
1 см высоты колонны ЯуД, от ее производительности [28]:
/ — трубчатая щелевая колонна диаметром 2,5 см и шириной щели 0,4 см;
2 — колонна диаметром 2,5 см с насадкой Стедмана; 3 — колонна диаметром 2,4 см с насадкой из проволочных спиралей 4X4 мм.
Метод перегонки, основанный на принципе многократного увеличения эффекта разделения, был разработан Куном путем обширного аналитического исследования 124, 25]. Эти теоретические расчеты были проведены для колонны с кольцевым поперечным сечением, образованной из двух концентрических труб (рис. 253). Подобные колонны называют колоннами с кольцевой щелью или трубчатыми щелевыми колоннами (см. рис. 257а).
Метод, предложенный Куном, можно успешно применять при разделении таких близкокипящих изомеров, как ксилолы (рис. 254) и изомерные амиловые спирты. Колонна Куна пригодна также для проведения процессов перегонки, требующих более 100 теоретических ступеней разделения, так как эта колонна имеет исключительно малую ВЭТС.
Методы расчета подобных колонн для разделения идеальных смесей были обсуждены в разд. 5.1.4.2. Крэйг [23] описал щелевую колонну, предназначенную для микроперегонки при загрузке исходной смеси в объеме 0,25 мл. Эта колонна представляет собой канал кольцевого сечения, образованный капилляром и соосно размещенным внутри его стержнем. Теоретические основы расчета этих колонн разрабатывали также Вестхавер [26], Доннелл и Кенеди [27 ], а также Янцен и Викхорст, проводившие сравнение данной колонны с колоннами, заполненными насадкой Стедмана и насадкой из проволочных спиралей [28 ]. Как видно из рис. 255, щелевая трубчатая колонна при производительности менее 10 мл/ч 340
имеет бдльшую эффективность, чем обе описанные выше колонны.
Перепад давления в трубчатых щелевых колоннах можно рассчитать по формуле:
Pi — Pi = Зт)1/<2/(26а2) (188а)
где рг — р2 — перепад давления, дин/см2; л — динамическая вязкость пара, П; / — высота колонны, см; Q — расход пара, рассчитанный по среднему давлению в колонне (p-i + р3)/2, см3/с; b — длина средней окружности кольцевого сечения колонны, см; а — половина ширины кольцевого сечения, см.
Гидравлическое сопротивление трубчатой щелевой колонны в сопоставимых рабочих условиях примерно на порядок меньше, чем у вышеупомянутых насадочных колонн (рис. 256). Кох и Файнд [29] определяли в изотермических условиях гидравлическое сопротивление трубчатых щелевых колонн с шириной щели, изменяющейся от 4,05 до 19,68 мм, при одинаковом диаметре наружной трубы, равном 49,96 мм. Ими был также исследован процесс теплопередачи в' подобных колоннах при внешнем обогреве наружной трубы. Бек [30 ] исследовал эффективность теплообмена и гидравлическое сопротивление в трубчатых щелевых колоннах, образованных коаксиально или некоаксиально расположенными трубами, при вынужденном движении и свободной конвекции.
Доннелл и Кенеди [27] показали, что при перегонке на трубчатых щелевых колоннах можно достигнуть ВЭТС 0,5—0,6 см при соблюдении следующих условий: обеспечение одинаковой ширины щели по поперечному сечению и высоте колонны (достигается путем применения калиброванных труб); равномерное распределение пленки жидкости по поверхности стенок (достигается путем спиралевидного травления стенок); адиабатический режим перегонки (обеспечивается хорошей теплоизоляцией колонны или использованием обогревающего кожуха); постоянная нагрузка колонны по пару (достигается с помощью автоматического регулирования скорости потока паров).
В ряде работ Фишера [31 ] описана разработанная им стеклянная трубчатая щелевая колонна новой конструкции, а также приведены полученные на ней экспериментальные данные. Эта колонна предназначена, главным образом, для ректификации высококипящих веществ; она обладает гидравлическим сопротивлением от КГ1 до 10“3 мм рт. ст. при удерживающей способности по
Рис. 256.
Зависимость гидравлического сопротивления от производительности колонны при атмосферном давлении, рассчитанная на 50 теоретических ступеней разделения 1281: 1, 2, 3 — см. рис. 255.
341
Рис. 257.
Автоматическая лабораторная установка для полумикроректификации, разработанная Фишером.
Установка содержит системы регулирования мощности электронагревателя масляной банк по температуре масла или перепаду давления потока пара в колонне, мощности электронагревателя кожуха колонны, флегмового числа путем деления потока пара, стабилизации вакуума, объема отбираемых фракцйй дистиллята. Предусмотрена регистрация всех параметров процесса. Установка снабжена предохранительными устройствами против превышения заданной температуры н выхода из строя системы подачи охлаждающей воды.
а — схема участка трубчатой щелевой колонны:
1 — наружная труба со спиралевидной канавкой на внутренней стенке; 2 — внутренняя труба со спиралевидной канавкой иа наружной стенке; <3 — кольцевой канал для прохода паров; *
б — общий вид установки:
1 — масляная баня на подъемной платформе; 2, 8, 10 — термометры сопротивления масляной бани, обогревающего кожуха и головки колонны соответственно; 3 — куб; 4 — контрольный термометр куба; 5 — электромотор мешалки; 6 — трубчатая щелевая колонна; 7 — кожух для компенсационного обогрева колонны; 9 — головка колонны с дефлегматором; 11 — сердечник клапана; 12 — магнитная катушка; 13 — холодильник дистиллята; 14 — вакуумированный сосуд; 15 — сборник фракций; 16 — устройство для установки пробирок; 17 — магнитная муфта; 18 — автоматическое приводное устройство; 19 — холодильник прибора, измеряющего перепад давлений; 20 — шкаф управления; 21 — шесть компенсационных самописцев; 22 — прибор «Миннтрон 5» для регулирования мощности электронагревателей куба и кожуха колонны, а также для установки электронного реле головки колонны; 23 — регулятор перепада давления в колонне и подачи охлаждающей воды; 24 — стабилизатор вакуума; 25 — регулятор сборника фракций; 26 — шкаф для запасных частей н принадлежностей.
Изготовитель: фирма Фишера «Лабораторная и исследовательская техника», Бонн—курорт Годесберг.
жидкости 0,1 — 1,5 мл. Ректификационная установка с этой колонной, оснащенная большим числом автоматических приборов и регуляторов, предназначена для работы при атмосферном давлении и под вакуумом (остаточное давление до 10~3 мм рт. ст.), причем давление в кубе не превышает 0,1 мм рт. ст.
Высокая эффективность массопереноса достигается в колонне Фишера благодаря гидродинамически оптимальному конструктивному выполнению стенок кольцевого канала (рис. 257я). Оптимальную скорость потока пара в колонне шипт можно рассчитать по формуле:
гиопт = 2£> 1^2/а (189)
где D —коэффициент диффузии низкокипящего компонента в паре при рабочих условиях; а — ширина щели.
На рис. 2576 схематически показана автоматическая лабораторная установка Фишера для полумикроректификации. Эта установка может снабжаться семью различными колоннами с числом теоретических ступеней разделения от 10 до 80; объем загружаемой исходной смеси составляет от 0,5 до 4000 мл. Масляная баня с регулируемым подводом тепла позволяет поддерживать постоянную температуру куба с точностью около 0,1 °C. Сведения о других элементах и узлах установки приведены в разд. 8.1.2.
343
7.3.2. НАСАДОЧНЫЕ КОЛОННЫ
Насадочная колонна представляет собой трубу, в которой размещены слои из нерегулярно уложенных элементов насадки (см. разд. 4.10.2). Насадку укладывают на решетки различной формы (рис. 258) или на перфорированные воронки (рис. 259), применяемые например, в ректификационных колоннах Штаге для установок «Лабодест» и в колоннах по нормалям «Дестинорм», разработанных автором. Свободное сечение решеток не должно быть меньше свободного сечения уложенных на них слоев насадки. Обычно сначала засыпают слой более крупной насадки, на которой затем укладывают основную насадку, размеры которой определяют из следующего соотношения:
Внутренний диаметр колонны Ю
Эквивалентный гидравлический диаметр насадки
Чтобы предотвратить растекание жидкости к стенкам (см. разд. 4.2), необходимо неоднократно собирать флегму по высоте колонны и снова распределять ее, возвращая к центральной части колонны. Гроссе-Етрингхауз [32] пытался достичь этого, исполь-зуя колонну, в корпусе которой были кольцевые вмятины глубиной 2 мм на расстоянии 30 мм друг от друга по высоте колонны (рис. 260). С этой же целью колонну можно разделить на отдельные участки и перераспределять флегму в конце каждого участка (см. разд. 4.10.2), а также использовать специальные вставки из металлической сетки с коническими углублениями. Их преимущество состоит в том, что они практически не имеют мертвых объемов [33].
Рис. 258.
Решетки для размещения насадки: спиральная (а); плоская (б);
1 — стеклянный стержень; 2 — носок для стекания капель.
Рис. 259.
Перфорированная воронка для поддержания насадки.
Рис. 260.
Колонна Гроссе—Етрннгхауза с кольцевыми вмятинами в корпусе.
344
Выбор диаметра насадочной колонны определяется требуемой пропускной способностью (см. разд. 4.11). В лабораторных ректификационных установках используют обычно колонны диаметром 10—50 мм. Колонны диаметром 50—200 мм относятся к полупромышленным. Пилотные установки снабжаются ректификационными колоннами диаметром от 150 до 400 мм.
Рабочую высоту слоев насадки обычно выбирают в интервале от 500 до 1000 мм, чтобы легко было сравнивать результаты испытаний насадок различных типов. В зависимости от цели перегонки и необходимого числа теоретических ступеней разделения применяют колонны, составленные из отдельных царг, что особенно выгодно при аналитической дистилляции, так как можно без особых трудностей собрать колонну требуемой высоты. Кроме того, подобные колонны удобны при сравнительной ректификации, моделирующей промышленный процесс, когда рабочая высота насадки точно определена по числу теоретических ступеней разделения, а также по параметрам гидродинамического режима процесса. В этом случае может оказаться, что необходимая высота колонны не совпадает с высотами стандартизованных колонн.
7.3.3. ТАРЕЛЬЧАТЫЕ КОЛОННЫ
Тарельчатые колонны применяют в лабораторных и промышленных установках для специальных задач разделения, например для получения дистиллята с высокой степенью чистоты методом непрерывной ректификации. Эти колонны используют также в лаборатории для проведения сравнительной ректификации, моделирующей промышленный процесс. Колонны с ситчатыми тарелками пригодны для аналитических разгонок при атмосферном давлении. Тарельчатые колонны используют в промышленных ректификационных установках непрерывного действия [34].
Новые типы лабораторных и промышленных тарельчатых колонн подробно рассмотрены Штаге [35]. В этой работе обсуждены также вопросы испытаний эффективности подобных колонн. Преимущества колонн с колпачковыми тарелками для непрерывных процессов перегонки разобраны в разд. 5.2.2.3 (см. также рис. 167). Коэффициент полезного действия колпачковых тарелок при циклическом способе ведения процесса превышает 150%.
По конструктивному выполнению различают фонтанирующие, колпачковые и ситчатые тарелки *. Ректификацию в тарельчатых колоннах проводят в основном при атмосферном давлении, вслед-ствии их высокого гидравлического сопротивления потоку паров, которое существенно превышает гидравлическое сопротивление колонн других типов.
* Как эти, так и ряд других тарелок принципиально сходной конструкции (провальные, клапанные и др.) в отечественной технической литературе принято называть барботажными. — Прим. ред.
345
Рис. 261.
Колонна Кеезома с фонтанирующими тарелками:
1 — ванны; 2 — трубы.
Рис. 262.
Колонна Брууна с колпачковыми тарелками:
1, 3 — трубы для подвода и отвода жидкости; 2 — колпачок; 4 — штуцер для прохода пара.
Принцип действия тарельчатых колонн заключается в барботаже восходящего потока пара через перемещающийся по тарелке слой жидкости определенной высоты. Флегма перетекает с вышележащей тарелки на нижележащую по переливным трубам, расположенным как внутри, так и снаружи колонны (см. разд. 4.2 и 4.7).
Колонна с фонтанирующими тарелками Кеезома [36] показана на рис. 261. Жидкость находится в ваннах 1, а флегма стекает по трубам 2. Во всех колоннах с фонтанирующими и колпачковыми тарелками поток пара при входе в слой жидкости на тарелке меняет направление движения и барботирует через жидкость, двигаясь в виде отдельных пузырьков радиально вверх. Колонны с фонтанирующими тарелками Кеезома применяются сравнительно редко из-за низкого коэффициента полезного действия тарелки, не превышающего 50%. Колонны с колпачковыми тарелками различных конструкций находят широкое применение прежде всего для определения необходимых параметров масштабного перехода.
Штаге [35 ] предложил для непрерывных процессов разделения лабораторные и промышленные колонны с колпачковыми тарелками, предназначенные для работы в широком интервале нагрузки по жидкости. Эффективность этих колонн не зависит от нагрузки по жидкости и соотношения нагрузок паровой и жидкой фаз, а также от кратковременных нарушений технологического режима процесса. В лабораторных установках широко применяют тарельчатые колонны Брууна [37] (рис. 262). По конструктивному выполнению они приближаются к промышленным колоннам с колпачковыми тарелками. Их недостаток заключается в том, что переточ-ные трубы для флегмы размещены снаружи колонны, и это может приводить к значительному переохлаждению жидкости. Степень охлаждения можно уменьшить, поместив колонну в заполненный воздухом или вакуумированный кожух (рис. 263). Трубы для подвода 1 и отвода 3 жидкости (см. рис. 262) расположены таким об-346
разом, чтобы на тарелках образовывались слои жидкости толщиной около 10 мм. На штуцер для прохода паров 4 свободно насажен колпачок 2, имеющий в нижней части прорези для дробления потока паров на пузырьки возможно меньших размеров.
Колонна Креля с колпачковыми тарелками (см. рис. 139) и внутренними переточными трубами для флегмы выполнена подобно промышленным колоннам. Диаметр колонны 50—70 мм; она хорошо зарекомендовала себя при сравнительных разгонках, моделирующих промышленные процессы. Эту колонну можно снабдить пробоотборниками с кранами и шлифами для термометров. Адиабатичность процесса обеспечивается с помощью разъемного обогревающего кожуха.
Многокамерная колонна Клейна, Штаге и Шульце также имеет внутренние переточные трубы для подачи флегмы [381. Распределение пара на тарелках обеспечивается с помощью отверстий, расположенных по окружностям. Через эти отверстия пар выходит в виде небольших пузырьков, интенсивно перемешивая
Рис. 263.
Колонна Брууна с колпачковыми тарелками:
а — колонна с 40 реальными тарелками и съемным изоляционным кожухом, заполненным воздухом; б — то же с 20 реальными тарелками и вакуумированным кожухом.
Изготовитель: народное предприятие «Комбинат технического стекла», Ильменау.
Рис. 264.
Колонна с колпачковыми тарелками, снабженными отражательными перегородками для пара:
а — схема движения потока пара на тарелке; б — схема колонны с тарелками.
347
Рис. 265.
Колонна с колпачковыми тарелкам» Штаге, снабженными защищенными переточными трубами.
слой жидкости, находящийся на тарелке. Штаге разработал колонну с колпачковыми тарелками диаметром от 40 до 130 мм, снабженными отражательными перегородками для пара. На каждой тарелке поток пара через кольцевую щель поступает в перемещающийся по окружности слой жидкости и затем движется радиально от периферии к центру тарелки, где отражается от пластины, установленной напротив кольцевой щели (рис. 264 а). Возникающее при этом вихревое движение фаз существенно ин
тенсифицирует процесс массообмена. Значительная длина пути вынужденного движения жидкости по тарелке от подводящей до выводящей трубы также способствует повышению эффективности разделения (см. рис. 2646 и 167). Коэффициент полезного действия тарелки в интервале применяемых рабочих нагрузок практически не изменяется и лежит в пределах от 80 до 90% [35, 39]. Колпачковая тарелка Штаге с защищенными переточными трубами (рис. 265) обладает высокой пропускной способностью по жидкости.
На рис. 266 представлены сравнительные данные по гидравлическому сопротивлению и к. п. д. тарельчатых колонн различных типов.
В табл. 50 приведены геометрические параметры этих колонн, а на рис. 267 показаны их некоторые конструктивные особенности.
Ситчатые тарелки обычно выполняют в виде перфорированных пластин. Невысокий слой жидкости на этих тарелках поддерживается благодаря динамическому напору восходящего потока паров. Очевидно, что такие тарелки характеризуются минимально допустимой нагрузкой по пару, ниже которой жидкость стекает через отверстия в тарелках.
Колонны с ситчатыми тарелками наиболее пригодны для аналитической ректификации низкокипящих углеводородов. Ситчатые тарелки Олдершоу—Гроля [41 ] выполнены в виде пластин с вертикально просверленными отверстиями диаметром 0,75— 1 мм, расположенными по окружностям (рис. 268). Флегма поступает на нижележащую тарелку по переточной трубе, расположенной в центральной части тарелки (о гидравлическом сопротивлении колонн этого типа см. разд. 4.11, табл. 30). У колонны с ситчатыми тарелками Зигварта переточные трубы установлены 348
снаружи, а тарелки выполнены в виде перфорированных цилиндрических обечаек с глухими днищами (рис. 269).
Коэффициент полезного действия колонн с ситчатыми тарелками зависит от различных факторов. Ван-Вийк и Тиссен [43] исследовали работу колонны с восемью ситчатыми тарелками внутренним диаметром 38 мм при высоте уровня жидкости на тарелке 120 мм. При ректификации бинарной смеси «-гептан — метилциклогексан было установлено, что к. п. д. тарелок значительно снижается, если концентрация любого компонента в смеси ниже 0,1% (мол.) В интервале скоростей потока пара от 10 до 27 см/с к. п. д. изменялся в пределах от 86 до 75%. При постоянной скорости пара 17 см/с и концентрации низкокипящего компонента в кубовой жидкости хв = 50% (мол.) к. п. д. увеличивался от 65 до 85% с ростом флегмового числа от 0,54 до 1,0.
Умхольц и Ван-Винкле [44] изучали работу колонны с ситчатыми тарелками диаметром 25 мм при высоте уровня жидкости на тарелке 50 мм. Свободное сечение тарелки составляло 16,2%. Оказалось, что наибольшей эффективностью обладают тарелки с диаметром отверстий 1,9 мм. При ректификации смеси о-кси-лол —м-кс и лол было установлено значительное снижение к. п. д.
Рис. 266.
Зависимости коэффициента полезного действия и гидравлического сопротивления от нагрузки по флегме для тарельчатых колонн (размеры см. в табл. 50):
1,2 — колонны типа «Лабодест» с колпачковыми тарелками, снабженными отражательными перегородками для пара; 3 — колонны типа «Лабодест» с колпачковыми тарелками, оборудованными защищенными переточными трубами; 4 — колонна с колпачковыми тарелками Шмюклера —Фрица; 5 — колонна с колпачковыми тарелками Бранда; 6 — колонна с колпачковыми тарелками фирмы Нормшлиф.
349
Таблица 50.
Площади прохода пара и флегмы для шести типов колонн с колпачковыми тарелками, испытанных Штаге [35]
(Описание конструкций колонн см. в подписи к рис. 266)
Порядковый номер колонны
Геометрический параметр колонны 1 2 3 4 5 6
Наружный диаметр, мм 55 55 55 55 62 58
Внутренний диаметр, мм 51,5 51,5 51,5 51,5 57,5 54
Площадь поперечного сечения колонны, см2 21 21 21 21 26 23
Площадь поперечного сечения штуцера для прохода паров, рассчитанная по наружному диаметру, см2 3,51 7,8 2,58 3,31 7,9 3,29
Отношение к площади сечения колонны, % 16,7 37 12,3 15,7 30,3 14,4
Площадь поперечного сечения штуцера для прохода паров, рассчитанная по внутреннему диаметру, см2 2,44 5,2 2,12 1,13 3,5 1,76
Отношение к площади сечения колонны, % 11,6 24,7 10,1 5,4 13,7 7,7
Площадь прорезей в колпачке, см2 5,58 3,1 3,1 2,25 1,82 2,1
Отношение к площади сечения колонны, % 26,65 14,7 14,7 10,7 7,0 9,2
Площадь поперечного сечения колпачка, рассчитанная по внутреннему диаметру, см2 5,58 1,83 1,83 0,85 4,33 1,0
Отношение к площади сечения колонны, % 26,65 8,7 8,7 4,05 16,6 4,36
Общая площадь для прохода пара, см2 11,16 4,93 4,93 3,10 6,15 3,10
Отношение к площади сечения колонны, % 53,3 23,4 23,4 14,8 23,6 13,6
Площадь кольцевой щели для входа пара, см2 9,7 4,15 13,45 10,0 8,83 16,3
Отношение к площади сечения колонны, % 46,3 20,0 64,0 47,5 33,9 72,5
Площадь поперечного сечения отводной трубы, см2 9,74 7,17 6,38 6,6 . 7,22 6,4
Отношение к площади сечения колонны, % 46,2 34,1 30,4 31,5 27,8 28,0
Площадь сечения труб для перетока флегмы, см2 1,04 0,67 0,29 0,6 0,39 0,5
Отношение к площади сечения колонны, % 4,93 3,15 1,38 2,38 1,5 2,19
350
тарелки с 90 до 65% при уменьшении абсолютного давления в колонне с 750 до 50 мм рт. ст. С ростом нагрузки коэффициент полезного действия сначала увеличивается, затем в интервале нагрузки от 200 до 250 г/(см2-ч) остается практически постоянным. В работе [45] указывается, что эффективность колонны зависит также от толщины перфорированной пластины тарелки и физико-
Рис. 267.
Схемы новых типов колонн с колпачковыми тарелками:
а — колонна Шмюклера — Фрица [401; б — колонна Бранда [35], нзго товнтель: фирма Р. Бранд, Вертхайм; каталог фирмы Бранда 68; в — ко лонна фирмы Нормшлнф, изготовитель: фирма Фридрнхса—Мачке «Сте клянные приборы Нормшлнф», Вертхайм.
Рис. 268.
Колонна с ситчатыми тарелками Олдершоу—Гроля:
а — колонна в вакуумированном кожухе, снабженном линзовыми ком пенсаторами; б — вид тарелок в работе.
Рис. 269.
Колонна с ситчатыми тарелками ЗиГварта.
Изготовитель: фирма «Йенский завод стекла Шотт», Майнц.
Рис. 270.
Колонна с ситчатыми тарелками модели «Пер-фо-дрип» [46 ].
351
химических свойств разделяемой смеси (поверхностного натяжения жидкости, плотностей паровой и жидкой фаз).
Колонна с ситчатыми тарелками модели «Перфо-дрип» [46] состоит из калиброванной трубы, внутри которой на стержне укреплены перфорированные пластины из коррозионностойкой стали (рис. 270). Расстояние между тарелками можно изменять. Тарелки могут быть выполнены из тефлона. Эта колонна обладает более высокой разделяющей способностью, чем колонна Олдер-шоу—Гроля, и позволяет обеспечивать высокую степень очистки разделяемых компонентов.
При пульсирующей подаче пара в колонну Олдершоу диаметром 32 мм (каждая тарелка имела 82 отверстия диаметром 0,85 мм, высота уровня жидкости на тарелке была равна 30 мм) Мак-Гуре и Маддокс [47] достигли существенного повышения эффективности разделения, которое зависит от амплитуды и частоты пульсаций скорости потока пара.
Основное требование к конструктивному оформлению всех тарельчатых колонн состоит в том, что выбранное межтарельчатое расстояние должно практически исключать унос жидкости с тарелки на тарелку. При размещении тарелок на небольшом расстоянии друг от друга поток пара уносит частицы жидкости на вышележащую тарелку, что значительно снижает к. п. д. тарелок.
Вагнер с сотр. [48] определял с помощью радиоактивных индикаторов степень загрязнения дистиллята высококипящими соединениями при вакуумной ректификации. Невитт с сотр. [49] проводил теоретическое и экспериментальное исследование механизма каплеобразования и распределения капель по размерам. -
Исследованию гидродинамических характеристик колонн с колпачковыми и ситчатыми тарелками посвящена работа Меликяна [50].
Некоторые параметры описанных тарельчатых колонн приведены в табл. 28, составленной Штаге [35, 38].
Значения к. п. д. тарелок различных колонн, приведенные в табл. 28, дают лишь приближенное представление об эффективности данных колонн, так как они были определены в несопоставимых рабочих условиях.
7.3.4. КОЛОННЫ С НЕПОДВИЖНЫМИ КОНТАКТНЫМИ УСТРОЙСТВАМИ
В отличие от насадочных колонн, заполненных беспорядочно уложенной насадкой, и колонн с тарелками, жестко крепящимися к корпусу, к колоннам с неподвижными контактными устройствами, относят аппараты со свободно вставленными элементами, выполненными из непроницаемого или перфорированного материала. В промышленности такие контактные устройства называют насадками *. Жидкость стекает по элементам насадки в виде
* В отечественной литературе такие колонны называют колоннами с регулярной насадкой. — Прим. ред.
352
пленки, причем степень смачиваемости насадки, выполненной из перфорированного материала, выше (см. разд. 4.2). Поскольку эффективность разделения в подобных колоннах зависит от степени смачиваемости материала насадки, их называют «колоннами со смоченными стенками».
Различают следующие виды колонн с неподвижными контактными устройствами: со стеклянной спиралью; со спиралью из металлической сетки; с проволочной спиралью; с насадкой Стедмана и с наклонными орошаемыми пластинами.
Прототипом колонны со стеклянной спиралью Видмера [51 ] (рис. 271) является колонна Голодца [52], снабженная сифонными гидрозатвором и концентрическими трубами. Основной недостаток таких колонн состоит в том, что в них не выдерживается принцип противотока фаз [38], поэтому они не могут быть рекомендованы для использования. Колонну Дафтона со спиралью переменного шага, уменьшающегося по ходу потока паров (рис. 272), целесообразно применять в процессах разделения, при которых не требуется высокой эффективности и необходимо небольшое время пребывания фаз в колонне. ВЭТС у подобных колонн обычно составляет около 10 см.
Значительно более эффективной является колонна Леки и Эвела с перфорированным контактным устройством, выполнен-
Рис. 271.
Колонна Видмера с концентрическими трубами и стеклянной спиралью.
Рис. 272.
Колонна Дафтона со спиралью переменного шага.
Рис. 273.
Колонна Леки—Эвела со спиралью из металлической сетки.
Рис. 274.
Спиральное контактное устройство Бауэра—Кука, выполненное из металлической сетки.
12 Э. Крель
353
Таблица 51.
Характеристика колонны Бауэра и Кука со спиральным контактным устройством из металлической сетки
Диаметр колонны 5 мм; высота колонны 430 мм.
Исследованная смесь: н-гептан —метилцнклогексан
Нагрузка, мл/ч ВЭТС, см Удерживающая способность по жидкости, приходящаяся на 1 теор. ступень разделения, мл
38,5 1.88 0,045
63,0 2,28
84,0 2,54
110,0 2,79 0,066
ным в виде спирали из металлической сетки [53]. В этой колонне полоса металлической сетки намотана винтообразно на стеклянный стержень (рис. 273). ВЭТС у данной колонны лежит в интервале от 1 до 5 см, а удерживающая способность примерно составляет 0,5 мл на одну ступень разделения. Сравнительно сложный способ изготовления контактного устройства Леки и Эвела описан в работе Сталкупа с сотр. [54]. Более просто изготовляется специальная спиральная насадка из металлической сетки, предложенная Бауэром и Куком [55]. Этой насадкой обычно снабжают колонны, диаметр которых не превышает 5 мм. Проволочную сетку 50 меш из монель-металла сгибают под углом 90° таким образом, что она образует расположенные друг за другом вертикальные плоские пластины, между которыми располагаются горизонтально пластины, образующие два открытых сегмента (рис. 274). Такую спираль удобно вставлять в колонну, выполненную из калиброванной трубы, втягивая ее внутрь с помощью медной проволоки, прикрепленной к одному из концов спирали. Стенки колонны предварительно смачивают маслом, которое удаляют после установки насадки с помощью растворителя. Медную проволоку растворяют в концентрированной азотной кислоте. Характеристика колонны этого типа приведена в табл. 51.
Следует особо отметить, что подобные колонны обладают чрезвычайно малой удерживающей способностью по жидкости и сравнительно низким гидравлическим сопротивлением. Эти преимущества позволяют применять данные колонны для вакуумной ректификации при остаточных давлениях до 1 мм рт. ст. и загрузке исходной смеси от 6 до 15 мл.
Сложной в изготовлении, но более эффективной является колонна Подбильняка с проволочной спиралью [56]. Проволочную спираль укладывают по винтовой линии вокруг центрального стержня. Эта спираль, известная как насадка «Хэли-грид», должна плотно прилегать к стенкам колонны, чтобы жидкость не стекала 354.
Таблица 52.
Характеристика колонн Подбильняка с насадкой «Хэли-грид»
Диаметр колонны, мм Нагрузка, мл/ч ВЭТС, см Удерживающая способность по жидкости, приходящаяся на 1 теор. ступень разделения, мл
11,0 200 0,9 0,07
245 1,14
315 1,4
375 1,64 Предел захлебывания
25,0 500 1,0 0,33
1000 1,25 0,53
1500 1,58 0,76
2000 1,90 1,07
по ним (рис. 275). Насадку «Хэли-грид» изготовляют диаметром 8—30 мм из проволоки толщиной 0,25 мм, оставляя зазор между соседними витками до 0,25 мм. Характеристика этих колонн приведена в табл. 52.
Модификация насадки Подбильняка, предложенная Брезина, выполнена в виде трубы из стеклоткани, спирально намотанной на центральный стержень, также обернутый стеклотканью. ВЭТС для данной насадки лежит в интервале от 1,6 до 2,37 см. Высокая эффективность этих колонн обусловлена образованием тонкой пленки жидкости между отдельными витками за счет капиллярных сил [57 ].
Чрезвычайно малая ВЭТС даже при сравнительно больших нагрузках позволяет рекомендовать колонну Подбильняка для аналитической ректификации. Подобные колонны наряду с много-
Рис. 275.
Колонна Подбильняка с проволочной спиралью (насадка «Хэли-грид»).
Рис. 276.
Насадка Стедмана из металлической сетки (а) и модифицированная насадка Коха—Ваи-Рэя (б).
12*
355
Таблица 53.
Характеристика колонн с насадкой Стедмана и с насадкой, модифицированной Кохом и Ван-Рэем
Диаметр колонны 25 мм, высота насадки 1 м
Тип насадки Удельная нагрузка, мл/(см2 • ч) Флегмовое число ВЭТС, см
Насадка Стедмана 30 ОО 1,25
100 1,96
200 2,42
220 2,54
Модифицированная насадка 25 40 1,43
50 100 1,59
120 100 2,35
трубчатыми и трубчатыми колоннами с кольцевой щелью являются наиболее эффективными из всех известных колонн.
Насадка Стедмана [58], выполненная в виде конусов из металлической сетки (рис. 276а), обладает высокой эффективностью, малой удерживающей способностью по жидкости и значительной пропускной способностью. Однако при сборке такой насадки трудно обеспечить правильное размещение конусов, чтобы избежать растекания жидкости к стенкам колонны. Отверстия для прохода пара А в смежных конусах смещены относительно друг друга. Эту насадку можно использовать в колонне, выполненной только из калиброванной трубы.
Кох и Ван-Рэй [59 ] предложили более простую модификацию насадки Стедмана, которая состоит из сферических элементов, придающих насадке большую эластичность (рис. 2766). Отбортованная пружинящая кромка элемента плотно прижимается к стенкам колонны, поэтому можно использовать трубы с отклонением размера диаметра от 0,5 до 1 мм. Значения ВЭТС у оригинальной насадки Стедмана и ее модификации практически равны между собой, что было установлено при исследовании процесса ректификации смеси «-гептан — метилциклогексан (табл. 53). Удерживающая способность этих колонн по жидкости при нагрузках 190— 500 мл/(см2-ч) составляет 0,2—0,75 мл на одну теоретическую ступень разделения.
В соответствии с работой Брэгга [60] число теоретических ступеней разделения для насадки Стедмана диаметром 10— 3300 мм можно определить по следующей формуле
пул = 9,2 + 31/(0,277?) + 5,04/(0,97?) (190)
где Луд — удельное число теоретических ступеней разделения, равное числу теоретических ступеней разделения, приходящихся на 1 м высоты слоя насадки; 7? — расход флегмы при рабочей температуре, л/ч.
Еще более простой в изготовлении является колонна с насадкой из наклонных орошаемых пластин [61 ]. Элементы насадки 356
Таблица 54.
Характеристика колонны с насадкой из наклонных орошаемых пластин
Диаметр, мм Число наклонных пластин, шт. Расход флегмы, мл/ч ВЭТС, см Удерживающая способность по жидкости, приходящаяся на 1 теор. ступень разделения, мл
15 230 250 1,82 0,4
30 115 400 2,40 0,83
в виде овальных пластин располагают в колонне зигзагообразно друг за другом. Материал насадки — проволочная сетка из нержавеющей стали V4A. Отверстия для прохода пара на смежных элементах насадки смещены относительно друг друга (рис. 277). Характеристики колонн с такой насадкой диаметром 15 и 30 мм приведены в табл. 54. •
Благодаря сравнительно высокой эффективности и малого гидравлического сопротивления колонны с регулярными насадками находят все более широкое применение в пилотных и полупромышленных установках для вакуумной ректификации при остаточных давлениях до 1 мм рт. ст. Некоторые насадки обеспечивают практически неизменяющиеся газо- и гидродинамические условия движения потоков фаз (см. разд. 4.2), вследствие чего разделяющая способность насадки остается постоянной при широком интервале диаметра колонны.
На основе насадки Стедмана был разработан ряд насадок, изготовляемых из различных тканей. Сведения об этих насадках можно найти в работе Эллиса и Варяванди [62]. Юхайм [63] испытывал ректификационную установку с колонной, снабженной насадкой «Мультифил». Пакет насадки представляет собой металлическую сетчатую ленту (сталь V4A), свернутую в архимедову спираль. Такое выполнение насадки позволяет равномерно распределить жидкость по ней за счет капиллярных сил и обеспечивает значительное свободное значение для прохода газа (рис. 278). Стандартизованные пакеты насадки
Рис. 277.
Колонна с насадкой из наклонных орошаемых пластин.
Изготовитель: фирма Эрнст Хааге, Мюльхайм (Рур).
357
имеют длину 100 мм и диаметр от 25 до 87 мм. Пакеты насадки устанавливают в колонне друг на друга до достижения слоя необходимой высоты. При электрическом компенсационном обогреве стбнок колонны слой насадки высотой 1 м имел эффективность, равную 60 теоретическим ступеням разделения, что соответствует ВЭТС 1,66 см (разделяли эталонную смесь о-кси-лол—лг-ксилол).
В промышленных установках обычно применяют насадку типа «Гиперфил» с высотой пакетов 100 мм и диаметром от 100 до 375 мм (максимально 900 мм). Эта насадка хорошо распределяет жидкость при диаметре колонны до 300 мм. В колоннах большего диаметра необходимо устанавливать специальные распределители жидкости (рис. 279). Хубэр с сотр. [64] разработал преимущественно для промышленных колонн пакетную насадку типа «Зульцер», которая изготавливается из металлических или из стекловолокнистых сеток и имеет систему регулярно распределенных проточных каналов (разд. 4.2), наклоненных к оси колонны.
Тимофеев и Аэров [65] провели систематическое исследование колонн с сетчатыми насадками диаметром 28 и 150 мм для скорости
Рис. 278.
Насадка из металлической сетки типа «Мультнфил» .
Изготовитель: фирма Юхайм, Золинген.
Рис.. 279.
Колонна с насадкой «Гиперфил» .
Изготовитель; фирма Юхайм. Золинген.
Рис. 280.
Схема распределителя жидкости лабораторной пленочной колонны.
358
Рис. 281.
Пакеты пленочной насадки ACV диаметром 200 мм.
потока пара от 0,07 до 0,47 м/с. В своих исследованиях они изменяли угол наклона гофров, их глубину, а также удельную объемную поверхность насадки.
Весьма близки к рассмотренным сетчатым насадкам некоторые другие пленочные насадки (часто очень дорогостоящие). Необходимым условием их успешного применения является обеспечение равномерного распределения жидкости по стенкам насадки. Насадки выполняют из металла, стекла или керамики. По данным Штаге, сотовые насадки для лабораторных и промышленных колонн изготавливают в виде пакетов высотой 200 и диаметром 25—150 мм с размерами каналов 6 X 6 и 10 X 10 мм; стандартная высота слоя насадки составляет 1000 мм. На рис. 280 представлена схема распределителя жидкости лабораторной пленочной колонны, а на рис. 281 показан пакет пленочной насадки ACV диаметром 200 мм.
Квасняк [65а] предложил насадку, состоящую из металлических пластин, в которых выштамповано большое число уступов, расположенных зигзагообразно.
Колонны, работающие при прямотоке паровой и жидкой фаз, разработаны Жаворонковым и Малюсовым [66]. Они предложили следующие контактные устройства: насадки с вертикальными каналами круглого или прямоугольного сечения, в которых потоки жидкости и пара движутся снизу вверх, и насадки с каналами, снабженными направляющими устройствами, обеспечивающими перекрестное движение потоков фаз за счет спиралеобразного течения и диспергирования потока жидкости. В работе этих авторов приведены экспериментальные данные и методы расчета колонн с такими контактными устройствами. Указано также, что наибольшая трудность при эксплуатации данных колонн заключается в необходимости предотвращения уноса жидкости с одной ступени разделения на другую.
7.3.5. КОЛОННЫ С ВРАЩАЮЩИМИСЯ КОНТАКТНЫМИ УСТРОЙСТВАМИ
Колонны с вращающимися контактными устройствами, или так называемые роторные колонны, известны с 1925 г. В работе Иоста с сотр. [73] рассматривается вопрос о перспективах раз-
359
Рис. 282.
Установка Абегга с колонной, снабженной вращающейся лентой.
а ~ принципиальная схема колонны; б — общий вид автоматизированной лабораторной установки.
Изготовитель: фирма В. Бюхи, Флавиль, Швейцария.
работки роторных колонн и о преимуществах колонн этого типа по сравнению с тарельчатыми, насадочными и колоннами других типов.
Разработка роторных колонн была вызвана необходимостью повышения эффективности разделения без увеличения удерживающей способности по жидкости и гидравлического сопротивления проходу потока паров. Такие высокоэффективные колонны особенно необходимы при микроперегонках с загрузкой смеси 1—5 г.
По конструктивному выполнению роторные колонны можно подразделить на следующие группы (см. разд. 5.1.1): а) колонны с вращающейся металлической лентой (рис. 282), которая может иметь плоскую, спиральную или крестовидную форму; б) колонны с вращающимся цилиндром (ширина щели между цилиндрическим корпусом колонны и вращающимся цилиндром составляет 1—2 мм [см. рис. 2861); в) колонны с цилиндрическим конденсатором, вращающимся внутри обогреваемого цилиндрического корпуса колонны (рис. 283); г) колонны с вращающимися и не
360
подвижными пластинами (или конусами), соединенными соответственно с валом и корпусом колонны (рис. 284); д) колонны с вращающейся спиралью * (рис. 285).
Первая лабораторная роторная колонна с вращающимися и неподвижными пластинами разработана Мейерсом и Джонсом. Урей и Хафмэн предложили для проведения процесса обогащения изотопа кислорода 18О роторную колонну диаметром 15 см и высотой 10,7 м, основанную на том же принципе. Эффективность колонны Урея и Хафмэна составила 500 теоретических ступеней
* Модификацией колонны этого типа является пленочная роторно-спиральная колонна, в которой на вертикальном валу установлены одно- или многоза-ходные рулоны отбортованных по краям лент. Пар проходит между витками рулона перпендикулярно отбортованным лентам (см. Авт. свидетельство СССР № 203621, 1967 г., патент США № 4038353, 1977 г. или патент ГДР № 126495, 1977 г.). В СССР колонны этого типа диаметрами от 240 до 2000 мм применяются для вакуумной ректификации термически нестойких веществ. — Прим. ред.
Рис. 283.
Колонна Байрона, Баумана н Коула с вращающимся конденсатором:
1 — электропривод конденсатора; 2 — холодильник конденсатора; з — охлаждаемая ловушка; 4 — обогреватель стенок колонны; 5 —вращающийся конденсатор, охлаждаемый водой; 6 — куб; 7 — обогреватель куба.
Рис. 284.
Колонна Мейерса—Джонсона с вращающимися и неподвижными пластинами.
361
Рис. 285.
Суперректнфнкатор Подбильняка со спиральным ротором центрифужного типа.
разделения. Лезесне и Лохте, разместив в трубчатой лабораторной колонне вращающуюся металлическую ленту, создали первую модель роторных колонн этого типа, которую затем неоднократно модифицировали различные исследователи.
На рис. 282а показана принципиальная схема колонны Абегга с вращающейся лентой. В этой колонне ВЭТС равна 2,5—3 см, что достигается благодаря диспергированию жидкости на мелкие капли и обеспечению противотока паровой и жидкой фаз. Лента вращается с частотой 1000—3500 об/мин. Удерживающая способность по жидкости этой колонны с внутренним диаметром 6 мм и высотой 375 мм составляет лишь 0,2 мл, а перепад давления потока пара, приходящийся на 1 м высоты колонны, равен 0,5 мм рт. ст. Благодаря таким характеристикам колонны этого типа удобно применять при ректификации очень высококипящих веществ для предотвращения их разложения. Технологические и геометрические параметры колонн различных типов с вращающейся лентой приведены в табл. 33, 55—57. Необходимо отметить, что производительность подобных колонн при работе под вакуумом значительно уменьшается, например при снижении абсолютного рабочего давления от 760 до 10 мм рт. ст. она уменьшается примерно на 70% [67].
Автоматизированная лабораторная ректификационная установка Абегга (см. рис. 2826) имеет колонну диаметром 6,5 мм и высотой 550 мм, снабженную спиралевидной лентой из нержавеющей пружинной стали или тефлона, частота вращения которой равна 1400 об/мин. Регулирование подачи флегмы в этой колонне 362
Таблица 55
Характеристика колонн с вращающимися контактными устройствами (см. также табл. 33)
[73] [25] [71] [69] о [25] [741
Примечания ’ Ширина кольцевого зазо- pa 1 мм Ширина кольцевого за- : зора 1—2 мм То же, 1 мм Ширина кольцевого за- i эора 1,09 мм Ротор высотой 1 м и диа- метром 25 мм. Соотноше- ние количеств тепла, под- веденных к стенкам ко- лонны и к кубу, 20 : 1 621 вращающихся эле- ментов 1 125 вращающихся и 124 неподвижных элементов 175 пар конических сег- ментов
Удерживающая способность по жидкости. Гидравлическое сопротивление 0,17 мл на 1 теор. ступень раз- деления. 0,5 мм рт. ст. на 43 теор. I ступени разделения 17,5 мл 1 2,0 мм рт. ст. Гидравлическое 1 сопротивление 0,2—0,5 мм рт. ст.
ним/ро ‘edoiod KHHatneda BlOlOBh 2600 800 6000 4000 250/1500 700/1700 320
(по 'ме он) 'о
О1ГЭИЬ аоаонаап’ф 8 8 ю О «О фле! 280 •е
h/irw 'хкин -В11Ч11ЭИ HdLI BMeAdjen 069 001 О о о о со О СО 210
wo *эхея ем 0,9 0,935 4,45 2,56 СМ 1,28 0,93
KHHairatfeed ВэиэиЛхэ •doax oiroHh СМ □О 38,9 500
‘1ЧИНО1ГОЯ diawuHl/ см Fw. OS ю 200— 240
WO ‘1ЧННО1Г -ом eiooiqg 58,4 О 00 001 1070 451
О fe О
я я
Тип ротора Цилиндр CU =1 Я 5 Я Д’ Си Я Я я д »я я £ 2 я я CU CQ си о я я <и =1 я о я Вращающиш CU о я я о КС я о я я я 8 я S я о 2 я <и я я <и я я я о 2 я <и я я 8 я я я о н я <и
Таблица 56.
Оптимальная частота вращения ротора в колоннах с вращающимися контактными устройствами
Тип колонны Тип ротора Нагрузка, мл/ч Рабочий интервал частоты вращения ротора, об/мии Оптимальная частота вращения ротора, об/мин ^Литера» тура
С вращающим- Цилиндр со щет-
ся цилиндриче- ками
ским ротором 36 690 400—2600 2000 [73] *
36 1330 400—2600 2000
4с 690 800—3200 2600
4с 950 400—3000 2600
4с 1170 400—2400
4с 1330 400—1600
Гладкий цилиндр; 1550 0—4000 2600 [71] **
ширина кольце- 2000 0—4000 2600
вого зазора 3000 0—4000 2600
1,09 мм 4000 0—4000 2600
С вращающей- Звездообразный 150 0—2000 1400 [75] ***
ся лентой
* Эффективность разделения при превышении оптимального числа оборотов ротора резко снижается. ♦♦ В интервале 1400—2000 об/мин эффективность разделения остается постоянной. *** При превышении 2600 об/мин эффективность разделения не уменьшается. В интервале 2200—2600 об/мин кривые эффективности поднимаются очень полого, превращаясь затем в прямые линии. Теоретически при нагрузке 3000 мл/ч кривая эффективности при превышении 2600 об/мии должна подниматься более полого.
Таблица 57.
Перепад давления в роторных колоннах
Тип ротора колонны Высота колонны, см Диаметр колонны, мм Рабочее давление, мм рт. ст. Перепад давления, Др-103 мм рт. ст. Литература
иа 1 см высоты колонны иа 1 теор. ступень разделения
Ленточный 545 6,7 1 1,3 [75]
82 20,0 1 2,0 34,4— 710
145 15,0 0,5 2,0 4,3
Цилиндр 11,5
Конденсатор 80 0,005— 1,5 4,4 20,0 [69]
осуществляется с помощью качающейся капельницы. Эффективность колонны составляет 30 теоретических ступеней разделения при нагрузке 70 мл/ч, удерживающая способность по жидкости равна 1,2 мл.
Автоматизированная установка, выпускаемая фирмой Эрнст Хааге (Мюльхайм/Рур) предназначена для микроперегонки в вакууме; она имеет колонну с вращающейся лентой. Колонна рассчитана на загрузку разделяемой смеси в объеме от 2 до 100 мл. Она может быть выполнена из кварца, высота ее составляет400 мм (^25 теоретических ступеней разделения) или 1000 мм («^50 теоретических ступеней разделения).
Суперректификатор Подбильняка центрифужного типа со спиральным ротором [68 ] имеет более высокую производительность по сравнению с другими роторными колоннами (см. рис. 285).
Во всех описанных выше колоннах жидкость за счет центробежных сил, создаваемых при вращении ротора, отбрасывается на стенки корпуса колонны в виде аэрозоля. Мелкодисперсный аэрозоль жидкости имеет большую поверхность, что обусловливает интенсивный массо- и теплообмен между жидкостью и восходящим потоком паров. Очевидно, что в таких колоннах несколько возрастает гидравлическое сопротивление проходу потока паров *.
В отличие от рассмотренных колонн Беннер с сотр. [69] предложил другое конструктивное решение. Он поместил вращающийся металлический конденсатор в колонну с обогреваемыми стенками. Эффект разделения в этой колонне возникает как за счет противоточного взаимодействия фаз в поле центробежных сил, так и за счет ряда последовательно протекающих и взаимосвязанных процессов парциальной конденсации и испарения. Байрон с сотр. [70] описал колонну аналогичной конструкции, он разработал лежащую в основе этого принципа разделения теорию «термической ректификации» (см. рис. 283 и разд. 5.4.3).
Прототипом колонн с вращающимся цилиндром являются трубчатые колонны с кольцевой щелью (см. разд. 7.3.1). В этих колоннах также имеется канал кольцевого сечения шириной 1— 2 мм с той, однако, разницей, что внутренний цилиндр вращается для обеспечения интенсивного перемешивания потока пара. Колонны данного типа были предложены Виллингэмом с сотр. [71 ]. Позднее разработкой подобных колонн занимался, главным образом, Пост [72]. На рис. 286 показана схема предложенной им колонны. Пост создал теорию о том, что эффект разделения при перегонке в меньшей степени зависит от степени турбулизации потока пара и обусловлен в основном равномерным распределе-
* Предлагаемый автором механизм интенсификации массообмена не является исчерпывающим, так как не во всех роторных колоннах образуется аэрозоль (капли) жидкости. Кроме того, автором не учитывается значительная турбулизация паровой фазы, что в подавляющем большинстве случаев имеет решающее значение. — Прим. ред.
365
Рис. 286.
Колонна Иоста с вращающимся цилиндром:
/ — куб; 2 — шарикоподшипники; 3 — цилиндрический ротор; 4 — штуцер для отбора дистиллята; 5 — вал привода;
6 — охлаждаемый кожух; 7 — карман для термопары; 8 — кольцевой канал; 9 — фланцевое уплотнение; 10 — дифференциальная термопара; // — компенсационный электронагреватель; 12 — слой изоляции.
нием пленки жидкости по стенкам колонны. На основе этой теории Иост предложил [73 ] снабжать цилиндрические роторы щетками (см. рис. 201), которые обеспечивают более равномерное распределение жидкости *. Из работ Куна [24 ] и Иоста [73 ] следует, что наибольший эффект разделения в роторных колоннах можно получить при равномерном
распределении жидкости по стенкам колонны и небольшой ширине кольцевого зазора, обеспечивающей снижение сопротивления массо- и теплопереносу в паровой фазе.
Необходимым условием эксплуатации роторных колонн всех типов является адиабатичность процесса и незначительное колебание частоты вращения ротора**. Имеются различные точки зрения относительно оптимального числа оборотов ротора и зависимости эффективности от частоты вращения ***. Из данных табл. 55 видно, что частота вращения ротора изменяется в широких пределах от 250 до 6000 об/мин. Согласно работе Иоста [73] число теоретических ступеней разделения зависит как от производительности роторной колонны, так и от частоты вращения и формы ротора. В табл. 56 приведены оптимальные числа оборотов ротора при различной производительности колонн. Соответствующие зависимости представлены на рис. 287—289.
Иост показал, что число теоретических ступеней разделения резко уменьшается при превышении оптимального числа оборотов цилиндрического ротора (см. рис. 288 и 289). Виллингэм с сотр. [71 ], напротив, установил, что число теоретических ступеней разделения непрерывно возрастает при увеличении частоты вращения ротора выше оптимальной (см. рис. 287а). В США
* Необходимо отметить, что работы по созданию роторных колонн с использованием принципа одновременной турбулизации жидкой и паровой фаз с обеспечением равномерного смачивания стенок и обновлении поверхности жидкости были впервые проведены в СССР в Государственном научно-исследовательском и проектном институте азотной промышленности и продуктов органического синтеза и Физико-химическом институте им. Л. Я. Карпова в период 1948— 1952 г. — Прим. ред.
** Это противоречит описанному выше (с. 365) принципу термической ректификации. — Прим. ред.
*** Правильнее связывать эффективность с окружной скоростью вращешя ротора, так как при этом будет учтен диаметр колонны. — Прим. ред.
366
Рис. 287.
Зависимости эффективности роторных колонн от нагрузки по жидкости и частоты вращения ротора:
а — для колонны с вращающимся цилиндром [71]; б — Для колонны со звездообразным ротором [75];
/, 2, 3, 4 — опытные кривые, полученные при различных нагрузках; 5 — теоретическая кривая
а), об/мин
Рис. 288.
Зависимость числа теоретических ступеней разделения от нагрузки и частоты вращения цилиндрического ротора (тип ЗЬ), полученная Иостом.
Рис. 289.
Зависимость числа теоретических ступеней разделения от нагрузки и частоты вращения цилиндрического ротора (тип 4с), полученная Иостом.
должна быть создана колонна с вращающимся цилиндром, которая при частоте вращения ротора 8000 об/мин и пропускной способности 4 л/ч будет иметь эффективность в 500 теоретических ступеней разделения. Оптимальное число оборотов ротора зависит также от конструктивных особенностей колонны и, особенно, ротора (см. табл. 56). Гельперин и Хаценко [74] установили, что в колонне с вращающимися и неподвижными конусами увеличение частоты вращения ротора выше 320 об/мин не влияло на эффективность разделения. Эта скорость вращения является оптимальной для колонн подобного типа; Гафмэн и Юри также нашли, что в интервале 250—1500 об/мин частота вращения ротора не оказывает заметного влияния на эффективность колонны *
Для колонн с вращающейся лентой оптимальная частота вращения ротора по различным данным лежит в интервале от 1000 до 35Q0 об/мин, причем на нее оказывает значительное влияние конструктивное выполнение ротора. Так, Мюррей [75], показал, что для колонн со звездообразным ротором оптимальная частота вращения равна 1400 об/мин при нагрузке 150 мл/ч. При дальнейшем увеличении числа оборотов ротора эффективность разделения остается постоянной (см. рис. 2876).
Следует отметить, что время установления стационарного режима работы колонны составляет для колонн с вращающейся лентой 0,5—1 ч, для колонн с цилиндрическим ротором не превышает, поданным Иоста [73], 1 ч. ИрЛин и Брунс [76] установили, что для колонны с вращающимся цилиндром при ВЭТС, равной 1 см, время выхода на стационарный режим составляет всего 6 мин **.
Как уже указывалось выше, гидравлическое сопротивление проходу паров для насадочных колонн зависит от типа и размеров насадки, нагрузки и давления и лежит в интервале 0,03— 1,3 мм рт. ст. на одну теоретическую ступень разделения, что соответствует 0,005—0,2 мм рт. ст. на 1 см высоты колонны. Для удобства сравнения в табл. 57 приведены данные по гидравлическому сопротивлению различных роторных колонн. Приведенные данные достаточно отчетливо подтверждают преимущество роторных колонн, гидравлическое сопротивление которых, по сравнению с насадочными и, особенно, тарельчатыми колоннами на несколько порядков ниже.
Другие характеристики роторных колонн примерно совпадают с соответствующими параметрами трубчатых колонн с кольцевой щелью, многотрубчатых колонн, а также колонн с проволочной сетчатой и спиральной насадками (см. разд. 7.3.1 и 7.3.4).
Из сравнения различных роторных колонн однозначно следует, что наиболее пригодной для лабораторных установок яв
* См. третье примечание па с. 366. — Прим. ред. 5
** Указанное время является, по-видимому, заниженным.—Прим. ред.
368
ляется колонна с цилиндрическим ротором (см. рис. 287а) *. При использовании этих колонн обычно необходимо обеспечивать относительно большую частоту вращения ротора. При работе в вакууме возникает затруднение, связанное с необходимостью обеспечения уплотнения быстро вращающегося вала. Этого затруднения можно избежать, если использовать электромагнитный привод. Для колонны с вращающейся спиралью Хёхер [77 ] исследовал зависимости числа теоретических ступеней разделения и удерживающей способности по жидкости от частоты и направления вращения спирали.
Нейман [78] анализировал пропускную способность промышленной роторной колонны диаметром 500 мм, состоящей из восьми контактных ступеней. Исследования промышленных роторных колонн, предназначенных для работы в вакууме, провел Мамин с сотр. [78а].
7.4. ХОЛОДИЛЬНИКИ И ДЕФЛЕГМАТОРЫ
В лабораторных и пилотных ректификационных установках холодильники применяют для следующих целей: полной конденсации паров (конденсаторы), частичной конденсации паров (дефлегматоры), охлаждения потоков жидкостей (холодильники).
Во всех этих случаях хладоагент, в качестве которого часто используют воду, служит для приема и отвода тепла. Таким образом, холодильник является теплообменником, который при необходимости может использоваться и для нагревания потока жидкости. Как и в промышленных установках, подобные теплообменники используют для подогревания исходной смеси. Если температура затвердевания дистиллята выше температуры охлаждающей воды из водопровода, то в качестве хладоагента следует использовать воду из термостата, температура которой должна быть выбрана таким образом, чтобы исключалось выпадение твердых частиц дистиллята в холодильнике. Дефлегматором называют такой холодильник, в котором путем регулирования расхода охлаждающей воды конденсируют лишь часть потока пара. Образующийся конденсат подают в качестве флегмы в колонну, а не-сконденсировавшийся остаток паров полностью конденсируют в конденсаторе и отбирают в качестве дистиллята (см. разд. 5.2.3).
Важнейшие типы лабораторных холодильников, стандартизованные по конструктивному выполнению и размерам, приведены в табл. 58.
* На основании долголетнего опыта мы считаем, что наиболее простыми в изготовлении и надежными в работе являются лабораторные роторные колонны с бессальниковым экранированным электромотором и ротором в виде гибкого «ерша». Они позволяют плавно регулировать число оборотов и не нуждаются в подшипниках благодаря свойству ротора самоцентрироваться (см. Голубев И. Ф., Мещеряков Н. В., Олевский В. М., Труды ГИАП, вып. V, 1956, с. 316—329;. — Прим. ред.
369
Таблица 58.
Стандартные холодильники для ректификационных установок
Стандарт Наименование
TGL 40-344 группа 7 Холодильники, интенсифицированные холодильники
8339 группа 1 Холодильники, технические условия изготовления
группа 2 Холодильники Либиха
группа 3 Шариковые холодильники
группа 4 Холодильники Димрота (Все указанные холодильники выполняются со стандартными шлифами или без них)
DIN 12575/53 Холодильники Либиха с кожухом, припаянным к охлаждаемой трубе
12576/56 Холодильники Либиха со стандартными шлифами
12580/53 Шариковые холодильники
12581/56 Шариковые холодильники со стандартными шлифами
12585/57 Змеевиковые холодильники
12586/55 Змеевиковые холодильники со стандартными шлифами
12591/54 Холодильники Димрота со стандартными шлифами
В основном различают холодильники двух типов. Это холодильники с наклонной охлаждаемой трубой (см. рис. 238, 5) и холодильники с вертикальной охлаждаемой трубой (см. рис. 238, 11), причем пары или охлаждающую жидкость можно вводить в холодильник как сверху, так и снизу. При вводе паров в холодильник сверху происходит наиболее интенсивное охлаждение, поскольку при этом пары проходят вдоль всей охлаждающей поверхности. Однако в этом случае возникает опасность, что конденсат переохладится. Этот вариант ввода паров применяют преимущественно при обезвоживании органических жидкостей, так как в отличие от варианта с нижним вводом сконденсировавшиеся капли воды не будут зависать в холодильнике, а будут смываться последующими порциями конденсата. В свою очередь вертикальные холодильники с нижним вводом паров обладают теми преимуществами, что в них конденсат не переохлаждается и дистиллят не смешивается с восходящим потоком несконденсировавшихся паров. Охлаждающую воду и пары обычно подают в холодильник противотоком.
В зависимости от формы выполнения охлаждающей поверхности различают следующие типы холодильников.
1) Холодильники с гладкой трубой: а) воздушные холодильники (см. рис. 238, 3 и 9) для жидкостей с высокими температурами кипения и затвердевания; б) холодильники Либиха с рубашкой 370
сравнительно большого диаметра (см. рис. 238, 5); в) холодильники Веста с рубашкой малого диаметра (рис. 290).
Интенсифицированный воздушный холодильник (рис. 291), используемый при перегонке для охлаждения жидкостей с температурой кипения по меньшей мере на 25 °C выше температуры окружающей среды, описал Тёльдте [79]. Пары поднимаются по трубе 1 и конденсируются на поверхностях кожуха 2 и трубы 4. В результате разогревания в трубе 4 возникает вертикальный ток воздуха, обеспечивающий непрерывный подвод более холодного воздуха из окружающего пространства. Гидрозатвор 5 заполняют на V5 высоты высококипящей жидкостью. Давление выравнивается путем подсасывания воздуха через штуцер 3. В холодильнике, состоящем из трубы и соосного охлаждающего кожуха, целесообразно, чтобы между трубой и кожухом был возможно меньший зазор, как в холодильнике Веста. Данные холодильники изготавливают также с охлаждаемыми шлифами (см. рис. 290).
Рис. 290.
Холодильник Веста с вмятинами на внутренней трубе небольшого диаметра и охлаждаемыми шлифами.
Рис. 291.
Воздушный холодильник Тёльдте:
/ — труба для подвода паров дистиллята; 2 — кожух; 3 — штуцер для подвода воздуха; 4 — внутренняя труба; 5 — гидрозатвор.
Рис. 292.
Холодильник Шотта с волнистой поверхностью охлаждаемой стенки.
Изготовитель: народное предприятие «Йенский завод стекла Шотт», Иена.
371
Рис. 293.
Холодильник Штаеделера с кожухом для твердых хладоагентов.
Рис. 294.
Интенсифицированный холодильник, составленный из холодильников Либиха и Димрота.
Рис. 295.
Холодильник и охлаждаемая приставка к кубу в виде колбы, разработанные Хенкелем.
Рис. 296.
Дефлегматор с изменяющейся охлаждаемой поверхностью [80 1.
2) Холодильники с развитой охлаждаемой поверхностью: а) холодильник Веста с вмятинами на охлаждаемой стенке (см. рис. 290); б) холодильник Шотта с волнистой поверхностью охлаждаемой стенки (рис. 292); в) шариковый холодильник Аллина (см. рис. 238, 8); г) холодильник Фридрихса с винтовой поверхностью охлаждаемой стенки (см. рис. 238, 7).
Большую охлаждающую поверхность на сравнительно небольшой длине можно получить, если применять охлаждаемую трубу с вмятинами или выполнять поверхность охлаждаемой стенки волнистой, шаровой или винтообразной.
3) Змеевиковые холодильники: а) змеевиковый холодильник с подачей хладоагента в кожух (см. рис. 238, 10)', б) холодильник Димрота с подачей хладоагента в змеевик (см. рис. 238, 11)', в) холодильник Штаеделера с кожухом для твердых хладоагентов (рис. 293); г) интенсифицированный холодильник с комбинированными охлаждаемыми поверхностями.
Змеевиковые холодильники наиболее пригодны для охлаждения потоков жидкости или пара при атмосферном давлении. При вакуумной ректификации вследствие значительного увеличения скорости потока паров легко возникают очаги подвисания капель жидкости. Поэтому при вакуумной перегонке лучше использовать холодильники Димрота, обладающие высоким коэффициентом теп-372
лопередачи. Холодильники Штаеделера преимущественно применяют для конденсации низкокипящих веществ, поскольку они позволяют использовать в качестве хладоагента лед или твердый диоксид углерода. Для этой же цели предназначены интенсифицированные холодильники с комбинированной охлаждаемой поверхностью, в которых паровой поток охлаждается как изнутри, так и снаружи. Такие холодильники изготавливают из трубы с волнистыми стенками, которую вставляют в гладкостенную трубу (см. рис. 292), или из комбинации холодильников Либиха и Димрота (рис. 294).
К холодильникам с развитой охлаждаемой поверхностью относится также холодильник Хенкеля [81 ], который представляет собой холодильник Либиха или Веста, свернутый в многовитко-вую спираль (рис. 295). Этот интенсифицированный проточный холодильник особенно пригоден для работы под вакуумом. При высоте 20 см он обладает охлаждающей поверхностью 500 см2 (см. для сравнения характеристики других холодильников в табл. 59).
Проточные холодильники обычно применяют в качестве дефлегматоров, однако при использовании этих холодильников возникают трудности в точной установке требуемого флегмового числа. Охлаждаемую поверхность дефлегматора, показанного на рис. 296, можно изменять, перемещая поршень путем вращения гайки с накаткой [80].
Для стеклянных холодильников минимальное значение коэффициента теплопередачи равно 0,5 кал/(см2-мин-°С). Необходимая охлаждаемая поверхность определяется по формуле:
f = Q/(Wcp) (191)
где Q — передаваемое количество тепла, кал/мин; k — коэффициент теплопередачи, кал/(см2-мин-°C); А/Ср — средняя разность температур между паром и хладоагентом, °C.
Среднюю разность температур Д/ср рассчитывают по соотношению:
(192)
где t — температура насыщенного пара, °C; и /2 — соответственно температура охлаждающей воды на входе в холодильник и выходе из него, °C.
Обширные исследования эффективности холодильников проведены Фридрихсом [83], Фридрихсом и Крузкой [84], а также Махом и Германом [85]. По данным Эйхгорна [86], коэффициенты теплопередачи для стеклянных холодильников длиной 200— 1000 мм изменяются в интервале от 1,5 до 0,5 кал/(см2-мин-°С). Известно, что коэффициент теплопередачи уменьшается с увеличением длины холодильника, поэтому целесообразно вместо одного длинного холодильника применять два холодильника меньших размеров. При этом воду в каждый холодильник необходимо подавать раздельно, поскольку коэффициент теплопередачи
373
Таблица 59.
Характеристики различных холодильников по данным Эйхгорна [861
(Э0-НИН)/1ГВЯ ‘вминчииИ1 -О1ГОХ КЭЧ-ООЕПЭХ OSOlS — ’frf CO 04 r-io io eo —, —, СЧ —• 04 04 — — 04 Tf 04 —. —« CO 04 04
(Эо • HHW • zWO)/LfBM ‘HhBtfadauoiruai хнаи'пиффео’Я 00 04 1Л LOLO LO CO 00ООЮШО4О4^Ь- 04 05 h- Г-- LO CO О О О Ф О О Г-- 04 CO Ю Tf О О 00 lO O4~ lO -Г о o d -" —" cd —- о —1 — о о о о — о
ww ‘MpAdx yowgBtfJKBirxo имнахо BHHtnLfoX 05 05 04 04 04 04 04^ Ф СЛ СО СО О СЭ О О4~
ни ‘rnpAdi HowgBtfJKBirxo dxaw -виЯ UHHHadiAHH 00 LO LO LO Ю lO СО cocococo"coo4cob>h^t^-b-ir^oo о Ь-О4Ь- со
ww ‘вхЛжоя dxawBHtf UHHJKAdBH LO 04 04 СО СО СО СО М" СО СО СО СО О О О 05 Tf 04
ww ‘вхКжоя oxaTn -енв^жв^хо вншг'П' LO О О О О О Ф о О О О О О О—1 LO LO OOOO — OJOOO’tlHlOO 04 LO Tf rf —СОГ'-004СОМ"04С004С00404 —
гиэ ‘qxooHXdaa -он UBWae^Birxo ю ООЮЮ сооооою о о о ф о 00 Ю 00 04 04 04 00 04 О оо —05 04 О— — oq Tf — CW-« 04 —< СО 04 04 04 ’Ф —* 04
WW ‘ВНИ1Г1/ 200 400 700 1000 200 300 400 200 300 200 300 200 1137 205 435
Mid BidBtfuBio dawoH L0 о Ю —' о ь- 00 ОО СП СП L0 L0 Ю LO ю 04 04 04 04 04
Тип холодильника Холодильник Либиха Шариковый холодильник Змеевиковый холодильник Холодильник Димрота Холодильник Штаеделера Интенсифицированный холодильник Шариковый холодильник Сокслета Йенский холодильник Шотта с волнистой поверхностью охлаждаемой стенки Холодильник Фридрихса с винтовой поверхностью охлаждаемой стенки Спиральный холодильник Бахмана
в длинном холодильнике уменьшается вследствие повышенного нагревания охлаждающей воды в нем и снижения вследствие этого средней разности температур между паром и водой *. Наиболее эффективными являются холодильник Димрота и холодильник Фридрихса с винтовой поверхностью, имеющие коэффициенты теплопередачи от 1,5 до 1,25 кал/(см2-мин-°С). Высокий теплосъем холодильника Шотта с волнистой поверхностью обусловлен его весьма большой охлаждаемой поверхностью, равней 1137 см2, в то время как коэффициент теплопередачи этого холодильника очень низок и составляет 0,575 кал/(см2-мин-°С).
Брезина [82] исследовал влияние конструктивных особенностей холодильника Либиха на их эффективность. Он предложил новые математические соотношения для определения эффективности таких холодильников и разработал аппаратуру для их испытаний.
Пользуясь значениями коэффициентов теплопередачи, приведенными в табл. 59 и формулой (191), можно рассчитывать геометрические характеристики холодильников всех типов. Следует иметь в виду, что при выборе холодильников по величине охлаждаемой поверхности необходимо, чтобы она превышала расчетное значение примерно на 50%. Геометрические характеристики лабораторных холодильников, выпускаемых промышленностью, изменяются в широком интервале.
Для правильного выбора охлаждаемой поверхности теплообменники полупромышленных и пилотных установок следует рассчитывать по возможности более точно. При расчете и изготовлении стеклянных элементов подобных холодильников можно пользоваться значениями коэффициентов теплопередачи, приведенными в табл. 60.
Для удобства сборки холодильников с любой охлаждаемой поверхностью стекольная промышленность выпускает охлаждающие элементы в виде пакетов змеевиков диаметром от 40 до 450 мм.
Таблица 60.
Коэффициенты теплопередачи для стеклянных холодильников
Вид теплообмена Коэффициент теплопередачи, ккал/(м2 ч °C)
Конденсация паров в холодильнике 250—300
Охлаждение жидкостей 150
Охлаждение газов 50
* Неточная формулировка, так как при снижении средней разности температур уменьшается не коэффициент теплопередачи, а теплосъем, т. е. количество тепла, отводимого в единицу времени. При увеличении высоты конденсатора снижение коэффициента теплопередачи может происходить из-за возникновения на нижней части поверхности теплообмена относительно толстой пленки стекающего конденсата. — Прим. ред.
375
7.5. ФОРШТОСЫ, ПРИСТАВКИ
ДЛЯ ДИСТИЛЛЯЦИОННЫХ АППАРАТОВ и головки КОЛОНН
Для отвода паров, образующихся в кубе при дистилляции, их конденсации и измерения температуры к кубу присоединяют фор-щтосы или приставки, использующиеся в качестве связующих элементов. При ректификации в колонну возвращают определенное количество флегмы и измеряют при этом флегмовое число. Для подобных измерений колонну снабжают специальной головкой.
При проведении непрерывного процесса ректификации требуются специальные патрубки для ввода газов и жидкостей в колонну. Конструкции форштосов, приставок и головок колонн весьма многочисленны, поэтому ниже рассмотрены лишь те, которые обладают принципиальными отличиями.
7.5.1. ФОРШТОСЫ
Форштосы являются связующим звеном между холодильником и сборником дистиллята (TGL9970—9972). Обычно они снабжаются штуцерами для присоединения к вакуумной линии (см. рис. 238, 6 и 12).
Особенно удобным является вакуумный форштос Аншюца— Тиле (см. рис. 238, 13). Этот форштос предпочтителен также при работах в условиях атмосферного давления, поскольку позволяет отбирать произвольное число фракций (см. разд. 7.2).
7.5.2. ПРИСТАВКИ для дистилляционных
АППАРАТОВ
При дистилляции приставки служат связующим звеном между кубом (в виде колбы) и конденсатором. Приставки обычно снабжаются термометрическими карманами. Приставки простейших видов показаны на рис. 238, 2 и 4. Наиболее предпочтительны приставки со стандартным шлифом NS14,5 для термометра. Они выполнены таким образом, что ртутный шар термометра при измерениях оказывается постоянно в одном и том же месте пароотводной трубы. Это особенно важно при сравнительной перегонке. Некоторые приставки стандартизованы. В табл. 61 приведены стандартные приставки. Некоторые из них показаны на рис. 297.
Приставка Кляйзена (TGL9979) снабжена двумя штуцерами со шлифами NS14.5. Правый штуцер предназначен для установки термометра со стандартным шлифом, а левый может использоваться для установки капилляра (TGL9978, DIN12404/63), мешалки или воронки для ввода жидкости (см. рис. 236). Эта приставка хорошо зарекомендовала себя при дистилляции под вакуумом и при атмосферном давлении. Правый штуцер можно удли-376
Таблица 61.
Стандартные приставки для дистилляционных аппаратов
Стандарт
Наименование
TGL 9974 Приставка с одним боковым штуцером
9975 Приставка с одним параллельным штуцером
9977 Перемычка со шлифом для термометра
9979 Приставка Кляйзена с боковым шлифом
9980 Приставка с припаянным холодильником Либиха
9981 Приставка с припаянным холодильником Димрота
13840 Приставка Праля
40—353 Приставка, применяемая при перегонке с водяным паром
DIN 12594/54 Приставки со стандартными шлифами
пять, выполняя его в виде короткой колонны с насадкой или спиралями Видмера, а также елочного дефлегматора (рис. 298). В этом случае колонна служит в качестве брызгоотделителя, как приставка Райтмайра (см. рис. 239, а) или сферическая приставка с кольцами Рашига (рис. 299). Для сильно вспенивающихся жидкостей разработаны специальные приставки (см. рис. 239,6). При перегонке с водяным паром применяют приставку Праля с трубой для ввода пара (рис. 300). Изготовляют также ряд при-
Приставки для дистилляционных аппаратов с одним боковым штуцером, TGL9974 (а), с одним параллельным штуцером, TGL 9975 (б) и перемычка со шлифом для термометра, TGL 9977 (в).
1 — муфта шлифа В по TGL 9967; 2 — конус шлифа В по TGL 9967; 3 — муфта шлифа A NS 14,5/23 по TGL 9967; 4 — конус шлнфа В NS 29/32 по TGL 9967.
Рис. 298.
Приставка Кляйзена с коротким елочным дефлегматором.
377
Рис. 299.
Сферическая приставка с кольцами Раш ига.
Рис. 300.
Приставка Праля с трубой для ввода пара.
Рис. 301.
Приставка с припаянным холодильником Либиха со штуцером для присоединения к вакуумному насосу и со шлифом для присоединения сборника дистиллята.
ставок с припаянными вертикальными (см. рис. 295) или наклонным холодильниками (рис. 301). Аппараты с наклонными холодильниками занимают сравнительно много места, поэтому чаще используют вертикальные холодильники. При использовании в качестве приставки перемычки (см. рис. 238, 4 и рис. 297в) в сочетании с винтовым холодильником или холодильником Дим-рота дистилляционный аппарат значительно компактнее.
Очень компактна приставка Хенкеля (см. рис. 296), изготавливаемая в различных модификациях [81 ]. Приставки других типов описаны Геммекером и Штаге [87].
Для полупромышленных и пилотных установок приставки в основном собираются из отдельных деталей, применяемых для монтажа трубопроводов (см. разд. 7.2). Для ввода газов и жидкостей в колонну применяют вставки с входным штуцером или кольцевым разбрызгивателем (см. рис. 138 и 142).
7.5.3. ГОЛОВКИ КОЛОНН
Головки колонн являются связующим звеном между колонной и конденсатором или дефлегматором. Поскольку их применяют в основном при ректификации, они снабжаются дополнительными устройствами для регулирования и измерения флегмового числа. В зависимости от способа получения флегмы различают головки частичной и полной конденсации [88].
Головка колонны частичной конденсации подробно описана выше (см. рис. 171). При низкотемпературной ректификации обычно используют дефлегматоры, поэтому дистиллят часто получают в виде пара. В этом случае применимы различные головки
378
частичной конденсации потока паров. Как указывалось в разд. 5.2.3, полная конденсация паров дистиллята при ректификации более предпочтительна. В лабораторной и промышленной практике в основном проводят перегонки с полной конденсацией, особенно в тех случаях, когда необходимо обеспечить высокие флегмовые числа, которые практически не удается регулировать посредством парциальной конденсации в дефлегматоре.
Торман [19], Геммекер и Штаге [87], а также Шнайдер и Шмид [89] показали, что головка колонны должна удовлетворять следующим требованиям: обеспечивать легкость регулировки и измерения флегмового числа; обладать минимальной удерживающей способностью по жидкости; иметь простую и механически прочную конструкцию, применимую как для работы при атмосферном давлении, так и под вакуумом; обеспечивать герметичность аппаратуры при распределении флегмы; предотвращать «подви-сание» жидкости; обеспечивать точность измерения температуры паров и подачу флегмы в колонну при температуре кипения или с небольшим переохлаждением. Кроме того, головка должна позволять регулировать и измерять нагрузку и флегмовое число в любой момент времени. Подобные измерения необходимо проводить в тех случаях, когда нагрузка колонны превышает 500 мл/ч, при которой визуальным путем уже нельзя подсчитать число образующихся капель. Особенно важно беспрепятственно измерять температуру паров. При этом необходимо следить за тем, чтобы на термометрический карман не попадали капли переохлажденной жидкости и давление в точках измерения температуры и давления было одинаковым.
Методы распределения небольших количеств жидкости в массообменных колоннах разобраны Клоссом [89а].
Для точного измерения и регулирования флегмового числа головки колонн снабжаются следующими устройствами и приборами: капельницами; трубками с косым срезом и капиллярами, служащими для подсчета падающих капель флегмы и дистиллята; двумя параллельно соединенными и независимо охлаждаемыми конденсаторами флегмы и дистиллята; регуляторами расхода флегмы и дистиллята в виде капилляров различной длины и диаметра, выбираемыми в соответствии с заданным флегмовым числом; механическими или электронными реле времени, обеспечивающими автоматическое регулирование расхода флегмы путем деления потока пара или жидкости (электронное реле времени обычно выполняется в комплекте с электромагнитным регулятором).
Метод подсчета капель в головках колонн не потерял своего значения до настоящего времени как способ контроля за расходом флегмы и отбираемого дистиллята, особенно при микроперегонках.
Шнайдер и Шмид [89] систематически исследовали работу различных капельниц (рис. 302). Данные, полученные ими при использовании бензина, приведены на рис. 303. Из рис. 303 отчет-
379
Рис. 302.
Капельницы:
а — формы выполнения выходного отвер стия капельниц для подсчета капель по Шнайдеру— Шмиду.
б — наконечник с несколькими капельницами, применяемый при нагрузках по жидкости, превышающих 500 мл/ч.
№ 8234, аналог—патент ФРГ №
ливо видно, что размер капель для различных капельниц существенно зависит от нагрузки по жидкости.
В головке колонны, запатентованной автором (патент ГДР 1011177) и описанной в нормалях
«Дестинорм», в качестве счетчика капель используются капилляры. Эта головка позволяет дополнительно измерять объемные расходы флегмы и отбираемого дистиллята. Плоскошлифованные и полированные капилляры обеспечивают хорошее формирование капель и вследствие небольшой их высоты не вызывают значитель-
ного подъема жидкости в них (см. рис. 128, 162).
Для большинства использующихся в лабораторной и промышленной практике органических жидкостей даже при температурах, приближающихся к температуре кипения, поверхностное натяжение на границе с воздухом составляет от 20 до 40 дин/см. Поэтому капилляры, описанные выше, будут образовывать капли объемом (4—5)-10-2 мл, т. е. объем 20—25 капель будет примерно составлять 1 мл. При повышении поверхностного натяжения объем образующихся капель увеличивается, исходя из этого можно
оценивать значение поверхностного натяжения жидкости.
Зависимости числа капель от нагрузки по, жидкости — бензину, полученные для различных капельниц. Обозначения кривых см. на рис. 302а.
•//--l. число капель, суммарный объем которых равен 1 мл.
380
Другими примерами конструкций головок ректификационных колонн, основанных на методе подсчета капель, являются йенская головка и головка «Нормаг» [91 ]. У йенской головки (рис. 304) штуцер для ввода пара расположен на высоте нижней трети конденсатора, который может быть дополнительно соединен с холодильником. Кран для отбора дистиллята имеет круговые шкалы на корпусе и на тефлоновом сердечнике, с помощью которых можно точно устанавливать воспроизводимые от опыта к опыту флегмовые числа.
Головка «Нормаг», разработанная Реном и Тайлигом (рис. 305), имеет надежно работающий «воротник» для сбора дистиллята. Конструктивной особенностью этой головки является размещение двух кранов на линии отбора дистиллята. Левый кран является регулирующим и служит для установки заданного флегмового числа, а правый кран — запорный. Регулирующие краны обычно являются кранами точной регулировки. Для этой цели они снабжаются установочными коническими углублениями или несколькими каналами. Примером подобного регулирующего крана является многоходовой кран Шульца [92]. Для повыше-
Рис. 304.
Головка лабораторной ректификационной колонны, предназначенная для работы при атмосферном давлении и под вакуумом.
Изготовитель: народное предприятие «Йенский завод стекла Шотт», Иена.
Рис. 305.
Головка ректификационной колонны Рена—Тайлига:
Рис. 306.
Головка ректификационной колонны Шнайдера—Шмида со стеклянным игольчатым клапаном.
381
Рис. 307.
Головка ректификационной колонны Хюбнера, имеющая соединенные конденсатор и холодильник дистиллята, а также клапан с коническим сердечником.
ния точности регулирования флегмового числа Шнайдер и Шмид [89] предложили головку колонны со шлифованным игольчатым клапаном, изготовленным из стекла (рис. 306).
Головку ректификационной колонны, изображенной на рис. 305, также изготавливают со стеклянным игольчатым клапаном, конструкция которого подробно описана Крамером [90]. В головках других конструкций используются клапаны со шпинделем из тефлона (см. рис. 247) или клапаны с сильфонами (см. рис. 248). Для измерения флегмового числа применяют трубки, нижний торец которых срезан под углом 60° к вертикали. Такая форма среза выбрана на основе многочисленных экспериментов (см. рис. 302 и 303). Головка колонны, предложенная Хюбнером [93], имеет соединенные друг с другом конденсатор и холодильник дистиллята. Охлаждаемый конический сердечник клапана (рис. 307) в этой головке служит для регулировки расхода отбираемого дистиллята. Благодаря установке холодильника под некоторым углом к горизонтали головка имеет небольшую высоту и в ней практически отсутствуют «мертвые» зоны. Устройство для замены сборников дистиллята, имеющееся в данной головке, обладает существенными преимуществами по сравнению со сборником Аншюца—Тиле.
Головка ректификационной колонны типа «Корад-хид», предназначенная для работы с постоянным флегмовым числом, имеет независимые конденсаторы флегмы и дистиллята [94]. Принцип 382
работы головки состоит в том, что вращающийся на шлифе конденсатор имеет зубцы 1 различного размера, с которых по каплям стекает конденсат (рис. 308). С помощью шлифа конденсатор можно установить таким образом, чтобы выбранный зубец, с которого стекает определенное количество жидкости, оказался над воронкой 2, служащей для приема дистиллята. Правильно выбрав зубец, можно получить необходимое флегмовое число. Для прекращения отбора дистиллята конденсатор устанавливают таким образом, чтобы конденсат капал с зубцов мимо воронки 2.
Автоматические головки ректификационных колонн обычно работают на принципе регулирования объема отбираемой фракции по времени отбора. В этих головках с помощью механического или электронного реле времени (см. разд. 8.4) устанавливают необходимое отношение промежутка времени включения реле (подача флегмы в колонну) к промежутку времени его выключения (отбор дистиллята), соответствующее заданному флегмовому числу. При этом необходимо, чтобы скорость выкипания жидкости в кубе поддерживалась постоянной, например, с помощью специальных устройств, описанных в разд. 8.4. Подобные головки могут работать по двум методам. По первому из них паровой поток разделяется в определенном соотношении и полученные потоки направляются в раздельно работающие конденсаторы для флегмы и дистиллята. Второй метод заключается в полной конденсации паров с последующим делением образовавшегося конденсата в определенном соотношении.
Автоматические головки колонн обладают тем преимуществом, что они не имеют кранов, вследствие чего исключается возможность загрязнения дистиллята и флегмы жировой смазкой шлифов. Кроме того, они позволяют быстро и надежно устанавливать любое флегмовое число. Удерживающая способность по жидкости у этих головок очень мала. При использовании подобных головок можно работать с флегмовыми числами от 30 до 100, в то время как при применении головок с самыми точными регулировочными н кранами обеспечение флегмового числа бо-
лее 30 представляет значительные труд-fE'l ности.
На рис. 309 (см. также рис. 2576) показана автоматическая головка Коллинза и
I Лэнца [95], основанная на делении паро-
7 вого потока. Восходящий поток паров об-
текает термометр 1 со стандартным шлифом
Щ ПТ Рис. 308.
JI Головка Ллойда— Горнбахера типа «Корад-хид» с незави-
симыми конденсаторами для флегмы н дистиллята:
J 1 — зубцы для стекания капель дистиллята; 2 — воронка
_______________________ для отбора дистиллята.
383
Рис. 309.
Автоматическая головка Коллинза—Ленца с делением парового потока: 1 — термометр со стандартным шлифом; 2 — соленоид; 3 — дефлегматор; 4 — трубчатый стержень клапана; 5 — клапан; 6 — труба для отвода паров дистиллята.
Рис. 310.
Головка Штаге [97], работающая по методу деления парового потока, снабженная двумя магнитными клапанами и выпускным сифоном.
и поступает в дефлегматор 3. Соленоид 2, соединенный с временным регулятором, поднимает через определенные промежутки времени трубчатый стержень 4 с клапаном 5, размещенном на небольшом сферическом шлифе. При поднятом клапане 5 определенное количество паров в соответствии с заданным флегмовым числом поступает по трубе 6 в холодильник дистиллята.
Описанная головка колонны особенно предпочтительна, если дистиллят является расслаивающимся гетероазеотропом, так как благодаря делению гомогенного потока паров удается сохранить соотношение компонентов в азеотропе. На принципе деления потока паров работают также обе головки типа «Нормаг» (см. рис. 232, а, б). Коллинз и Ленц [95] опытным путем установили, что при одинаковых соотношениях промежутков времени между включением и выключением регулирующего прибора метод деления потока паров обеспечивает меньшее флегмовое число, чем метод деления потока конденсата. Кроме того, при работе по методу деления потока паров требуется надежная термоизоляция головки колонны с помощью вакуумированного кожуха для предотвращения образования «дикой» флегмы. В связи с этим перед проведением ректификации необходимо определить поправки, зависящие от конструктивных особенностей головки колонны и свойств разделяемой смеси.
384
Штаге (97] предложил головку ректификационной колонны с двумя магнитными клапанами, работающую по методу деления парового потока, для которой флегмовое число точно соответствует отношению промежутков времени включения и выключения регулирующего прибора (рис. 310). Промежутки времени, в течение которых клапаны находятся в открытом и закрытом состояниях у обоих клапанов различны.
В головках колонн, работающих по методу деления потока конденсата, широко применяются сферические или конические клапаны, а также качающиеся стержни или воронки с электромагнитным приводом. На рис. 311 показана принципиальная схема головки с клапанами [96]. Восходящий поток паров конденсируется в конденсаторе 2. При закрытом коническом клапане 4
Рис. 311.
Головка Кизельбаха, работающая по методу деления потока конденсата: 1 — соленоид; 2 — конденсатор; 3 — стержень конического клапана; 4 — конический клапан; 5 — запорные клапаны.
Рис. 312.
Автоматическая головка Креля с качающейся воронкой по нормалям «Де-стинорм»:
1,5 — шлифы; 2 — корпус; 3 — регулирующее устройство; 4 — железный сердечник приводного механизма; 6 — кожух; 7 — воронка для приема дистиллята.
Изготовитель: народное предприятие «Комбинат технического стекла», Ильменау.
13 Э. Крел:
385
конденсат полностью стекает обратно в колонну. С помощью соленоида 1 стержень 3 клапана 4 поднимается через определенные промежутки времени, соответствующие заданному флегмовому числу. При этом часть конденсата через клапан 4 проходит в сборник дистиллята. Оба клапана 5 служат для прекращения подачи жидкости (вместо кранов) и могут использоваться при рабочих давлениях до 10 мм рт. ст. Штаге [97] применял простые и двойные клапаны, а также скользящие шайбы.
Головка с качающимся стержнем использовалась в колонне Абегга с вращающейся лентой (см. рис. 282). Конденсат, образующийся на поверхности конденсатора, сначала стекает на пол движный стержень, снабженный железным сердечником, а затем поступает в виде флегмы в колонну. При включении электромагнита стержень отклоняется и конденсат начинает поступать в сборник дистиллята.
При высоких нагрузках более целесообразно применять головку с качающейся воронкой. Подобная автоматическая головка, разработанная автором, показана на рис. 312. В этой головке поток паров проходит мимо делительного устройства. Конденсатор расположен таким образом, чтобы пары не проникали в пространство корпуса 2 и не создавали помех работе регулирующего устройства 3. Качающаяся воронка и приводной механизм с железным сердечником 4, крепящиеся на шлифах 1 и 5 соответственно, могут быть легко демонтированы. Это обеспечивает возможность легко устранять возникающие неполадки. Кожух 6, установленный за приемной воронкой дистиллята 7, служит для охлаждения низкокипящих веществ или для нагревания жидкостей с высокой температурой плавления. Данная головка позволяет регулировать флегмовое число с точностью ±2%.
В головках с механическим регулированием, снабженных медленно вращающимся стержнем, стекающий конденсат, попадая на вращающийся стержень, поступает далее либо в колонну, либо в сборник дистиллята. Благодаря периодическому экранированию высоконапорной струи воздуха направление вращения стержня через определенные промежутки времени меняется.
Головки ректификационных колонн для промышленных установок собирают из отдельных частей конденсаторов и делительных устройств. В этих головках преимущественно применяют клапаны с электромагнитным регулированием. Регулировка клапанов осуществляется также, как и в головках лабораторных колонн (см. рис. 306, 247). С помощью электромагнитов, электромоторов или пневматического привода можно автоматически регулировать работу клапанов (см. рис. 248). Для автоматического деления потока конденсата в основном применяются головки с качающейся воронкой (см. рис. 142). В этих головках колеблющийся сердечник движется с помощью электромагнитов, размещенных на наружной стенке колонны. Отбор дистиллята через боковой штуцер проводят при включенных магнитах.
386
7.6. КУБЫ, ПРИЕМНИКИ И СБОРНИКИ ФРАКЦИЙ ДИСТИЛЛЯТА
Кубы и приемники или сборники фракций дистиллята являются начальными и конечными элементами ректификационной установки. В куб загружают разделяемую смесь, а в приемнике или сборнике дистиллята получают очищенный или разделенный на фракции дистиллят.
7.6.1. КУБЫ
При лабораторной перегонке в качестве кубов применяют обычные круглодонные колбы с коротким или длинным горлом емкостью до 500 мл, снабженные стандартными шлифами. Для стандартизованных методов дистилляции используют колбы специальных конструкций, например колбы Энглера (см. рис. 235) и колбы Сэйболта (TGL 0-12363). Стандартизованы также дистилляционные колбы (рис. 313) емкостью от 25 до 1000 мл (TGL 0-12364).
Стандартные длинногорлые колбы (см. рис. 313) применяют в качестве кубов преимущественно при дистилляции. Благодаря длинному горлу предотвращается унос брызг. Плоскодонные колбы не следует использовать при вакуумной дистилляции, так как они недостаточно прочны. В качестве кубов ректификационных колонн наиболее подходящими являются круглодонные трехгор лые колбы. Два боковых штуцера со стандартными шлифами NS14,5 используют для установки термометра, загрузки и выгрузки веществ, а также для отбора проб, подачи газа или пара через подводящую трубу и ввода капилляра, предотвращающего пульсации давления при кипении. Боковые штуцеры должны составлять с вертикальной осью колбы угол 20°, чтобы оставалось достаточно места для размещения необходимых деталей при установке обогревающего кожуха. Целесообразно снабжать колбы крючками для стягивания пружинами шлифовых соединений. Это необходимо потому, что при ректификации в кубе возникает избыточное давление, которое может при отсутствии стягивающих устройств вытолкнуть детали, вставленные в куб на шлифах. Если в ходе ректификации необходимо часто менять термометры или желательно использовать термометры без шлифов, применяют колбы с термометрическими карманами, в которые для улучшения теплопередачи заливают немного масла (рис. 314). Для ректификации при атмосферном давлении куб заполняют разделяемой смесью на 2/3 объема, а для ректификации под вакуумом — на х/2 объема.
Для микро- и полумикродистилляции как правило необходимы небольшие колбы, которые в основном изготавливают с длинным горлом (см. рис. 127), что позволяет проводить отгонку с минимальным кубовым остатком.
Для проведения перегонки удобны колбы с плоским дном (рис. 315). Их преимущество состоит в том, что поверхность испа-
13*
387
Рис. 313.
Стандартные дистилляционные колбы (TGL 0-12364).
Рис. 314.
Трехгорлая колба с пробоотборником и термометрическим карманом.
Рис. 315.
Плоскодонная колба со сферическим шлифом.
рения в них практически не изменяется до конца отгонки, в то время как у круглодонных колб поверхность испарения постепенно уменьшается с понижением уровня жидкости. При непрерывной ректификации применяют колбы с сифонной трубкой, благодаря которой поддерживается постоянный уровень жидкости и обеспечивается непрерывный отбор кубового продукта.
При низкотемпературной ректификации используют в основном кубы цилиндрической формы, которые удобнее помещать в сосуды Дьюара (см. рис. 173, 175, 176, 180). Другие формы выполнения кубов обуславливаются конструкцией обогревателя (см. разд. 7.7.2).
Для промышленных ректификационных установок, выполненных из стекла, применяют круглодонные колбы емкостью до 4 л с коротким горлом и стандартными шлифами и трехгорлые круглодонные колбы емкостью до 10 л (TGL10102). Стекольная промышленность освоила выпуск стеклянных кубов круглой формы емкостью до 200 л и цилиндрических сосудов объемом до 375 л со штуцерами, расположенными сверху, сбоку и снизу сосуда. Цилиндрические сосуды снабжаются также крышками со штуцерами.
На рис. 316 показаны подвесные сосуды емкостью от 16 до 150 л [1,5]. Переход к сферическому или коническому шлифу может быть осуществлен с помощью промежуточной вставки. Такие большие сосуды можно нагревать с помощью водяного пара, теплоносителей или погружного электрического кипятильника. При перегонке легковоспламеняющихся и взрывоопасных веществ применяют кубы из нержавеющей стали V2A. Штаге [99] разработал металлический куб емкостью от 10 до 200 л (рис. 317) с двойным металлическим кожухом для обогрева куба при помощи масляной бани. Переход от плоских металлических уплотнительных поверх-388
ностей фланцев штуцеров к стеклянным сферическим шлифам в этом случае также можно осуществить с помощью промежуточных вставок. Наиболее безопасным стеклянным кубом является стеклянная колба с защитным металлическим кожухом и баней из расплавленного металла (рис. 318). Такие колбы изготавливают емкостью от 1 до 20 л [99]. Между колбой и металлическим кожухом имеется свободное пространство, которое заполняют расплавленным висмутом или каким-либо металлическим сплавом.*
* Например, сплавом Вуда. — Прим. ред.
Рис. 316.
Подвесной сосуд емкостью 16 — 150 л, используемый в качестве куба ректификационной установки.
Изготовитель: народное предприятие «Йенский завод стекла Шотт», Иена.
Рис. 317.
Металлический куб Штаге с приваренным кожухом для масла [98 ]:
1 — штуцер для присоединения ректификационной колонны; 2, 3, 13 — штуцеры для установки датчиков температуры; 4 — штуцер для установки датчика давления; 5 — штуцер для слива кубовой жидкости; 6 — штуцер для присоединения бака с маслом; 7, 10 — штуцеры для ввода и вывода охлаждающей воды; 8,9 — штуцеры для присоединения к циркуляционному насосу; 11, 14 — штуцеры для вывода и ввода масла; 12 — штуцер для присоединения нагревателя.
389
Рис. 318.
Куб в виде стеклянной колбы с защитным металлическим кожухом и баией из расплавленного металла (по патенту Германии).
Изготовитель: фирма Эрнст Хааге, Мюльхайм (РУР).
Кожух с расплавленным металлом обеспечивает равномерный подвод тепла к колбе. Для наблюдения
за процессом кипения и уровнем жидкости в колбе в металличе-
ском кожухе имеется два смотровых окна, размещенные напротив.
7.6.2. ПРИЕМНИКИ И СБОРНИКИ ФРАКЦИЙ ДИСТИЛЛЯТА
В качестве приемников дистиллята можно использовать практически любые колбы (см. рис. 238). Очень удобны цилиндрические градуированные приемники (см. рис. 238, 15), которые позволяют непрерывно следить за объемом отбираемого дистиллята. Если получаемый дистиллят имеет высокую температуру плавления, то его собирают в приемник с обогревающим термостатирующим кожухом, который охватывает и сливной кран (рис. 319). При аналитических разгонках необходимо также устанавливать холодильник дистиллята и термостатировать приемник, чтобы можно было измерять объем дистиллята при постоянной температуре. При обычной перегонке приемники должны сообщаться с окружающей атмосферой, а при перегонке под вакуумом их соединяют с вакуумной линией. В приемном сосуде, изображенном на рис. 320, предусмотрено охлаждение штуцера, присоединяемого к вакуумной линии [100].
Для отбора четырех фракций дистиллята без нарушения процесса вакуумной дистилляции применяют вращающийся на шлифе приемник Бредта (см. рис. 238, 14 и рис. 321), а для отбора семи фракций — приемник Брюля (см. рис. 238, 16) с семью сборниками размером 120 X 30 мм (см. рис. 127, 130). Любое число фракций можно отбирать с помощью форштоса Аншюца—Тиле (см. рис. 238, 13) со сменными приемниками (см. разд. 7.5.1). На рис. 322 показан вакуумный приемник Штаге с переключаемыми сборниками, а на рис. 137 и 142 — приемники для сбора больших количеств дистиллята, снабженные штуцерами для продувки и присоединения к вакуумной линии, с помощью которых можно работать без нарушения процесса перегонки. Приемник Рока и Янца [101 ] выполнен в виде «коровьего вымени». Он (рис. 323) снабжен магнитным регулирующим устройством. Магнит 1, укрепленный на кольце 2, притягивает к соответствующему сборнику штифт 4, подвешенный на крючке 3. Преимущество данного 390
Рис. 319.
Приемник дистиллята с термостатирующим кожухом.
Рис. 320.
Приемник дистиллята с термостатирующим кожухом и охлаждаемым штуцером А для присоединения к вакуумному насосу.
Рис. 321.
Приемник Бредта с градуированными сборниками.
Рис. 322.
Приемник Штаге с переключаемыми сборниками.
приемника заключается в том, что дистиллят не соприкасается с жировой смазкой шлифов.
Для отбора газообразного дистиллята при низкотемпературной ректификации необходимы специальные устройства.
В лабораторных ректификационных установках, работающих при атмосферном давлении, предпочтительно использовать автоматические сборники фракций, аналогичные применяемым при хроматографических работах. Эти сборники обычно имеют вра-
щающиеся диски, на которых по окружности или по спирали
Архимеда установлены пробирки. Регулирование объема отбирае-
мой пробы дистиллята осуществляется в этих сборниках следующими способами: подсчетом капель, применением сифона или фотоэлемента,
Рис. 323.
Приемник Рока—Янца с магнитным регулированием:
1 --- магнит; 2 — кольцо для крепления магнита; 3 — крючок; _____________ 4 - - штифт,
391
Рис. 324.
Сифонный приемник Креля с точной регулировкой объема отбираемых фракций дистиллята методом погружения груза.
1 — мерный сосуд с погружным грузом; 2 — фотоэлемент; 3 источник света; 4 — измерительная бюретка.
Рис. 325.
Фотоэлектрический регулятор сборника фракций Грассмана—Дефнера:
1 — источник света; 2 — мениск жидко сти; 3 — фотоэлемент.
заменой пробирок через определенные промежутки времени. Подобные принципы регулирования используют также и в специальных устройствах для вакуумной перегонки.
Сборники фракций, в которых осуществляется подсчет капель, работают таким образом, что падающие капли дистиллята приводят в действие контактное устройство или фотоэлемент; получаемые при этом импульсы электрического тока передаются через реле на счетчик капель. На шкале счетчика можно устанавливать любое заданное число капель, по достижении которого приводное устройство поворачивает вращающийся стол на определенный угол, подставляя следующую пробирку к месту приема дистиллята. При этом счетчик капель возвращается в нулевое положение.
При аналитических разгонках обычно отбирают фракции одинакового объема, фиксируя температуру в момент отбора. Объем фракции определяется загрузкой куба и составляет обычно 1 — 20 мл. Для отбора небольших фракций объемом 1—5 мл целесообразно применять метод подсчета капель, а для отбора фракций объемом свыше 5 мл лучше использовать метод регулирования, основанный на принципе сифонирования (рис. 324). Мерный сосуд 1 снабжен изогнутой трубкой, через которую осуществляется сифонирование жидкости. Поток жидкости проходит при этом мимо фотоэлемента 2 с источником света 3, дающего импульс на 392
реле. Реле срабатывает с запаздыванием на несколько секунд, при этом включается поворотное устройство стола с пробирками. Мерный сосуд с сифонной трубкой снабжен подвижным погружным грузом. Меняя глубину погружения этого груза, можно устанавливать требуемый объем отбираемой фракции с точностью =±=0,1 мл, чем устраняется необходимость замены сосудов с сифоном при изменении объема фракций.
Процесс отбора дистиллята в полностью автоматизированном сборнике фракций Грассмана и Дефнера [102] регулируется с помощью фотоэлемента. Благодаря тому, что в данном сборнике объем дистиллята непосредственно измеряется в пробирке, можно устанавливать любой объем отбираемых фракций от 0,5 до 0,8 мл с точностью до одной капли (около 0,03 мл). Регулирование объема осуществляют, поднимая или опуская источник света 1 (рис. 325). Фотоэлектрическое устройство приводит в действие поворотный механизм в тот момент, когда мениск жидкости 2 в пробирке пересекает световой луч, при этом световой пучок, преломляется в вертикальном направлении. Преломленный световой пучок воспринимается фотоэлементом 3, расположенным под приемной пробиркой. Необходимо отметить, что вогнутый мениск жидкости значительную часть светового луча отражает вниз, т. е. в глубь жидкости. В результате повторного отражения от стеклянных стенок пробирки световой пучок достигает ее выпуклого дна, которое служит своеобразной собирательной линзой, обуславливающей формирование направленного луча света.
Для регулирования отбора малых объемов около 1 мл в качестве импульса, который приводит в действие механизм поворотного стола, используют ток, возникающий при контакте между двумя электродами [103]. При этом работают с очень слабыми токами около 0,1 мА, что позволяет применять такой метод для перегонки всех обычных растворителей за исключением углеводородов. Погрешность регулирования не превышает 1 %.
При регулировании объема пробы дистиллята по времени вращающийся стол с пробирками поворачивается при помощи электрических контактных часов через заданные промежутки времени. Для отбора фракций одинакового объема по этому методу необходимо обеспечивать строгое постоянство нагрузки ректификационной колонны. Данный метод применяется в основном при микроректификации (см. рис. 2576).
7.7. НАГРЕВАТЕЛЬНЫЕ ПРИБОРЫ
И ТЕПЛОИЗОЛЯЦИЯ
Для проведения процессов перегонки необходим непрерывный подвод тепла. Это тепло в основном используется для нагревания и испарения перегоняемого вещества. Остальная часть подводимого тепла идет на термоизоляцию аппаратуры, которая должна
393
работать по возможности в адиабатических условиях. В тех случаях, когда перегонку ведут при температуре до 100 °C для предотвращения самопроизвольной конденсации паров зачастую бывает достаточно хорошей термоизоляции, в то время как при более высоких рабочих температурах в слой изоляции необходимо помещать нагревательные приборы.
Нагревательные системы в установках для перегонки предназначены для обогрева куба, для нагревания исходной смеси и кубовой жидкости и для термоизоляции.
Нагревательные приборы различных конструкций, предназначенные для нагревания и испарения перегоняемых веществ, подробно рассмотрены в работе Штаге и Геммекера [104].
7.7.1. ОБОГРЕВ КУБОВ
Дистилляционные кубы обогревают с помощью газовых горелок, электронагревателей или теплообменников. Горелку Бунзена применяют в основном только для обогрева небольших кубов, например при дистилляции по Энглеру (см. рис. 235) и при микродистилляции. Этот метод обогрева особенно удобен при перегонке сильно вспенивающихся жидкостей, поскольку уменьшением пламени горелки можно предотвратить чрезмерное вспенивание жидкости в кубе. Кубы больших размеров редко обогревают непосредственно газовым пламенем, так как это связано с опасностью перегревания и затрудняет точное регулирование температуры. Для предотвращения перегрева при работе с газовыми горелками куб помещают на металлической сетке с асбестом или применяют воздушную баню [105]. В последнем случае куб обогревается в мягких условиях более равномерно нагретыми отходящими газами. Выполнение нагревателя в виде дымовой трубы позволяет эффективно использовать тепло пламени (рис. 326).
Более надежное регулирование обогревания кубов достигается с помощью электрических нагревательных приборов. Различают электронагреватели следующих типов: открытые, закрытые, фасонные, инфракрасные, погружные кипятильники.
Плоские нагревательные элементы с открытой спиралью позволяют легко регулировать нагрузку ректификационной колонны по перепаду давления потока пара в ней (см. разд. 8.4), в то время как плоские нагревательные элементы с закрытой спиралью обладают слишком большой тепловой инерцией, что приводит в отдельных случаях к захлебыванию колонны при выходе ее на заданный температурный режим. Рекомендуется между кубом и плоским нагревательным элементом оставлять воздушный зазор и обеспечивать термоизоляцию куба для уменьшения теплоизлучения (рис. 327).
Кипятильники с зеркальными отражателями, заменяющие горелки Бунзена, являются модификацией нагревателей, работающих по принципу обогревания куба горячим потоком воздуха, 394
Действие подобных кипятильников конструкции Клейна [106] основано на концентрировании теплового излучения, испускаемого нагревательным элементом, в одной точке над выходным окном нагревателя с помощью отражательных зеркал. Наличие регулирующего устройства (электропереключателя) позволяет поддерживать требуемую температуру. Электрогорелка Хофмана [107] обеспечивает нагрев куба до 800 °C.
К фасонным нагревателям относятся нагревательные элементы с электроспиралью, которым придают форму обогреваемой поверхности (см. рис. 149, 184). При необходимости перемешивать разделяемую смесь с помощью электромагнитных мешалок можно рекомендовать куб с нагревателем, показанный на рис. 328. Для обеспечения вращения мешалки нижний тубус куба имеет плоское дно и обогревается с помощью коаксиального цилиндрического подогревателя с ленточным элементом. Безопасность при перегонке легковоспламеняющихся и взрывоопасных веществ достигается при использовании эластичных обогревающих кожухов, пригодных для колб'различных размеров. С помощью этих нагревателей, в которых спирали вплетены в стекловолокно, можно получить температуру до 400 °C. Система пружинных колец обеспечивает плотное прилегание стекловолокна к стенкам колбы (рис. 329). Наличие круглых отверстий в днище нагревателя предотвращает образование воздушных «мешков» и позволяет применять данный прибор для обогревания колб с нижним отводящим штуцером и воронок [108]. Расположение нагревательных эле-
Рис. 326.
Обогревательное устройство Хюбнера, работающее по принципу воздушной бани.
Рис. 327.
Колбонагреватель по нормалям «Дестинорм» .
Изготовитель: народное предприятие «Комбинат технического стекла», Ильменау.
395
Рис. 328.
Перегонная колба с тубусом, обогреваемым ленточным электронагревательным элементом.
Рис. 329.
Универсальный обогревающий кожух для обеспечения температур до 450 °C.
Рис. 330.
Колба с наружным кипятильником.
Рис. 331.
Колба с внутренним погружным кипятильником Юиге.
ментов непосредственно у стенок колбы обеспечивает высокую эффективность теплопередачи и уменьшает возможность перегрева при кипении, особенно при перегонке вязких жидких смесей.
Значительная эффективность теплообмена достигается при использовании погружных кипятильников (см. разд. 7.6.1). В приборах этого типа нагревательный элемент помещается в трубу. На рис. 147 показан аппарат для перегонки воды с нагревательным элементом, изготовленным из кварца. В некоторых случаях применяют колбы с карманом в днище, в который вставляют нагревательный элемент соответствующей формы (рис. 330). В подогревателе «Интус»», предложенном Юнге (рис. 331), погружной кипятильник вводят в колбу сверху и вставляют в карман на дне колбы, предназначенный для обеспечения более полной отгонки. Следует иметь в виду, что применение термоизоляционного кожуха мешает наблюдать за процессом кипения и уровнем жидкости в кубе. Непрерывное перемешивание кубовой жидкости достигается в колбе с циркуляционным обогревом (рис. 332). На тубус с вставленным нагревательным элементом наплавлен стеклянный порошок, что обеспечивает равномерное кипение кубовой жидкости. Для полупромышленных и пилотных ректификационных установок можно использовать погружные нагревательные 396
элементы типа THQ 6 мощностью б кВт (см. разд. 5.1.3.3), выпускаемые «Йенским заводом стекла Шотт» (ГДР).
Наряду с нагревателями, работающими по принципу передачи тепла теплопроводностью, в последнее время все более широкое применение находят подогреватели, передающие тепло излучением. К подогревателям такого типа, обеспечивающим равномерный подвод тепла в мягких условиях, относится инфракрасный излучатель. Различают светящиеся теплоизлучатели, в качестве которых используют лампы накаливания мощностью 150, 250 и 500 Вт, и теплоизлучатели ИК-лучей мощностью до 1000 Вт, выполняемые из металлических труб. Применяя несколько светящихся теплоизлучателей, можно получать температуры до 300 °C. ИК-теплоизлучатель можно сравнительно легко выполнить в форме обогреваемой поверхности. Эти излучатели особенно пригодны для микродистилляции. Преимущество применения инфракрасных нагревателей заключается в том, что они обеспечивают очень малую разность температур между наружной и внутренней стенками обогреваемого стеклянного аппарата. Методы использования в лаборатории высокочастотных нагревателей описаны в работе Клиса [ПО].
Обогрев кубов с помощью теплоносителей применяют в тех случаях, когда непосредственный обогрев аппарата невозможен и требуется особенно мягкий и равномерный режим нагревания. Например, применение теплоносителей необходимо при отгонке низкокипящего компонента от основной высококипящей фракции, поскольку при непосредственном обогреве в этом случае после отгонки низкокипящего компонента температура резко повышается.
Рис. 332.
Колба с циркуляционным обогревом.
Рис. 333.
Куб с нагревательным змеевиком по нормалям «Дестииорм» .
Рис. 334.
Куб с обогревающим кожухом по нормалям «Дестннорм> .
397
Рис. 335.
Погружные теплообменники для пилот* иых и промышленных аппаратов без мешалок (а) и с мешалками (б).
Изготовитель: фирма «Стеклянная
техника», Висбаден — Шнрштайн.
зуют перегретый пар (см. разд, тели — парафиновые масла,
Обычно теплоносители пропускают через открытые жидкостные бани (см. рис. 203), змеевики (рис. 333) или кожухи (рис. 334), которыми снабжается куб колонны. В тех случаях когда для получения температур выше 100 °C нельзя применить пар высокого давления, исполь-6.1). Жидкие теплоноси-
ицерин или триэтиленгликоль — нагревают в замкнутом контуре с помощью обогревающего змеевика (см. рис. 317) или термостата. Для обогрева пилотных и промышленных стеклянных аппаратов в качестве теплоносителей в основном используют водяной пар и нагретое масло. На рис. 335 показаны погружные теплообменники для пилотных и промышленных аппаратов с мешалками и без них. В качестве открытых жидкостных бань используют водяные бани для температур до 80 °C, масляные бани для температур до 330 °C (см. табл. 39), бани из расплава солей для температур 150— 550 °C (см. табл. 39); песчаные бани для любых температур, бани с расплавленным металлическим сплавом для температур выше 70 °C (см. рис. 318).
Следует иметь в виду, что при применении песчаных бань трудно регулировать температуру, а при использовании бань из расплава солей или металлов стеклянный куб необходимо вынимать из бани до начала затвердевания расплава. В противном случае куб можно разбить. Наиболее пригодными металлическими сплавами для бань являются: сплав Вуда с температурой плавления 71 °C, состоящий из 1—2 ч. кадмия, 2 ч. цинка и 7—8 ч. висмута, и сплав Розе с температурой плавления 95 ° С, состоящий из 2 ч. висмута, 1 ч. свинца и 1 ч. цинка. Применять ртуть и сплавы с более высоким содержанием свинца не рекомендуется вследствие токсичности их паров.
7.7.2. НАГРЕВАНИЕ ИСХОДНОЙ СМЕСИ И КУБОВОЙ ЖИДКОСТИ
ПРИ НЕПРЕРЫВНОЙ РЕКТИФИКАЦИИ
При проведении непрерывных процессов ректификации необходимо, чтобы исходная смесь, поступающая в колонну, была нагрета до температуры, соответствующей температуре жидкости внутри
398
колонны в месте ввода исходной смеси (см. рис. 142, 167). Для нагревания исходной смеси до температур, не превышающих 100 °C, обычно достаточно змеевикового теплообменника (см. разд. 7.4), в котором в качестве теплоносителя используется термостатированная жидкость. Через змеевик, погруженный в масляную баню, можно также пропускать кипящую жидкость. При этом температура масляной бани регулируется с помощью контактного термометра.
В полупромышленных и пилотных ректификационных установках преимущественно применяют стеклянные змеевиковые пакеты с предохранительным устройством против выталкивающего действия жидкости (рис. 336). В качестве теплоносителей в этих случаях используют водяной пар (избыточное давление 3 кгс/см2 и температура 147 °C) или горячее масло (максимальная температура 220 °C). Пар подают в верхний штуцер, а конденсат выводят из нижнего штуцера, расположенного под углом к горизонтали. Левый верхний штуцер предназначен для установки термометра.
На рис. 138 показан нагреватель, выполненный в виде наружной нагревательной обмотки, а на рис. 337 изображен нагреватель типа «Лабодест VD2», снабженный погружным нагревательным элементом, крепящимся с помощью фланца. Мощность этих нагревателей можно регулировать с помощью контактных термометров и реле (см. разд. 8.2.2).
При непрерывной перегонке кубовую жидкость можно нагревать с помощью устройств, показанных на рис. 328—335. При использовании циркуляционных испарителей количество жидко-
Рис. 336.
Пакет обогревающих змеевиков с предохранительным устройством против выталкивающего действия жидкости. NW — диаметр условного прохода. Изготовитель: фирма «Стеклянная техника», Висбаден — Ширштайн.
Рис. 337.
Нагреватель для исходной смеси типа «Лабодест VD2» с погружным электронагревательным элементом.
Изготовитель: фирма Фишера «Лабораторная и исследовательская техника», Бонн —курорт Годесберг,
399
Рис. 338.
Циркуляционные испарители системы «Нормшлнф» (а), системы «Нор-* маг» (5) и системы «Шмидт» (в) [1041.
Рис. 339.
Проточный куб типа «Лабодест» с раздельным вводом пара и флегмы, разработанный Штаге.
Изготовитель: фирма Фишера «Лабораторная и исследовательская техника», Бонн—курорт Годесберг.
ста, находящееся в кубе колонны, и, прежде всего, среднее время пребывания ее в кубе могут быть значительно уменьшены [104].
На рис. 338 показаны некоторые циркуляционные испарители с вертикальными и наклонными погружными нагревательными элементами. Горизонтальный циркуляционный испаритель типа «Лабодест» (рис. 339), предназначенный для подогрева высоко-кипящих жидкостей под вакуумом, имеет раздельный подвод греющего пара и кубовой жидкости. Аппараты, работающие с этим испарителем, обычно применяют в крупных установках. Данный испаритель целесообразно использовать также в пилотных аппаратах, показанных на рис. 141 и 142.
400
7.7.3. ТЕПЛОИЗОЛЯЦИЯ
Пары, выходящие из куба, обычно должны достигать конденсатора в адиабатических условиях, т. е. без подвода и отвода тепла. При температурах до 100 °C эти условия могут быть обеспечены с помощью соответствующих теплоизолирующих веществ. При температурах, превышающих 100 °C, снаружи изоляционного слоя необходимо размещать дополнительные подогревающие устройства. Потери тепла в окружающую среду, вызванные конвекцией и излучением, должны быть компенсированы путем подвода соответствующего количества тепла. Обычно теплоизолируют куб и все части колонны, размещенные над кубом. Достаточная термоизоляция обеспечивает не только более хорошее и воспроизводимое разделение, но и лучшее использование подводимого тепла (см. разд. 4.12).
Для термоизоляции лабораторных, пилотных и промышленных аппаратов применяют теплоизоляционные материалы с малой теплопроводностью, вакуумированные кожухи, обогревательные системы с циркулирующим теплоносителем [111 ], электронагревательные элементы, размещенные в слое изоляции.
В обычных ректификационных установках, не требующих точного регулирования флегмового числа; в качестве теплоизоляционного материала широко используют асбестовый шнур. Следует отметить, что в большинстве случаев применяют слой изоляции недостаточной толщины, в то время как этот слой должен составлять 50—60 мм. Очень удобна термоизоляция в виде полуцилиндров из стекловолокна, которые легко накладываются на колонну любой длины (рис. 340), затем эти полуцилиндры дополнительно обматывают снаружи лентой из стеклоткани. Если в качестве теплоизоляционных материалов используют магнезию или минеральное волокно, то их помещают в кожух, изготовленный из тонкого листового металла. Эффективно также дополнительно обматывать стенки колонны алюминиевой фольгой. Описанные виды термоизоляции, а также изоляция с помощью кожуха, заполненного воздухом, применимы при температурах стенок аппаратов не превышающих 60—80 °C.
Вакуумированный кожух обеспечивает надежную термоизоляцию аппаратов, работающих при температуре до 250 °C, если остаточное давление в кожухе составляет от 10“4 до 10-6 мм рт. ст. [112]. Решающее значение при этом имеет способ покрытия серебром стенок кожуха.
При необходимости обязательного наблюдения за ходом процесса перегонки часто используют колонны с прозрачными вакуумированными кожухами. В этих случаях необходимо непрерывно вакуумировать кожухи с помощью диффузионных насосов, поскольку в выпускаемых промышленностью колоннах указанные выше значения остаточного давления соблюдаются очень редко. Для уменьшения теплопотерь излучением вакуумированные ко-
401
100
100 , 500 WOO 2000 3000
Нагрузка, мл/ч
Рис. 340.
Термоизоляционный кожух колонны в виде соединенных полуцилиндров нз стекловолокна.
Рис. 341.
Зависимость числа теоретических ступеней разделения п от нагрузки колонны по жидкости [141].
Колонна диаметром 25 мм н высотой 1200 мм с посеребренным вакуумированным кожухом.
Насадка колонны: 1 — сетчатая насадка из широкотканной сетки марки DNW1 с малым гидравлическим сопротивлением; 2 — сетчатая насадка из узкотканной сетки марки DNW2; 3 — то же с использованием дополнительного компенсационного электрообогревателя кожуха.
жухи колонн обычно серебрят изнутри. В слое серебра можно оставлять окно шириной 5—10 мм для наблюдения за процессом, протекающим в колонне. Однако наличие этого окна существенно снижает эффективность теплоизоляции. Теплопотери за счет излучения можно уменьшить также путем установки внутри вакуумированного кожуха цилиндров из тонколистового металла или алюминиевой фольги. Кожух с перфорированной фольгой обладает таким же изолирующим действием, как и полностью посеребренный кожух, при этом, в отличие от последнего, позволяет наблюдать за ходом процесса в различных частях колонны [113]. Различие тепловых расширений внутренней и наружной обечаек кожуха компенсируют, снабжая наружную обечайку кожуха линзовыми компенсаторами (см. рис. 257, б, 282) или выполняя внутреннюю обечайку, кожуха со спиральным участком. Колонны диаметром менее 10 мм и высотой более 500 мм, работающие при температуре выше 150 °C, рекомендуется снабжать теплоизолированной электронагревательной обмоткой, наматываемой на вакуумированный кожух. Качество теплоизоляции можно приближенно оценивать по температуре наружной стенки аппарата, которая не должна значительно превышать комнатную температуру-
Фишер и Вейанд [27 ] показали (см. гл. 8), что дополнительный электронагреватель, размещенный на вакуумированном кожухе 402
колонн, значительно повышает эффективность разделения (рис. 341). Не рекомендуется выполнять термоизоляцию в виде кожуха с проточным теплоносителем — паром или термостатированной жидкостью (см. разд. 7.7.1), поскольку в этом случае вследствие непосредственного контакта теплоносителя с колонной даже при небольшой разности температур имеет место значительный подвод или отвод тепла от стенок колонны [112]. В результате будет образовываться «дикая» флегма или может быть вызвано дополнительное испарение жидкости по высоте колонны. Во избежание этого Коллинг и Трамм [114] предложили помещать слой термоизоляции между стенкой колонны и кожухом с циркулирующим маслом, а также снаружи кожуха.
Надежную термоизоляцию лабораторных и пилотных аппаратов удается обеспечить только при использовании электрообогре-ваемого кожуха со слоем термоизоляции необходимой толщины. Такая термоизоляция особенно необходима при перегонке под вакуумом. На рис. 342 показаны нагревательные элементы со стекловолокном, которые были использованы для термоизоляции куба и колонны в установке молекулярной дистилляции [115]. Вебер [112] показал, что тепловой поток через стенку колонны не зависит от того, на каком расстоянии от стенки в слое теплоизоляции расположена электронагревательная обмотка. Намотка спирали непосредственно на корпус колонны не вызывает каких-либо нарушений технологического режима процесса. *
Очевидно, что вследствие наличия температурного градиента по высоте колонны, необходимо выполнять обогревающий кожух из отдельных секций, мощность которых целесообразно регулировать с помощью контактных термометров (см. разд. 8.2.2). Не рекомендуется регулировать температуру обогревающего кожуха по температуре кипения жидкости в головке колонны за исключением тех случаев, когда разность температур между головкой и кубом не превышает 1 —2 °C.
* Без особой необходимости наматывать спираль непосредственно на корпус колонны не рекомендуется во избежание местных перегревов и возможных поломок вследствие термических напряжений. — Прим. ред.
Рис. 342.
Нагревательные элементы со стекловолокном, устанавливаемые на аппарате для молекулярной дистилляции.
403
Рис. 343а.
Зависимость температуры первого нагревательного контура от длины колонны, полученные Кортюмом н Биттелем.
I — расстояние от верхнего конца колонны; N — число витков нагревательной спирали, приходящееся иа 10 см ее длины.
Рис. 3436.
Схема обогревающего кожуха с термопарами.
Кортюм и Биттель [116] предложили применять для обогрева колонны два нагревательных элемента, которые позволяют получать любую постоянную разность температур по длине обогревающего кожуха. Для этой цели они использовали два нагревательных контура, в первом из которых нагревательная спираль наматывалась таким образом, что расстояние между витками увеличивалось от нижней к верхней части колонны. Соответствующее понижение температуры регистрировали термопарами, установленными через каждые 10 см высоты колонны (см. рис. 343, а). Оказалось, что при любой силе тока в контуре обеспечивается линейное изменение температуры по высоте колонны.
Нагревательная спираль второго контура, изолированная бусами, наматывалась на первый контур, причем расстояние между витками во втором контуре было одинаковым и соответствовало расстоянию между витками в средней части первого контура. Снаружи спирали изолировали толстым асбестовым шнуром и кожухом, заполненным магнезией или кизельгуром. Два контактных термометра устанавливали таким образом, чтобы их сосуды для ртути соприкасались со стенкой колонны в местах, удаленных на 10 см от верхнего и нижнего ее концов. Верхний контактный термометр регулировал с помощью реле мощность второго контура, а нижний —мощность первого контура. При этом в каждом контуре регулировалась только часть общей мощности.
404
На рис. 343, б показана принципиальная схема обогревающего кожуха, регулируемого с помощью термопар [117]. Мощность обогрева 85 Вт (220 В; 0,39 А) достаточна для получения температур до 200 °C; для достижения более высоких температур необходима мощность обогрева 100 Вт (220 В; 0,46 А). При калибровке мощности подобного кожуха в него помещают колонну соответствующей длины с термометрами, введенными на шлифах в местах, удаленных от концов колонны на расстояниях, равных 1/3 её длины. При различных мощностях обогрева измеряют температуры, установившиеся внутри колонны, и берут среднее значение из показаний обоих термометров. Строят график зависимости температуры внутри колонны от мощности обогрева.
Втабл. 62 приведены опытные данные Вебера [112], полученные при калибровке обогревающих кожухов. Мощность обогрева соответствует теплопотерям, которые имели бы место при работе колонны без внешнего обогрева. Подводимая теплота отнесена к 1 м высоты колонны, а эквивалентное этому теплу количество конденсирующихся паров рассчитано на основе правила Трутона по скрытой теплоте парообразования при атмосферном давлении.
Используя график зависимости мощности обогрева от температуры внутри колонны можно регулировать реостатом мощность обогрева кожуха по показаниям ваттметра или амперметра. Более точное автоматическое регулирование можно обеспечить, применяя термопары или газонаполненные термометры (см. разд. 8.2.2).
Необходимо изолировать также и различные детали между кубом и колонной, колонной и конденсатором, и участки колонны между отдельными секциями обогревающего кожуха. Если эти участки не изолировать, то они начинают играть роль парциальных дефлегматоров, образующих дополнительное количество флегмы. При перегонке высококипящих веществ часто бывает затруднительно выводить пары из колонны. В этих случаях применяют паровые трубы с вакуумированным кожухом (см. рис. 309, 310), которые обеспечивают надежную термоизоляцию при температуре паров до 150°С. Для получения более высоких температур предпочтительно использовать дополнительный нагревательный элемент
Таблица 62.
Результаты калибровки обогревающего кожухв колонны
Температура, °C Мощность обогрева, Вт Подводимая теплота, кал/(м-ч) Количество конденсирующихся паров ректифицируемого вещества, эквивалентное подводимой теплоте, моль/(м«ч)
75 14 9 300 1,27
157 42 28 700 3,2
219 77 51 000 5,0
297 114 76 000 6,3
405
Рис. 344.
Нагревательный элемент в виде электропроводящей ленты типа «Хемотерм».
Изготовитель: народное предприятие «Электрическое тепло Бельциг», Бельциг.
в виде электропроводящей ленты (рис. 344). Такие ленты изготавливают длиной 600—2500 мм и шириной 6—90 мм [118]. Их потребляемая мощность составляет 36—850 Вт, что при удельной мощности 0,4—1,25 Вт/см2 позволяет получать температуры выше 400 °C. Для достижения более высоких температур используют гибкие проволочные спирали из нержавеющей жаропрочной стали. Диаметр проволоки равен 6 мм. Применяя спирали различной длины, получают максимальные температуры 450 и 800 °C. Удельная мощность спирали составляет 4,65 Вт/см2. Ленточный нагревательный элемент фирмы «Электротермаль энджиниринг» (Лондон) выпускается в виде ленты, наматываемой на барабан. По заданной мощности электрообогрева с помощью калибровочного графика определяют необходимую длину электронагревательной ленты, которую можно разрезать на участки любой протяженности. К концам отрезанного участка ленты прикрепляют вручную с помощью небольшого простого приспособления соединительные клеммы.
При использовании ленточных нагревательных элементов необходимо следить за тем, чтобы пары не перегревались, в противном случае возможно искажение температуры в головке колонны. Для предотвращения этого следует предварительно откалибровать нагревательный элемент тем же способом, что применяется для калибровки обогревающего кожуха колонны. При перегонке веществ с высокой температурой плавления легко поддерживать необходимую температуру соединительных коммуникаций перед приемником дистиллята и самого приемника с помощью ленточных нагревателей.
Для термоизоляции стеклянных трубопроводов применяют трубчатые кожухи из меди и свинца, плотно прилегающие к стенкам стеклянных труб. Эти кожухи обогревают снаружи паром и теплоизолируют. Эластичный профилированный шланг «Калорекс» [119] из синтетического каучука также можно использовать для термоизоляции стеклянных коммуникаций. Выпускают шланги с диаметрами условного прохода 8, 15 и 30 мм. Коэффициент теплопередачи от таких шлангов к стеклу толщиной 1,5 мм лежит в пределах от 165 до 260 ккал/(ч-м2 град).
406
7.8. НАСАДКИ
Нерегулярно уложенные насадки (см. разд. 4.10.2) применяют для получения в ректификационных колоннах возможно большей поверхности, по которой жидкость распределяется в виде тонкой пленки (см. разд. 4.2). * В некоторых случаях подобного эффекта достигают, используя устройство, симметрично размещенное в свободном пространстве колонны [118а]. Часто поверхность насадки, на которой происходит лассо- и теплообмен, называют активной поверхностью. Чем меньше по размеру элементы насадки, тем больше их суммарная поверхность, приходящаяся на единицу объема колонны. Однако при этом соответственно возрастает удерживающая способность насадки по жидкости, что снижает разделяющую способность колонны (см. разд. 4.10.5). Таким образом, приходится выбирать для конкретного процесса перегонки оптимальные форму, размер и материал насадки с учетом всех необходимых факторов. В работе Лева [120] приведены обширные сведения о характеристиках и методах расчета различных полупромышленных и промышленных насадочных колонн.
7.8.1. ФОРМЫ ВЫПОЛНЕНИЯ ЭЛЕМЕНТОВ НАСАДОК
Разработаны и испытаны насадки с элементами разнообразной формы, однако наиболее широко применяют насадки с цилиндрическими элементами. Из насадок с элементами нецилиндрической формы наиболее применимы в лаборатории шаровая и седловидная насадки. Исследованы насадки следующих видов: участки стеклянных спиралей, стекловолокно, цепная насадка, скобы, гвозди, полусферы из листового металла. Характеристики насадок различных видов приведены в табл. 28.
В систематической работе Керени [121] рассмотрены методы выбора высокоэффективных насадок для ректификационных установок. В данной работе на многочисленных диаграммах представлены зависимости эффективности разделения от формы и размера элементов насадок, а также приведены рабочие параметры массообменных процессов. Кроме того, в статье [121 ] предложено характеризовать эффективность разделения с помощью фактора G, определяемого из соотношений:
С, = nlVH~Ap ~AL~ri (193)
IgG =lg (п/1/'7ГДр)= IgA— В lg L (194)
. где ii — число теоретических ступеней разделения; Н — удерживающая способ-, ность колонны по жидкости; Др — перепад давления в колонне; L — пропускная способность колонны, мл/ч; А, В — константы.
г * Данисе положение не относится к насадочным колоннам, работающим ;В режиме эмульгирования с затопленной насадкой. — Прим. ред.
407
О 200 W ООО 800 1000 1200 1400 1600 1800 2000 Нагрузка, мл/v
Рис. 345.
Сравнительные характеристики различных насадок, полученные Керенн [121].
Результаты получены при разделении смеси н-гептан—метилциклогексаи в колонне с диаметром 24 и высотой разделяющей части 500 мм. Ректификацию проводили с бесконечным флегмовым числом при атмосферном давлении.
УС — удерживающая способность по жидкости, приходящаяся иа одну теоретическую ступень разделения; Др — перепад давления пара, приходящийся иа одну теоретическую ступень разделения.
В логарифмических координатах зависимость фактора эффективности G от нагрузки представляет собой прямую линию, поэтому сравнительно легко можно выбрать наиболее эффективную насадку. На рис. 345 показаны сравнительные характеристики новых насадок Керени (/—4), а также насадок из участков спира
408
лей (5 и 6), навитых с широким и узким шагом соответственно.
Райхельт [121а] провел сравнительные испытания -насадок с цилиндрическими элементами (рис. 346, а—и и м), а также насадок в виде пружин, перфорированных и разрезанных металлических колец. Эти исследования показали, что невозможно получить удовлетворительную сравнительную оценку их массообменных и гидродинамических характеристик, хотя многочисленные типы насадок были разработаны, исходя из различных химико-технологических требований к их конструктивному выполнению.
Насадки с элементами цилиндрической формы, основным типом которых являются кольца Рашига, показаны на рис. 346. Некоторые насадки нецилиндрической формы изображены на рис. 347. К ним относятся также проволочная насадка «Хэли-пак» [122], насадка из проволочной сетки «Окта-пак» (рис. 348, 349), разработанные Подбильняком, и насадка Кдннона из перфорированных полуцилиндров (рис. 350), обладающие высокой эффективностью.
Для лабораторной и пилотной перегонки применяют в основном следующие насадки с элементами цилиндрической формы: кольца Рашига с габаритными размерами 2—10 мм из гладкого или матированного стекла, из керамических материалов и металлов (см. рис. 346, а); кольца «Прим» из металлов —2—10 мм (см. рис. 346, 5); кольца Рашига из проволочной сетки с плоской поперечной перегородкой или без нее—3—10 мм (см. рис. 346, е);
Рис. 346.
Элементы насадок цилиндрической формы (типа колец Рашнга)
а — кольца Рашига гладкие и матированные; б — кольца Рашига с поперечными канавками; в -- кольца Рашига с продольными канавками; г — кольца Рашнга с перемычками; д — кольца «Прим»; е — кольца Рашига из проволочной сетки; ж — кольца «Иитос»; з — участки спиралей; и —-спирали Вильсона; к — шпульки; л — ролики; м — кольца Паля.
409
участки спиралей из стекла и металлов —1,4—10 мм (см. рис. 346, з); спирали Вильсона из стекла —3—6 мм (см. рис. 346, и).
Кольца «Прим» в основном изготавливают из металла; их используют при сравнительной ректификации, моделирующей промышленный процесс. Для этой насадки опубликовано весьма мало данных по измерению числа теоретических ступеней разделения. Согласно работе Тормана [19], ВЭТС для колец «Прим» 2,5 X2,2 мм соответствует ВЭТС для насадки из участков металлических спиралей 4X4 мм. Металлические кольца Рашига, также как и элементы насадки «Перфо-ринг», могут иметь отверстия круглой формы [123].
Все более широкое применение находят модифицированные Диксоном кольца Рашига с плоской поперечной перегородкой или без нее. Их изготавливают путем сворачивания проволочной сетки (примерно 1500—3000 отверстий наем2) в один или два слоя [124, 125]. Сравнительные характеристики различных насадок, выполненных из проволочных сеток, приведены в работе Эллиса и
Рис. 347.
Элементы насадок нецилиндриче-ской формы:
а — шары гладкие и матированные; б — сплошные тела Киршбау-ма; в — бусы гладкие и матированные; г — седла Берля; д — иасадка из сдвоенных элементов; е — звездообразная иасадка.
Рис. 349.
Насадка Подбильняка «Окта-пак» .
«Хэли-пак» .
Рис. 350.
Элемент насадки Каниона из металлических полуцилиндров с мелкой перфорацией.
410
Рис. 351.
Зависимость числа теоретических ступеней разделения от скорости пара в колонне, полученная для насадки из колец Рашига размером 3X3 мм без перегородки, выполненных из однослойной бронзовой сетки (данные Киршбаума).
Диаметр колонны 100 мм, высота слоя насадки 1000 мм. Разделяемая смесь н-гептан —метилциклогексан.
Варяванди [126]. Эти насадки состоят преимущественно из элементов с габаритными размерами, не превышающими 3 мм, и обладают ВЭТС лежащей в пределах 1,5—2,5 см. Существенным преимуществом подобных насадок явля-
; ется незначительное уменьшение эффективности разделения : с увеличением диаметра колонны. По данным Киршбаума [127] ВЭТС для слоя насадки длиной 1 м из сетчатых колец i Рашига 3x3 мм без перегородки даже в колонне диаметром f 100 мм находится в интервале 1,67—2,8 см при скорости i потока пара 0,02—0,27 м/с (рис. 351). Кольца Рашига были изго-товлены из однослойной бронзовой сетки. Опыты проводили 1 с использованием смеси «-гептан—метилциклогексан при бесконечном флегмовом числе.
Насадка Гроссе-Етрингхауза с элементами в виде участков : спиралей из тонкой проволоки, которые благодаря работам Шульце ; и Штаге стали известны под названием «брауншвейгские спирали», ‘ обладает очень высокой разделяющей способностью. Исследования Я автора показали, что эта насадка имеет также сравнительно Я высокую пропускную способность.
Я^^ Недостатком насадки Гроссе-Ётрингхауза является ее относи-тельно высокая стоимость, обусловленная тем, что изготовлять г спирали диаметром и высотой менее 2 мм весьма затруднительно.
Кроме того, вследствие возможной коррозии тонкой проволоки Я| в качестве материала насадки часто необходимо использовать доро-<Я гостоящую нержавеющую сталь V2A. Наибольшей эффективностью Я| разделения обладают насадки с элементами размером 1,3—2 мм. Я: Они имеют ВЭТС от 1 до 1,6 см. Если элементы спиральной насадки Я должны иметь диаметр и высоту 2 мм и менее, то применяют про-Я( волоку толщиной 0,15—0,2 мм. В зависимости от размеров участок ков спиралей для колонны объемом 1 л необходимо следующее Я| число элементов насадки:
Размер элемента, мм .................
Число элементов в 1 л ...............
2 4 20
115 000 12 500 100
| Предложено также изготавливать эти спирали из стекла. Автор I испытал насадку из плотнонавитых стеклянных спиралей разме-
411
Таблица 63.
Результаты испытаний насадки нз стеклянных
спиралей размером 4 и 7 мм
Колоииа диаметром 30 и высотой 1000 мм с длиной разделяющей части 920 мм. Разделяемая смесь: четырех хлор истый углерод — бензол. Концентрация четыреххлористого углерода в кубовой жидкости 40% (мол.); флегмовое число v = со.
Нагрузка, мл/ч ВЭТС (см) для спиралей Нагрузка, мл/ч ВЭТС (см) для спиралей
размер 4 мм размер 7 мм размер 4 мм размер 7 мм
100 5,2 5,4 400 5,6 8,0
200 6,4 800 6,1
ром4х4х0,3 и 7X7X0,5 мм при атмосферном давлении с использованием смеси четыреххлористый углерод —бензол. Результаты этих опытов при различных нагрузках приведены в табл. 63.
Спирали Вильсона являются стеклянной насадкой, элементы которой, в отличие от плотно навитых металлических спиралей, имеют 3—4 витка с шагом около 0,6 мм (см. рис. 346, и). Эта насадка имеет эффективность того же порядка, что и насадка из колец Рашига равного размера, однако она обладает значительно более высокой пропускной способностью.
Насадку с элементами в виде одновитковых проволочных спиралей изготавливают также, как и насадку из участков стеклянных спиралей. Значения ВЭТС, полученные Торманом [19 ] для насадки из одновитковых металлических спиралей, приведены ниже;
Диаметр витка, мм ... . 1,6 2,5 4,0 5,0
ВЭТС, см........... 2,5—3 4 4—8 12
Эффективность насадки из одновитковых проволочных спиралей соответствует эффективности насадок из стеклянных колец Рашига или шаров с элементами равных размеров.
Из группы насадок с элементами нецилиндрической формы в лаборатории и промышленности, в основном, применяют насадки, имеющие следующие элементы: шары из гладкого или матированного стекла размером 3—8 мм (см. рис. 347, а) и седловидные тела из фарфора, специальных материалов и проволочной сетки — 4—10 мм (см. рис. 347, а), а также насадки «Хэли-пак» из проволоки размером элементом 1,8—4,5 мм (см. рис. 348) и «Окта-пак» из проволочной сетки —5,0—7,5 мм (см. рис. 349).
Насадки из гладких и матированных шаров используют прежде всего для препаративных работ. Однако из-за большой массы сплошных шаров высота колонн с шаровой насадкой ограничена.
Насадка из элементов седловидной формы (см. рис. 347, а) обладает наилучшими гидродинамическими характеристиками.
412
Любая часть поверхности седловидного элемента, независимо от его положения, наклонена так, что создает благоприятные условия для контакта паров и жидкости. При центральной подаче потока жидкости угол ее растекания по седловидной насадке составляет 55—60°. Удельная поверхность этой насадки при размере элементов 4—10 мм превышает поверхность насадки из колец Рашига тех же размеров на 30—60%. Эта насадка имеет малое гидравлическое сопротивление потоку паров, которое, например, для насадки с элементами размером 8x8 мм примерно в два раза ниже гидравлического сопротивления насадки с кольцами Рашига того же размера. Хорошие эксплуатационные качества седловидной насадки обусловлены тем, что ее элементы не имеют острых кромок. Седловидную насадку сравнительно легко изготовлять. Следует отметить, что эту насадку выполняют также из латунной сетки 100 меш (насадка Мак-Магона) [128].
Модификацией насадки Мак-Магона является седловидная насадка «Инталокс» [129], которая отличается от обычной седловидной насадки большими поверхностью и свободным объемом, что обуславливает ее меньшее гидравлическое сопротивление (рис. 352) Для лабораторных и промышленных ректификационных колонн такую насадку изготавливают размером 6 и 12,5 мм. Видоизменением этой насадки является насадка «Гидронил-пластик-инталокс» [130], элемент которой имеет два отверстия (рис. 353). Коэффициент полезного действия данной насадки совпадает с коэффициентом полезного действия керамической насадки «Инталокс». При равном гидравлическом сопротивлении насадка «Гидронил-пластик-инталокс» обладает вдвое большей пропускной способностью по сравнению с насадкой из колец Паля. Другой модификацией седловидной насадки из металла является суперседловидная
Рис. 352.
Элемент насадки «Инталокс» .
Рис. 353.
Элемент насадки «Гидроннл-пластнк-ннталокс» .
Изготавливается из различных пластмасс размером 30,48—76,4 мм (1,2 — 3 дюйма).
413
Рис. 354.
Элемент суперседловидной насадки Кнршбаума.
Рис. 355.
Элемент насадки «Интерпак» из металла.
насадка Киршбаума [131], элементы которой имеют выпуклые с зазором седловидные поверхности (рис. 354).
Насадку «Интерпак» (рис. 355) можно изготавливать практически из всех металлов, а также из пластмасс. Эту насадку выпускают с размерами элементов 10x10x0,3; 20x25x0,4 и 30x35x0,8 мм [132]. Она обладает относительно низким гидравлическим сопротивлением.
Таблица 64.
Значения ВЭТС для насадок нз элементов нецилиндрической формы
Колония диаметром 30 мм с высотой разделяющей части 450 мм. Разделяемая смесь четыреххлористый углерод — бензол; флегмовое число v — ©о.
Тип иасадки Размер элемента, мм ВЭТС, см Нагрузка, мл/ч Плотность орошения, мл/(ч«см2) Примечание
Насадка Каннона из пер- 4 1,3—2 [19 к гл. 7]
форированных металлы-
ческих полуцилиндров.
Насадка «Хэли-пак» 1,8 0,5
Седловидная насадка нз 4 4,73 200 28,3 Данные
фарфора 6 6,00 200 28,3 автора
8 7,50 200 28,3
10 9,0 200 28,3
4 5,29 400 56,6
6 6,92 400 56,6
8 8,18 400 56,6
10 10,00 400 56,6
4 6,0 600 84,9
6 8,18 600 84,9
8 9,00 600 84,9
10 11,24 600 84,9
4 6,43 800 113,2
6 9,00 800 113,2
8 11,24 800 113,2
10 12,87 800 113,2
414
Значения ВЭТС для насадок из элементов нецилиндрической формы приведены в табл. 64. Характеристики фарфоровой седловидной насадки получены автором.
7.8.2. ВЛИЯНИЕ МАТЕРИАЛА НАСАДКИ НА ЭФФЕКТИВНОСТЬ РАЗДЕЛЕНИЯ
В разд. 4.2 сообщалось о влиянии химической природы материала насадки на разделяющую способность колонны. Насадки для лабораторных колонн в основном изготавливают из стекла, фарфора, глины, различных металлических сплавов и в последнее время также из пластмасс. Предпочтение обычно отдают стеклу и керамическим материалам благодаря их коррозионной стойкости в среде агрессивных жидкостей. Преимущество фарфора заключается в том, что он после обжига становится твердым и не содержит железа, которое может оказывать каталитическое воздействие на разделяёмые вещества. Проволочные или сетчатые насадки из нержавеющей стали V2A обеспечивают наибольшую эффективность разделения.
Различную эффективность сетчатых насадок, изготовленных из разных сплавов, Фухс [133] объясняет, основываясь на работах Форсайта с сотр. [134 ], неодинаковой смачиваемостью поверхности насадок и возможностью возникновения неодинаковых адсорбционных эффектов в зависимости от химической активности этих сплавов. Вольф и Гюнтер [135] разделяли эталонные смеси с различной полярностью веществ для определения влияния последней на эффективность насадок из различных металлов. В результате опытов было найдено, что в зависимости от материала насадки эффективность разделения изменяется на 10—12%.
Автором показано, что степень смачиваемости и, следовательно, эффективность насадки в значительной степени зависят также и от средней высоты выступов шероховатости поверхности элементов насадки [136]. Кроме того, установлено, что насадки из пластмасс с пористостью около 10% превосходят по степени смачиваемости сетчатые насадки.
Основные преимущества насадок из пластмасс по сравнению с насадками из других материалов заключается в строгой однородности размеров элементов, малой массе, большой доле свободного объема (вследствие малой толщины стенок), высокой механической прочности, относительно высокой коррозионной стойкости, а также в возможности легкой очистки [137].
Тэллер [138] объясняет сравнительно высокую
Рис. 356.
Насадка «Тэллерэттен» из полиэтилена, предложенная Тэллером [138].
415
эффективность насадок, изготовленных из плохо смачиваемых пластмасс, их низкой адгезионной способностью (см. разд. 4.2), которая обуславливает увеличение удерживающей способности жидкости свободным объемом насадки. Это приводит к возрастанию времени пребывания жидкой фазы в колонне. Насадка «Тэллерэт-тен» из полиэтилена (рис. 356), разработанная Тэллером, имеет долю свободного объема, равную 83%. Благодаря тому, что элементы насадки выполнены в форме розеток, газовая и жидкая фазы то вступают в контакт на относительно малых поверхностях элементов, то вновь разделяются на отдельные потоки.
Насадку из колец небольшого размера можно изготавливать как из металла или керамики, так и из полимеров [139]. Егбе-ронгбе [140] рассмотрел высокоэффективные компактные насадки из термопластичных пластмасс различных конструкций.
При использовании полиэтиленовых насадок в ректификационных колоннах из стекла или металла возникает опасность значительного растекания жидкости к стенкам колонны. Это растекание обусловлено разностью в силах адгезии, действующих в системах жидкость—стенки колонны и жидкость—элементы насадки (см. разд. 4.2). Согласно работе Штюрмана [141 ] растекание к стенкам можно уменьшить используя колонны с волнистыми стенками. Автором [137] показано, что путем специальной обработки внутренних стенок колонны можно также получить удовлетворительное распределение жидкости по насадкам, выполненным из пластмасс. Растекание жидкости к стенкам удается значительно уменьшить и с помощью покрытия внутренних стенок колонны полиэтиленовой или тефлоновой фольгой.
8
Автоматические установки, измерительные и регулирующие устройства
Высокую степень разделения и воспроизводимость результатов перегонки в лабораторных и пилотных ректификационных установках можно обеспечить, только применяя точные физические методы измерения.
Наряду с измерением температуры и давления при ректификации необходимо также определять флегмовое число и нагрузки колонны по пару и жидкости. Определение физико-химических свойств полученного дистиллята приобретает важное значение только в тех случаях, когда эти измерения проводят непосредственно в процессе перегонки.
Надежные измерения являются предпосылкой для регулирования и автоматизации процессов ректификации. При лабораторной и пилотной ректификации частично используют методы регулирования, применяющиеся в промышленности. Современное состояние техники регулирования рассматривается в специальных журналах.
8.1. АВТОМАТИЧЕСКИЕ УСТАНОВКИ
Основные стадии процессов ректификации во всех современных дистилляционных установках автоматизированы. Степень обогревания во многих случаях регулируют с помощью контактных термометров, скорость выкипания контролируют по дифференциальным манометрам. Для автоматического регулирования вакуума широко используют стенды с вакуумными насосами.
Современные исследования направлены на создание полностью автоматизированных ректификационных установок с одновременной регистрацией измеряемых величин. При работе на полностью автоматизированной установке вручную только загружают разделяемую смесь и через определенный промежуток времени, требуемый для проведения процесса ректификации, получают продукты разгонки; во время перегонки регистрируют кривые температуры в головке и кубе колонны и физико-химические свойства дистиллята.
Степень оснащения ректификационной установки периодического или непрерывного действия измерительными и регулирующими приборами зависит от конкретной задачи разделения и прежде всего от имеющихся экономических возможностей [12]. Различают полуавтоматизированныеи полностью автоматизирован
14 Э. Кррль
417
ные установки. Под полностью автоматизированной установкой понимают установку, в которой все рабочие процессы, включая пусковой период, протекают самостоятельно. Термин полуавто-матизированный целесообразнее заменить выражением «частично автоматизированный», под которым имеют в виду, что автоматизирована лишь часть рабочих процессов.
По степени оснащенности измерительными и регулирующими приборами различают следующие установки.
А. Частично автоматизированная установка, в которой осуществляется:
а) регулирование обогрева куба с помощью контактного термометра и контактного манометра посредством электронного пропорционального регулятора (см. разд. 8.2.2 и 8.4.2); обогрева кожуха колонны с помощью термопар, контактных или воздухонаполненных термометров (см. разд. 7.7, 8.2.1, 8.2.2); подогрева исходной смеси при непрерывной работе ректификационной установки с помощью контактного термометра (см. разд. 8.2.2); расхода исходной смеси при непрерывной работе с помощью дозирующих устройств (см. разд. 8.6); скорости выкипания и, следовательно, нагрузки колонны по пару, с помощью контактного манометра (см. разд. 8.4.2); вакуума с помощью регулятора давления или стенда с вакуумным насосом (см. разд. 8.3); расхода охлаждающей воды с помощью сосуда с постоянным уровнем и расходомера (см. разд. 8.6); а также предохранительного выключателя, отключающего установку при аварийном отключении системы подачи воды;
б) регистрация температур (см. разд. 8.2.1) верхней части колонны, исходной смеси и куба, а также температуры стенки обогревающего кожуха колонны (см. разд. 7.7.3) с использованием термопар или термометров сопротивления вместо - стеклянных термометров.
Б- Полностью автоматизированная установка, в которой осуществляется:'
а) регулирование тех же параметров, что и в частично автоматизированной установке; кроме того, регулируется расход исходной смеси с помощью небольших дозировочных насосов или посредством автоматического поддержания постоянного уровня в напорном баке (см. разд. 8.6); флегмовое число с помощью автоматической головки колонны, приводимой в действие механическим или электронным управляющим устройством (см. разд. 7.5 и разд. 8.4.1); объем отбираемых фракций дистиллята с помощью автоматического сборника фракций (см. разд. 7.6);
б) регистрация температуры (см. разд. 8.2.1) верхней части колонны в зависимости от количества дистиллята, а также температур исходной смеси, куба и обогревающего кожуха колонны с помощью термографа; флегмового числа косвенным путем посредством измерения разности температур между верхней и средней частями колонны; давления с помощью записывающего прибора.
418
8.1.1. АВТОМАТИЗИРОВАННЫЕ УСТАНОВКИ
ДЛЯ СТАНДАРТНОГО ДИСТИЛЛЯЦИОННОГО АНАЛИЗА
Примером полностью автоматизированной дистилляционной установки является установка D86, работающая по методу ASTM [1 ] и предназначенная для разгонки по Энглеру с автоматической записью диаграмм кипения. При работе на этой установке вручную приходится лишь загружать анализируемую пробу, устанавливать начальную мощность электрообогрева, извлекать зарегистрированную диаграмму разгонки и вынимать колбу с кубовым остатком. Электрическая схема данной установки представлена на рис. 357. Кроме указанных на рис. 357 устройств, в этой установке используют также следующие элементы: специальный электро-
Рис. 357.
Схема полностью автоматизированной дистилляционной установки D86, работающей по методу ASTM:
1 — термопара; 2 — баия с тающим льдом; 3 — нагревательное устройство; 4 — выключатель, служащий для отключения установки в момент отгонки 95% (об.) исходной смеси; 5 — потенциометр счетчика времени;
6 — потенциометр измерителя объема; 7 — индикатор отгонки 95% (об.) исходной смеси; 8 — мотор счетчика времени; 9 — регулятор обогрева; 10 — выключатель, служащий для отключения установки по окончании разгонки; 11 — включатель для пуска установки в работу; 12, 29 — пружины;
13, 14 — источники света для фотоэлемента; 15 — мотор для измерителя объема; 16 — мотор и пускатель для регистратора времени и температуры; 17 — фотоэлектрический усилитель в измерителе объема; 18 — усилитель;
19 — магнитная катушка термографа; 20 — фотоэлемент для регистрации первой капли дистиллята; 21 — фотоэлемент измерителя объема; 22 — электрический усилитель; 23 — мотор барабана самопишущего прибора;
24 — концевой выключатель; 25 — отключатель; 26 — потенциометр с реохордом; 27 — реле; 28 — включатель; 30 — барабан для миллиметровой бумаги.
14
419
Рис. 358.
Ректификатор-автомат для моторных топлив по DIN 51751 (февраль 1964 г.) в соответствии с A STM D86 или D158 с компенсирующим леиточиым самописцем (ширина леиты 210 мм).
Изготовитель: фирма Эрист Хааге, Мюльхайм (Рур).
нагреватель, который реагирует на резкие изменения температуры и работает таким образом, что первая капля дистиллята появляется через 5—10 мин после начала процесса перегонки; устройство, которое автоматически «наносит» отметки времени на диаграмму разгонки через 2 или 10 мин; прибор, автоматически фиксирующий момент отгонки 95% (об.) общего количества исходной смеси и обеспечивающий поддержание в дальнейшем постоянной мощности обогрева куба; приспособление для автоматического возврата всех частей установки в исходное положение после окончания разгонки; устройство, обеспечивающее автоматическое охлаждение приемного сосуда для дистиллята до 15±3 °C.
Точность измерения температуры составляет 0,5 °C, а объема — 0,1 мл. Установка работает при одной из двух предварительно установленных скоростях перегонки 4,5 или 9 мл/мин и рассчитана на круглосуточную эксплуатацию.
Подобная установка «Асда» типа 3 (фирма «Галленкамп» Лондон) может быть использована для проведения следующих операций: ASTM D86-JP 123 группы 1, 2 и 3, а также D1078, DIN 51751 и 51752; ASTM D86-JP 123 группа 4; скоростная перегонка (не стандартизированная).
В одной лаборатории, оснащенной 15 установками «Асда», в течение 8 месяцев было проведено 80000 дистилляционных анализов. Это соответствует производительности установки, равной 666 анализам в месяц. Один исследователь одновременно может обслуживать около 10 таких установок без дополнительных приспособлений. Преимущество данных установок состоит в том, что они 420
полностью исключают субъективные погрешности, возможные при обычной разгонке по Энглеру.
В Советском Союзе для дистилляционного анализа нефти используют автоматические ректификационные установки, предложенные Кантерманом с сотр. [3], с точностью определения температуры начала перегонки —2 °C *. Ректификатор-автомат Зальцера [4] по DIN 51751, предназначенный для разгонки моторных топлив и растворителей, выпускается предприятием АЕГ в Берлине.
На рис. 358 показана установка, описанная Ирманном [5]. Автомат состоит из пульта управления, снабженного компенсирующим линейным самописцем термометра сопротивления, и двух взаимозаменяемых ректификационных агрегатов. Установка предназначена для перегонки в температурных пределах от 0 до -|-285 °C и от -ф-100 до -j-385 °C. Ректификатор-автомат Герцога [6] одновременно с выдачей цифровых данных регистрирует результаты на бумажной ленте. Цаппе с сотр. [7] разработал установку для
* Кроме установок Кантер мана с сотр., в СССР разработаны и выпускаются лабораторные установки других типов, например |полуавтоматизированная установка для полумикроректификации типа ЛМР (СКТБ стеклянных приборов, г. Клин Московской обл.). — Прим. ред.
Рис. 359.
Установка для дистилляционного анализа под вакуумом с автоматическим регулированием температуры, разработанная Цаппе с сотр. [7 ]:
1 — самописец; 2 — термостат; 3 — усилитель; 4 — регулятор давления;
5 — наклонные трубчатые манометры; 6 — фотоэлементы; 7 — сборник с поплавком; 8 — форштос; 9 — стеклянный патрубок; 10 — капилляр для устранения пульсаций давления при кипении; 11 — обогреватель куба с регулятором; 12 — куб; 13 — ректификационная колонна с теплоизолирующим кожухом; 14 — счетчик пузырьков.
421
проведения дистилляционного анализа под вакуумом при стандартизованных условиях. Схема этой установки представлена на рис. 359 [6].
8.1.2. ПОЛНОСТЬЮ АВТОМАТИЗИРОВАННЫЕ
УСТАНОВКИ ДЛЯ ФРАКЦИОННОЙ ПЕРЕГОНКИ
В принципе степень автоматизации ректификационных установок периодического или непрерывного действия можно увеличить настолько, что их можно рассматривать как полностью автоматизированные установки. Полностью автоматизированная установка Улусоя [8] разработана специально для препаративных работ. Для низкотемпературной ректификации предназначена описанная выше автоматическая установка Подбильняка (см. рис. 178), схема включения измерительных и регулирующих приборов которой представлена на рис. 360.
Штаге [9] разработал ряд лабораторных ректификационных установок типа «Лабодест-робот». Примером такой установки является установка с трубчатой щелевой колонной, предназначенная для полумикроректификации (см. рис. 2576). Преимущество данных установок состоит в том, что их измерительную и регулирующую системы можно использовать для оснащения различных ректификационных колонн [10], например для трубчатых щелевых колонн с емкостью куба 5—100 мл, работающих при абсолютном давлении от 0,01 до 760 мм рт. ст. с эффективностью, равной примерно 40 теоретическим ступеням разделения, или для колонн с металлической сетчатой насадкой с емкостью куба 100—1000 мл, работающих при атмосферном давлении или остаточном давлении до 5 мм рт. ст. с эффективностью около 60 теоретических ступеней разделения.
На передней стороне установки для полумикроректификации (см. рис. 2576) расположены специальные вставки, выдвигаемые с помощью телескопических направляющих. Все функциональные элементы размещаются на штеккерной печатной плате, используемой в транзисторной технике. Установка имеет следующие системы регулирования: температуры масляной бани с помощью пропорционального электронного регулятора, работающего по предварительно заданной температуре в пределах от 0 до 300 °C, или переключаемого по предварительно заданной нагрузке, выражаемой числом капель в 1 мин; температуры компенсационного нагревателя кожуха колонны по предварительно заданной температуре от 0 до 300 °C, или с помощью переключения регулятора подстройки по температуре пара в верхней части колонны; флегмового числа путем деления парового потока с периодами включения и выключения реле времени от 0,5 до 200 с; других температур и объема отбираемых фракций; работы сборника фракций путем замены сосудов через заданный промежуток времени (в минутах). Кроме того, установка снабжена системами, обеспечивающими стабилиза
422
цию вакуума, а также предохраняющими от производственных неполадок, таких как прекращение подачи охлаждающей воды, превышение верхних пределов температуры и уровня заполнения сосудов в сборнике дистиллята.
В насадочных колоннах регулирование нагрузки и перепада давления потока пара осуществляется относительно просто, в то время как для трубчатых щелевых колонн, в которых перепад давления составляет порядка 10~3 мм рт. ст., требуется специаль-
ные. 360.
Электрическая схема автоматизированной ’низкотемпературной ректификационной установки Подбильняка «Термокон» (см. также рис. 178):
1 — электронный прибор для контроля количества дистиллята; 2 — трубопровод для паров дистиллята; 3 измерительная ячейка; 4 — измерительный контур для определения теплопроводности; 5 — передача привода устройства для передвижения бумажной ленты; 6 — регистрирующий манометр; 7 — прибор, контролирующий давление и степень охлаждения флегмы; 8 — прибор, регистрирующий теплопроводность; 9 — мотор; 10 — вывод паров; 11 — штуцер для подачи воздуха; 12 — штуцер для продувки; 13 — воздушный термостат; 14 — устройство для автоматического ввода проб газа.
423
| Рис. 361.
i: Полностью автоматизированная ректификационная установка «Аутома-
Ь; тик 62» :
г 1 — расходомер жидкости, стекающей в куб; 2 — капельница; 3 — пере-ходы; 4 — колонна; 5 — обогревающий кожух колонны; 6 — дифмаио-I метры; 7 — воздухонаполнениые термометры; 8 — головка колонны;
9 — термометры; 10 — холодильник; И — вакуумный соединительный V патрубок; 12 — охлаждаемая ловушка; 13 —магнитные клапаны; 14 — погружной груз; 15 — соединительный патрубок; 16 — заглушка; 17 — £ пробоотборник; 18 — дозировочное устройство; 19 — вакуумный прием-ник; 20 — электрические контактные манометры; 21 — трубопроводы; ; 22 — холодильник прибора, измеряющего перепад давления; 23 — рас-
пределитель дистиллята; 24 — подающие трубы; 25 — сосуд для уста-I. иовки сборника фракций; 26 — пробирки; 27 — сборник фракций; 28 — пробка со шлифом; 29 — куб; 30 — пробка; 31 — приемная бюретка; 32 — I вакуумный приемник Аншюца —Тиле; 33 — пульт управления.
к Изготовитель: народное предприятие «Комбинат технического стекла», £ Ильменау.
8 ная регулирующая система. Фишер и Вейанд [10] с помощью электронных приборов измеряли число капель флегмы, подаваемое | в единицу времени. Исходя из результатов этих измерений, регу-[ лировали мощность нагревателя куба по требуемой нагрузке пря-| мопропорционально разности между действительным числом капель [ и предварительно заданным значением. Оказалось возможным I точно и абсолютно воспроизводимо регулировать нагрузку в пре-f делах от 50 до 250 мл/ч при давлении в кубе от 760 до 10~3 мм рт. ст. [ В автоматизированную установку «Аутоматик 62», соответ-I ствующую нормалям «Дестинорм» (рис. 361), разработанную авто-I ром, входят следующие отличительные элементы [11 ]: куб с элек-i тронагревателем «Интус»; устройство для измерения расхода t жидкости, стекающей в куб, и устройство для отбора проб этой I жидкости перед кубом; автоматический электронный регулятор I обогрева кожуха колонны по средней температуре внутри колонны I (см. рис. 368); автоматическая головка колонны с качающейся | воронкой для регулирования флегмового числа с помощью элект-F ровного или механического реле времени (см. рис. 312); автомати-I ческий сборник фракций, содержащий 60 пробирок и управляемый I при помощи сосуда с сифоном и погружным грузом (см. рис. 324); | автоматический электронный регулятор вакуума с точностью I ^tzO, 1 мм рт. ст., работающий с помощью термостатированного ре-I гулятора давления (см. рис. 377); автоматический регулятор к скорости выкипания и, следовательно, нагрузки колонны по пару, |. снабженный контактным манометром (см. рис. 387, 388); оптиче-I ская система для визуального наблюдения за температурой и электронный компенсограф для регистрации температуры.
К Пары дистиллята (см. рис. 361) попадают сначала в термоста-I' тированную зону, где конденсируются низкокипящие компоненты, К при этом легко затвердевающие вещества можно разогревать К в термостате. Расположенный ниже магнитный клапан 13 служит Е для измерения объема жидкости по градуированной трубе. Пробо- отборное устройство 17 с капельницей позволяет отбирать пробы | для испытаний и промежуточных анализов при аналитической
425
ректификации. Можно установить также термометр или приемную воронку для калибровки сосуда с сифоном и погружным грузом. Этот сосуд снабжен вертикально перемещаемым погружным грузом 14, который позволяет регулировать объем фракций с точностью ±0,1 мл. При прекращении подачи дистиллята в сосуд манометр 20, заполненный органической токопроводящей жидкостью, с помощью электронного реле включает специальное устройство, которое устанавливает в сборнике фракций следующую пробирку. Одновременно импульс тока, поступающий от электронного реле, может приводить в действие перо с красителем, встроенное в электронный термограф, которое посредством магнитной катушки отмечает на полосе бумаги число отобранных фракций.
Сборник фракций имеет обычно 60 пробирок емкостью 20 мл каждая. Он помещен в эксикатор, защищенный кожухом из металлической сетки. Сборник фракций применяют при ректификации, протекающей как при атмосферном, так и остаточном (до 1 ммрт.ст.) давлении. После заполнения 30 пробирок следует звуковой сигнал или автоматически подается следующий диск с пробирками. При заполнении всех пробирок по соответствующему сигналу регулятор флегмы отключается и колонна начинает работать с бесконечным флегмовым числом, т. е. без отбора дистиллята.
Соединительная коммуникация между сосудом с сифоном и сборником фракций представляет собой легко разбираемую систему, в которой в значительной мере приняты предосторожности, чтобы дистиллят не соприкасался с жировой смазкой шлифов.
При препаративной ректификации используют сосуд с сифоном или применяют сборники, при достижении в которых определенного уровня жидкости подается звуковой сигнал и ректификационная колонна переключается на режим работы с бесконечным флегмовым числом.
Решая задачу автоматизации процесса ректификации по Мико для качественных испытаний при перегонке вин, Коуколь с сотр. [12а] разработал ректификационный автомат для фракционной перегонки. Эта установка обеспечивает регулирование мощности электронагревателя куба в зависимости от количества дистиллята, получаемого в единицу времени. При постоянной скорости перегонки можно разделить смесь на большое число фракций одинакового объема, получаемого в течение предварительно заданного постоянного промежутка времени, например 15 мин. Объем фракций измеряют с помощью термистора, который отрегулирован на определенную высоту уровня жидкости. Этот термистор обеспечивает получение фракции в объеме 25 мл с точностью ±0,1 мл.
Автоматические установки для непрерывной ректификации разработаны Коллингом и Траммом (см. рис. 166) и Штаге (см. рис. 167).
Рёк с сотр. [13] предложил ряд устройств для автоматизации лабораторных колонн, в частности предохранители, предотвращающие возможность прекращения подачи охлаждающей воды, и 426
Рис. 362.
- Экспериментальная установка непрерывного действия с колоннами, сна* i. бженнымн колпачковыми тарелками с отражательными перегородками для к пара, предназначенная для моделирования процесса перегонки сырой нефти: | 1 — дозировочные иасосы; 2. — пульт управления; 3 — регулятор вакуума; £4 — предохранительное реле.
t Изготовитель: фирма Фишера «Лабораторная и исследовательская тех* = ника», Бонн —курорт Годесберг.
противопожарные предохранители. Подробные обзоры по регулирующей технике для лабораторных колонн представлены в работах Аббега [14], Кесслера и Фоденаля [16], а также Кадлека [17].
Предпосылкой автоматизации непрерывно работающих пилотных ректификационных установок является решение задачи получения достоверных опытных данных, на основе которых можно разрабатывать промышленные установки. На рис. 362 показана экспериментальная установка, предназначенная для моделирования промышленного процесса перегонки сырой нефти. Установка работает непрерывно. Она состоит из одной основной и трех дополнительных колонн, предназначенных для отгонки низкокипящих фракций. Данная установка служит для разгонки многокомпонентных смесей, которые разделяются на четыре фракции. Кубовый продукт отбирается из куба основной колонны. Ректификационные колонны снабжены колпачковыми тарелками с отражательными перегородками для пара. По экспериментальным данным, получаемым при перегонке в этих колоннах, можно непосредственно разрабатывать установки больших размеров. Потоки паровой и жидкой фаз дозируются насосами 1 (см. разд. 8.6). Пульт управления 2 позволяет регулировать скорости выкипания, температуры обогревающих кожухов колонн и флегмовые числа. Регулятор вакуума 3 обеспечивает постоянную степень разрежения, а предохранительное реле 4 отключает установку, как только прекращается подача охлаждающей воды. Температуры на основных стадиях процесса непрерывно регистрируются электронным самописцем [17а].
8.2. ИЗМЕРЕНИЕ И РЕГУЛИРОВАНИЕ
ТЕМПЕРАТУРЫ
8.2.1. ИЗМЕРЕНИЕ ТЕМПЕРАТУРЫ
При ректификации прежде всего необходимо измерять температуру пара в верхней части колонны перед конденсатором и температуру кубовой жидкости. При работе с термолабильными веществами следует обязательно наблюдать за температурой в кубе. При непрерывной ректификации необходимо подогревать исходную смесь до температуры, соответствующей температуре жидкости в колонне в месте ввода в нее исходной смеси. Кроме того, требуется измерять температуру обогревающего кожуха колонны и жидкого теплоносителя (при использовании обогревающей бани) или потока греющего пара.
В лабораторных и пилотных ректификационных установках все еще преимущественно используют стеклянные термометры, однако при небольших интервалах измеряемой температуры предпочтительно применять термопары, полупроводниковые термометры или термометры сопротивления, которые позволяют авто-428
магически регистрировать и регулировать температуру *. О измерении температуры в лаборатории информирует обзорный доклад Фанфора [18].
В работе Лапорта [15] обсуждаются вопросы измерения, регулирования и стабилизации высоких и низких температур. Важно правильно выбрать точки измерения температуры в ректификационном аппарате, так, например, точка измерения температуры в верхней части колонны должна находиться примерно на 10 мм ниже штуцера для вывода пара. Температуру в кубе измеряют в самой нижней его части, чтобы можно было сразу же обнаружить перегрев. Датчики температуры, располагаемые в потоках жидкости или пара, тщательно изолируют и размещают таким образом, чтобы каждый датчик находился в средней части потока.
Стандартные стеклянные термометры, применяемые в ректификационных установках, представлены в табл. 65. Кроме указанных стандартных термометров выпускается большое число стеклянных термометров специального назначения, например угловые термометры со стандартным шлифом для измерения температуры внутри колонны. Термометры с присоединительным фланцем
Таблица 65.
Стандартные стеклянные термометры, применяемые в ректификационных установках
Стандарт Наименование
TGL 40-335 Стеклянный термометр для измерения температуры жидкости
40-336 Термометр для ректификационных установок Стеклянный термометр для измерения температуры жидкостей, термометр для измерения температуры минеральных масел и топлива, термометр типов А и В для измерения температуры кипения
40-339 Лабораторный термометр с коническим шлифом NS14,5/23
D1N 12779/71 12784/55 группа 1 Термометр для ректификационных установок Термометр со стандартным шлифом Термометр для ректификационных установок (эта-
12784/63 группа 2 лонный) Термометр со стандартным шлифом Термометр для измерения температуры в стеклянных
12785/58 группа 1 кубах-колбах (эталонный) Термометр для измерения температуры кипения
12788/63 группа 1 (эталонный) Карман для термометра со стандартным шлифом 14,5/23
* При наличии соответствующих измерительных приборов можно успешно использовать термопары и термометры сопротивления также при широком интервале температур. — Прим. ред.
429
Рис. 363.
1 2 Термометр для ректификационных установок со стандартным шлифом
2} АЛ 14,5 (TGL40-339, DI N12784/55):
ц г-Ц-| / — крепление шкалы по Рихтеру; 2 — крепление шкалы по Штифту;
п 3 — предохранительный расширительный участок; 4 — последнее де-
)JL QU леиие шкалы; /х — длина ножки погружения; /2 — глубина погруже-
ги п иия.
удобны при монтаже; термометры с капиллярами, L ]1 окрашенными с обратной стороны, облегчают визу-fS альное наблюдение за показаниями [19].
1Р В ректификационных установках в основном
тп[ используют термометры, у которых капилляр со шкалой запаян в трубчатый кожух (рис. 363). Тер-z, 1 мометры, предназначенные для измерения темпера-L 1г тур в интервале от —58 до +360 °C, заполняют ' у] ртутью или ее сплавами. Для низкотемпературной ректификации в области температур от —58 до —200 °C применяют термометры, заполненные пентаном или другой подходящей жидкостью. Следует принять во внимание, что кроме термометра со стандартным шлифом, показанного на рис. 363, все термометры градуируют при полном погружении, т. е. при измерениях термометр необходимо погружать в исследуемую среду на всю высоту столбика ртути.
При использовании термометров, устанавливаемых в спе-
циальные карманы, заполненные маслом, в результатах измерения могут возникать заметные погрешности. Пиати и Марти (20 ] показали, что погрешность измерения меньше, если ртутный шар термометра погружен в масло, служащее для передачи тепла. На практике термометр без стандартного шлифа помещают на всю его
Таблица 66.
Характеристики термометров для ректификационных установок со стандартным шлифом по TGL 40-339 DIN 12784/55
Форма выполнения Термометра Интервал шкалы, °C Цена деления, °C Интервал измеряемой температуры, °C Погрешность калибровки, °C
От 0 до +110 0,5 ±0,5
От 0 до +250 1,0 0—50 ±0,7
50—100 ±1,0
А или В 100—200 ±1,5
200—250 ±2,0
От 0 до +360 1,0 0—50 ±0,7
50—100 ±1,0
100—200 ±1,5
200—300 ±2,0
300—360 ±2,5
Длина ножки погружения Ц = 50 110 Глубина погружения /2 = 32 92 200 мм 182 мм
430
длину в поток пара или жидкости, хотя это и затрудняет визуальное наблюдение за показаниями. В других случаях необходимо вводить поправку на выступающий столбик ртути (см. разд. 4.13). Поэтому при ректификации предпочтительны термометры со стандартным шлифом, которые отградуированы при постоянной глубине погружения ножки (от нижнего края шлифа до ртутного шара). Характеристики термометров со стандартным шлифом представлены в табл. 66.
Термометры со стандартными шлифами выпускают с интервалом измерения от 0 до 50 и ценой деления 0,1 °C. Изготавливают также угловые термометры. Стеклянные термометры, с ценой деления 0,1 °C, отградуированные соответствующими метрологическим учреждением, служат в качестве эталонов для термометров со стандартными шлифами и применяются при аналитических разгонках. В паспорте термометра указывается глубина его погружения и средняя температура столбика ртути во время градуировки. Для термометров с ценой деления 0,1 °C погрешность измерения лежит в следующих пределах:
0—50 +0,05 °C 150—200 ±0,2 °C
50—100 ±0,05 °C 200—250 ±0,5 °C
100—150 ±0,2 °C
Поскольку погрешность измерения градуированных термометров изменяется в пределах от ±0,2 до ±6 °C, а стекло, из которого они изготовлены, подвержено процессу старения, необходимо периодически проводить проверку термометров. Для этой
цели на термометры наносят вспомогательные метки, которые позволяют установить’изменение погрешности измерения во времени. Термометры других типов необходимо проверять через определенные промежутки времени с помощью стандартного прибора. В качестве стандартного прибора'может быть использован прибор Юнге-Риделя, работающий по принципу аппарата Тиле, который
служит для измерения температуры плавления. Он пригоден для проверки термометров, предназначенных для измерения температур до 300 °C, которые’градуированы при полном погружении. Показания проверяемых термометров сравнивают с показаниями’подобных термометров, отградуированных метрологическим учреждением.
Для проверки термометров со стандартным шлифом автор применял прибор, показанный
Рис. 364.
Прибор Креля для проверки термометров со стандартным шлифом и термопар.
431
Таблица 67.
Вещества, рекомендуемые для проверки термометров в среде пара
Вещество Температура кипения при 760 мм рт. ст., °C Вещество Температура кипения при 760 мм рт. ст., °C
0—5 0 °C 150— 200 °C
Диэтиловый эфир 34,6 Бромбензол 155,6
Хлористый метилен 40,67 Декан 174,0
Ацетон 56,13 Анилин 184,4
50—100 °C Этиленгликоль 197,4
Метил ацетат 56,96 200—300 °C
Хлороформ 61,0 Бензиловый спирт 205,2
Метанол 64,72 Нитробензол 210,6
Четыреххлористый угле- 76,69 Нафталин 217,96
род Дифенил 254,9
Этилацетат 77,06 Бензофенон 305,9
Циклогексан 80,8
Вода 100
100- 150 °C
Толуол 110,6
н-Бутанол 117,75
н-Бутилацетат 126,2
Хлорбензол 131,69
изо-Амилацетат 142,0
на рис. 364, который также пригоден для калибровки термопар. Проверку термометров с помощью этого прибора проводят в среде пара. В табл. 67 указаны вещества, пригодные для проверки термометров в интервале температур от 34 до 300 °C. Для достижения более высоких температур используют глицерин или другое высококипящее термически стабильное вещество и работают под вакуумом. Прибор позволяет проверять одновременно три термометра со стандартным шлифом. При этом следует учитывать нормированные погрешности измерения четвертого термометра, отградуированного метрологическим учреждением. При проверке термометра следят за тем, чтобы температура столбика ртути этого термометра не сильно отличалась от температуры столбика ртути эталонного термометра, при которой проводили его градуировку. В других случаях вводят соответствующую поправку.
Как уже указывалось, для измерения температуры вместо стеклянных термометров можно использовать термопары или термометры сопротивления, которые имеются в продаже [181.
Электрические методы измерения температуры применяют в тех случаях, когда необходимо непрерывно автоматически регистрировать температуру. Преимущество термопар состоит в том, 432
| что с их помощью можно проводить измерения в очень небольшом | объеме, например на поверхности стеклянных стенок пленочной I ректификационной колонны (см. разд. 5.4.3) или других пленочных I ректификационных аппаратов. Кроме того, термопары и термо-I метры сопротивления характеризуются высокой чувствительностью г и широким интервалом измеряемых температур. Для термопар |- такой интервал лежит примерно в пределах от —200 до Д-1800 °C. | Для фиксации температуры холодного спая термопары целе-[‘ сообразно использовать термостаты, работающие по принципу | Пельтье (термоэлектрическое охлаждение) [211.
I Термометры сопротивления, изготовляемые преимущественно К из платины, предназначены для измерения температур в интервале | от —220 до 750 °C. Принцип действия платинового термометра | сопротивления основан на том, что омическое сопротивление термо-| метра, выполненного в виде пластины, цилиндра, прямолинейно К натянутой или свернутой в спираль проволоки, изменяется при- мерно на 0,4% при изменении температуры на 1 °C. Это означает, К что для обеспечения точности измерения в 0,01 °C требуется В фиксировать изменение омического сопротивления в несколько стотысячных долей от его первоначального значения при 0 °C. В Метод измерения температуры выбирают в зависимости от требуе-Е мой точности. Отметим, что при использовании моста Уитстона
можно измерять и регистрировать также разность температур и, следовательно, регулировать мощность электрообогрева кожуха ; колонны по температуре внутри нее и косвенно регистрировать ; флегмовое число. Применяя напыляемые термометры сопротивле-s ния [22 ], можно точно определить среднюю температуру поверх-( ности испарительных свечей или температуру теплопередающих [ поверхностей.
[.. Для измерения температуры паров и жидкости в пилотных I ректификационных установках часто применяют термометр сопро-| 1тивления, снабженный двумя отдельными измери-г тельными обмотками, служащими для одновре-
| l Т менной регистрации и регулирования измеряемой
д-к величины. Термометр Квикфита в полностью
I I---I стеклянном выполнении (рис. 365) имеет соедини-
I п—<===jw тельные штуцеры с диаметрами условного про-| п хода NW 25 и NW 40.
I — •— Для измерения давления паров и определения
I- характеристик фазовых равновесий применяют ус-
I тройства с полупроводниковыми датчиками, свя-
I занными с компенсографами, которые обеспечивают
Рис. 365.
Термометр сопротивления в полностью стеклянном выполнении.
Интервал измерения от -200 до +300 °C.
Изготовитель: фирма «Стеклянная техника». Висбаден —Шир-штайн.
433
точность регистрации температуры до ±0,001 °C. Таким прибором является термометр «Термофил», который особенно пригоден для измерения температур поверхностей, например в пленочных ректификационных колоннах, аппаратах для молекулярной дистилляции и в обогревающих кожухах колонн. В измерительный зонд этого прибора вмонтирован германиевый полупроводник диаметром 0,25 мм, который отбирает от измеряемого объекта только ничтожное количество тепла. Интервал температур, измеряемых этим прибором, лежит в пределах от —50 до Д45О°С. Измеряемый электрический ток усиливается электронным усилителем и фиксируется на градуированной шкале прибора. Кабельные провода, идущие к усилителю от нескольких датчиков, не искажают результатов измерений и к усилителю можно подключать до 10-ти измерительных зондов. Зонд выполняется из стекла или металла.
Для регистрации температур используют выпускаемые промышленностью термографы, которые снабжены 1, 2, 4, 6 и 12 измерительными зондами.
Лабораторную и пилотную ректификацию часто проводят при температурах примерно до 200 °C. Из этого следует, что для регистрации температуры с точностью, характерной для стеклянных термометров, необходимо пользоваться только теми приборами, которые имеют относительную точность не более 0,1% при абсолютной погрешности до 0,25% от интервала измерения. Этому условию удовлетворяют электронные приборы, записывающие показания, и компенсационные самописцы, которые можно подключать непосредственно к платиновому термометру сопротивления (100 Ом при 0 °C). Самописец можно использовать при непрерывной ректификации для регистрации отклонения температуры от заданного значения.
8.2.2. РЕГУЛИРОВАНИЕ ТЕМПЕРАТУРЫ
Для регулирования степени нагревания неподвижных и движущихся жидкостей, а также мощности электрообогрева различных частей аппаратов, например обогревающего кожуха колонны и нагревательных трубчатых змеевиков, широко используют контактные термометры. Эти термометры также имеют стандартные шлифы NS 14,5 (рис. 366). Они выполняются как с жестко впаянными контактами для любой фиксированной температуры, так и с магнитным регулятором. Термометры с впаянными контактами служат для поддержания требуемой температуры, а термометры с магнитным регулятором применяют в качестве датчиков температуры. Высокую чувствительность имеют контактные термометры со спиральным резервуаром для ртути (рис. 367а). Этот резервуар имеет оптимальную поверхность. Рис. 3676 иллюстрирует чувствительность ртутных термометров со спиральным и цилиндрическим резервуарами [23].
Контактные термометры изготавливают в виде максимальноминимальных Термометров, в которых электроконтакт замыкается 434
при переходе через верхний или через нижний предварительно установленные пределы температуры соответственно. С помощью ролика для точной регулировки можно устанавливать заданную температуру с точностью до 0,1 °C [23]. Температуру можно регулировать также и автоматически с помощью синхронного электромотора в зависимости от показаний задающего термометра.
Удобны контактные термометры с трехполюсным штепсельным разъемом и передвижным стандартным шлифом, благодаря которому можно ступенчато изменять длину погружной трубки [24 ].
Контактный термометр, описанный в работе [25], соединяют через реле с регулируемым потребителем электроэнергии, который включается или выключается при замыкании контактов термометра. В лабораторной практике предпочтительны реле с вертикальными ртутными резервуарами, в которых отсутствуют подверженные естественному износу подвижные детали, такие как рычаги, шарниры и гибкие провода [26].
Подобные транзисторные реле, используемые также и для регулирования мощности потребителей электроэнергии с помощью контактных манометров (см. разд. 8.3.1 и 8.4.2), выпускаются комбинированными в виде реле уровня, совмещенного с реле подстройки. Универсальные лабораторные реле снабжены переключателями для работы в нормальноразомкнутом и нормальнозамкнутом положениях. При замыкании контактов термометра реле, работающее в нормальноразомкнутом положении, включает потребитель электроэнергии, например сигнальные звонок и
Рис. 367.
Термометр со спиральным резервуаром для ртутн:
а — общий вид термометра; б — сравнительные данные о чувствительности термометров со спиральным (/) и цилиндрическим (2) резервуарами для ртути, полученные при одинаковом объеме резервуаров.
Рис. 366.
Контактный термометр с вращающимся магнитным регулятором.
435
лампу, а реле, работающее в нормальнбзамкнутом положении, наоборот выключает потребитель электроэнергии, например нагреватель куба, обмотку обогревающего кожуха колонны, клапан. Эти реле применяют и для фотоэлектрических коммутаторов, например для регулирования уровня жидкости или вакуума (см. разд. 8.6 и разд. 8.3.1.1). При использовании этих реле в качестве предохранительных, они отключают потребитель электроэнергии при выходе из строя контактного термометра.
Универсальные электронные приборы, которые можно использовать для настройки и регулирования ректификационной аппаратуры, описаны в работе Фишера [27]. Как указано в этой работе, наряду с электронным реле для регулирования температуры в интервале от —200 до -|-800 °C можно применять пропорциональные регуляторы. В этом случае в качестве измерительного зонда используют термометр сопротивления со стандартным шлифом или фланцем. Преимуществом этих приборов является возможность их использования для регулирования мощности электронагревателей с малой тепловой поверхностной нагрузкой, что особенно необходимо, если для обогрева применяют электронагреватель, который при замыкании цепи включается сразу же на полную мощность.
Особые сложности возникают при регулировании температуры обогревающего кожуха колонны в зависимости от температуры внутри нее. В разд. 7.7.3 были описаны различные способы тепловой изоляции колонн. Вследствие того, что при непрерывной ректификации, и особенно при ректификации многокомпонентных смесей температура внутри колонны постоянно, а часто и скачкообразно повышается, необходимо соответствующим образом регулировать мощность нагрева электроспиралей. При этом из-за тепловой инерции электроспиралей между температурой внутри колонны и температурой обогревающего ее кожуха может возникать градиент до 30 °C. Обеспечивая автоматическое регулирование мощности электроспиралей, удается существенно уменьшить этот температурный градиент. В этом случае в качестве температурных датчиков применяют воздухонаполненные термометры и термопары, или термометры сопротивления. При регулировании температуры с помощью термопар (см. рис. 343), установленных внутри колонны, а именно в ее верхней части и несколько выше куба, они воздействуют на показывающий прибор, который подает через короткие промежутки времени импульсы на коммутатор. При этом электрический контур, который включает электронагреватель кожуха колонны, замыкается [28]. В качестве температурных датчиков автоматических регуляторов мощности электронагревателей кожуха по температуре внутри колонны используются также и контактные термометры Хутла [29].
Автоматический регулирующий прибор для ректификационных установок «Минитрон 5» (см. рис. 386), разработанный Фишером [27], снабжен двумя электронными пропорциональными регуля
436
торами, которые соединяются с электронагревателями куба и кожуха’колонны. В качестве измерительного зонда служит термометр сопротивления Pt 100, работающий в интервале температур 0—350 °C. Температуру обогревающего кожуха колонны можно регулировать по температуре, предварительно установленной на приборе «Гелипот», или посредством автоматической подстройки по температуре внутри колонны. На термометре сопротивления, расположенном в верхней части колонны вместо прибора «Гелипот», устанавливается заданная температура. Автоматическая регулировка обогревающего кожуха колонны осуществляется по температуре пара внутри нее.
Применяя воздухонаполненные термометры, один баллончик которых размещен внутри колонны, а другой —в обогревающем кожухе, автору [11 ] удалось достигнуть точности регулирования ±0,5 °C. Эта точность была обеспечена при использовании контактного манометра с органической токопроводящей жидкостью, который посредством электронного реле включал и выключал электронагревательную обмотку (рис. 368). С помощью данного
Рис. 368.
Схема системы автоматического регулирования температуры обогревающего кожуха колонны по температуре внутри нее:
/ — колонна; 2 — обогревающая электрообмотка; 3 — воздухонаполненный термометр; 4 -- теплоизоляция нз стекловолокна; 5 — кожух колонны;
6 — регулирующий манометр; 7 — электронное реле; 8, 9 — стандартные шлифы NS29 н NS14.5 соответственно.
437
устройства можно обеспечить до 400 включений в 1 ч. Даже при резком повышении температуры внутри колонны температура обогревающего кожуха изменяется конформно температуре внутри колонны. Преимущество данного метода состоит в возможности регулирования температуры обогревающего кожуха, который целесообразно разделить на отдельные секции длиной 25—50 см, по средней температуре внутри соответствующего участка колонны. Если же регулировать обычно принятым способом, т. е. регулировать температуру верхней секции обогревающего кожуха колонны по температуре верхней части колонны, то при отгоне промежуточной фракции температура кожуха будет значительно ниже истинной температуры внутри колонны. В этом случае верхняя часть колонны будет работать как парциальный конденсатор, образующий дополнительное количество флегмы. Кроме того, при использовании этого метода регулирования можно поддерживать температуру обогревающего кожуха выше или ниже заданной величины по сравнению с температурой внутри колонны.
8.3. ИЗМЕРЕНИЕ И РЕГУЛИРОВАНИЕ
ДАВЛЕНИЯ
Перегонку проводят обычно в определенных интервалах давления. По рабочему давлению можно следующим образом классифицировать процессы перегонки: область давлений выше 760 мм рт. ст. — ректификация поддавлением; область давлений 760 —1 мм рт. ст. — вакуумная дистилляция и ректификация, расширительная перегонка, пленочная ректификация; область давлений 1—1(Г6ммрт. ст. — пленочная перегонка, молекулярная дистилляция. •
Для каждой из указанных областей применяют различные методы измерения и регулирования давления [2 ], которые рассматриваются в следующих разделах [30]. Подробный обзор методов измерения остаточного давления при перегонке под вакуумом представлен Леком [31 ].
8.3.1. ИЗМЕРЕНИЕ И РЕГУЛИРОВАНИЕ ДАВЛЕНИЯ
В ОБЛАСТЯХ ВЫШЕ 760 И ОТ 760
ДО 1 мм рт. ст.
При ректификации под давлением до 2 кгс/см2 (см. разд. 5.4.5) для измерения избыточного давления применяют U-образные манометры * высотой до 1 м (рис. 369). Давление в ректификационной колонне можно рассчитать по формуле:
рк = 6 4-Др (195)
где b — барометрическое давление с учетом необходимых поправок, мм рт. ст.; Др — избыточное давление по показаниям манометра.
* Манометры данного типа обычно заполняют ртутью. — Прим. ред.
438
При необходимости измерения более высоких давлений можно пользоваться выпускаемыми промышленностью мембранными манометрами или манометрами с трубчатой пружиной (Бурдона), которые пригодны также для измерения давления в интервале от 760 до - 0,1 мм рт. ст. Один из методов регулирования избыточного давления в процессе ректификации описан в разд. 5.4.5.
Анероид-регулятор абсолютного давления типа FA 149 (изготовитель: фирма «Баллас Тирнау-хлоратор», Гюнцбург) позволяет регулировать давление в интервале от 20 до 1520 мм рт. ст. с точностью до 0,25 мм рт. ст.
В ходе ректификации, протекающей при атмосферном давлении, необходимо также непрерывно регистрировать давление в колонне, которое в этом случае равно барометрическому давлению. Это давление нужно знать для того, чтобы привести значения температуры к давлению 760 мм рт. ст. (см. разд. 4.13).
Для измерения атмосферного давления с точностью ±0,1 мм рт. ст. следует применять прецизионные ртутные барометры, которые изготавливаются в виде рычажных, чашечных или комбинированных рычажно-чашечных моделей [32]. В лабораторной практике часто пользуются барометрами Гей-Люссака и Шродта-Кифера. Надежный’отсчет показаний с точностью ±0,1 мм рт. ст. обеспечивает нониус, или еще лучше —катетометр. Прибор для измерения давления жидкости, разработанный Никелем [32], учитывает влияние поверхностного натяжения исследуемой жидко
сти и позволяет корректировать смещение начала отсчета, возникающее, например,* при изменении температуры.
Для получения поправок в определенном интервале измерения
давления рекомендуется в течение длительного времени сравнивать показания барометра с' показаниями образцовых стационарных
манометров метеорологических станций. Подобным образом необ-
ходимо проверять также барографы, снабженные анероидами. Точность показаний таких приборов обычно составляет ±0,5 мм рт. ст. Эти приборы имеют устройства для регистрации показаний на бумажной ленте в течение суток или недели. Пользуясь такими лентами с записанными показаниями, можно позднее определить изменения давления в определенные промежутки времени, интересующие исследователя. Погрешность в измерениях
Рис. 369.
Манометр для измерения избыточного давления нлн вакуума в процессе ректификации:
1 — предохранительный сосуд; 2 — штуцер^для присоединения манометра; 3 —^шкала; 4 — сосуд для залива манометрической
жидкости.
439
Рис. 370.
Баровакуумметр
Изготовитель: народное предприятие «Комбинат технического стекла», Ильменау.
Рис. 371.
Укороченный манометр Беннерта для измерения остаточного давления от 0 до 180 мм рт. ст.
давления по анероиду, обусловленную изменением температуры в интервале от —40 до 4-40 °C, обычно устраняют с помощью биметаллического компенсирующего устройства.
При ректификации в условиях пониженного давления также применяют U-образные манометры высотой 1 м с одним открытым концом (см. рис. 369). Хорошо очищенную ртуть заливают в манометр, избегая попадания пузырьков воздуха, через предохранительный сосуд 4. Манометр присоединяют к аппарату с помощью штуцера 2. Сосуд 1 является также предохранительным на случай резкого возрастания вакуума в системе. При помощи шкалы 3, передвигающейся по вертикали, измеряют остаточное давление в системе Др. Абсолютное давление в колонне вычисляют по формуле:
рк=Ь —Др (196)
440
Преимущество этих манометров состоит в том, что они позволяют регистрировать показания во всем интервале давления от 760 до 1 мм рт. ст. с точностью ±0,5 мм рт. ст. Для более точного определения расстояния между менисками двух столбиков ртути необходимо применять вертикальные компараторы или катетометры.
Фаркварсон и Кермицле [331 предложили устройство для автоматической регистрации высоты столбика ртути. Прибор работает при высоте столбика ртути до 80 см с точностью, превышающей ±0,05 мм, и максимальной погрешностью ±0,11 мм.
Снятие показаний существенно облегчается при использовании баровакуумметров (рис. 370), в которых совмещено измерение остаточного и избыточного давлений. Приборы, выпускаемые промышленностью, пригодны для измерения барометрического давления в интервале от 600 до 820 мм рт. ст. и остаточного давления в пределах от 1 до 310 мм рт. ст. Высокая точность измерения обеспечивается благодаря применению калиброванных труб с отклонением внутреннего диаметра от заданного размера в пределах ±0,01 мм и устройства для автоматической поправки на отклонение уровня ртути от нулевого положения [32].
Укороченные манометры Беннерта (рис. 371) с закрытым вакуумированным коленом предназначены для измерения остаточного давления от 0 до 180 мм рт. ст. Точность отсчета показаний у этих приборов составляет ±0,5 мм рт. ст. Однако при измерениях в интервале давлений от 0 до 10 мм рт. ст. могут возникать значительные погрешности вследствие того, что воздух не полностью удаляется из закрытого колена манометра или проникает в него во время эксплуатации. Для предотвращения подобных ошибок измерения необходимо периодически сверять показания данного прибора с показаниями U-образных манометров полной длины (см. рис. 369). В модифицированном укороченном манометре Штрё-лейна на конце левого колена предусмотрен ртутный затвор, позволяющий удалять попавший в манометр воздух.
Наиболее вероятные источники погрешностей измерения при работе с жидкостными манометрами рассмотрены в работе Доша [34]. С помощью прецизионных масштабных линеек и нониусов можно определять высоту столба жидкости в манометре с максимальной погрешностью до 0,2 мм. Точный вакуумметр системы Голланд-Мертена [35 ] в интервале от 200 до 20 мм рт. ст. обеспечивает точность измерения ±0,5 мм рт. ст. Этот прибор позволяет измерять с помощью наклонного колена остаточное давление в пределах от 20 до 0 мм рт. ст. с точностью ±0,1 мм рт. ст. (рис. 372).
Точность измерения давления может быть значительно повышена путем замены ртути на силиконовое масло. Такой наполнитель используется в манометре Лапорта, предназначенном для измерения давления в пределах от 0,2 до 20 мм рт. ст., и в вакуумметре, работающем на принципе вытеснения наполнителя [35 ], с точностью 0,01 мм рт. ст. в интервале от 0 до 10 мм рт. ст. В каче
441
стве наполнителей манометров не следует использовать масло для вакуумных насосов, парафиновое масло и жидкость «амоил S». Повышенная способность к поглощению влаги, характерная для этих жидкостей, приводит к значительным погрешностям измерения. Бахман рекомендует при заполнении манометров водой добавлять к ней вещества, уменьшающие поверхностное натяжение воды. Для измерения давления в интервале 760— 1 мм рт. ст. можно использовать также манометр Мак-Леода. Этот прибор применим в основном для измерения остаточных давлений от 10-1 до 10-6 мм рт. ст., поэтому он описан ниже в соответствующем разделе. Для интервала давления от 0 до 60 мм вод. ст. можно применять указанные выше контактные манометры [36а].
Основное преимущество мембранных манометров заключается в том, что точность измерения с помощью этих приборов не зависит от химической природы исследуемых газов или паров [371. Измерительным устройством в данных приборах является тонкая металлическая мембрана. Мембранный вакуумметр VM-M (фирма Хэраеуза, Ханау) работает в интервале остаточных давлений от 50 до 0,1 мм рт. ст. с точностью ртутного манометра (рис. 373). Данный вакуумметр особенно пригоден для лабораторных и пилотных ректификационных установок. Существенное отличие этого прибора заключается в том, что его чувствительность не зависит от возможного проникновения в него воздуха. Прецизионные приборы для измерения давления, эталонные ртутные дифманометры и анероиды с круглой шкалой выпускает большими партиями фирма «Баллас Тирнау-хлоратор» в Вассербурге. В пилотных
Рис. 372.
Точный вакуумметр системы Голланд-Мертена.
Рис. 373.
Мембранный вакуумметр VM-M
Изготовитель: фирма Хэраеуза, Ханау.
442
Рис. 374.
Схемы систем регулирования давления методом подсоса воздуха (а) и методом частичного вакуумирования (6)'.
1 — вакуумный насос; 2 — буферная емкость; 3 — манометр; 4 — регулятор давления; 5 — электронное реле.
ректификационных установках целесообразно использовать также пружинные манометры различных конструкций [2 1.
При измерении давления с помощью любых приборов важно устанавливать перед манометром охлаждаемую ло-
вушку для задержки конденсирующихся паров. Необходимо также наблюдать за тем, чтобы между участком конденсации паров и точкой измерения давления не возникал градиент давления. С этой целью
до начала разгонки следует проводить сравнительные измерения давления, поочередно присоединяя манометр к шлифу для термометра и к штуцеру, расположенному за охлаждаемой ловушкой.
Для автоматического регулирования остаточного давления в интервале от 760 до 1 мм рт. ст. применяют механические и комбинированные электронномеханические приборы [38]. При
этом различают метод подсоса воздуха и метод частичного вакуумирования. При использовании метода подсоса воздуха вакуумный насос работает непрерывно, и в буферный сосуд с помощью крана тонкой регулировки подают такой объем воздуха, чтобы в системе установилось заданное давление. Основным условием успешного применения данного метода является равномерная работа вакуумного насоса и полная герметичность аппаратуры (рис. 374, а), поэтому этот способ используется только в исключительных случаях.
I 8.3.1.1. Методы частичного вакуумирования
| При регулировании давления методом частичного вакуумирования I вакуумный насос включается только тогда, когда давление в бу-I ферном сосуде превысит предварительно заданное давление, на J которое настроен регулятор. Схема системы регулирования давле-I ния по этому методу представлена на рис. 374, б.
I В интервале от 760 до 10 мм рт. ст. можно применять, например, I регулятор давления с пористой пластиной [39], показанный I на рис. 375. В качестве запорной жидкости в этом регуляторе I применяют ртуть. Количество ртути определяют из условия, что В ее уровень должен быть выше примерно на 3 мм нижнего края I пористой пластины. При открытых кранах 1—3 ректификационную
443
Рис. 375.
Регулятор давления с пористой пластиной;
1 — 3 — краны; 4 — сосуд с пористой пластиной; 5 — сосуд, находящийся под заданным остаточным давлением.
колонну вакуумируют до тех пор, пока давление, показываемое манометром, не будет превышать заданное на 1 —2 мм рт. ст., после
чего кран 3 закрывают. Давление в сосуде 5 начинает уменьшаться,
что вызывает соответствующее снижение уровня ртути в сосуде с пористой пластиной 4. Как только Пористая пластина 4 станет освобождаться от ртути, кран 1 перекрывают. При этом остаточное давление в сосуде 5 окажется меньше, чем давление в сосуде с пористой пластиной 4, и эта разность давлений будет определяться расстоянием между уровнями ртути в сосудах. При возрастании давления в ректификационной установке расстояние между уровнями ртути еще более увеличится и пористая пластина полностью освободится от ртути. В этот момент включается вакуумный насос, который вакуумирует систему до тех пор, пока давление в ректификационной установке не уменьшится до заданного значения и пористая пластина вновь покроется ртутью. При термостатировании данного регулятора давления, удается поддерживать заданный вакуум с точностью ±0,5 мм рт. ст.
Механический регулятор давления Джильмонта [40 ] работает на принципе поплавка с переменной глубиной погружения. Его можно использовать для регулирования остаточного давления не ниже 6 мм рт. ст. На рис. 376 показан этот регулятор, смонтированный на деревянном щите вместе с манометром Штока и обратным клапаном Фридрихса—Антлингера [41 ]. Точность регулирования данного устройства составляет ±0,5 мм рт. ст.
Применяя электронные приборы, можно повысить точность регулирования давления до ±0,1 мм рт. ст. Ртутный манометр можно заменить манометром, заполненным какой-либо высоко-кипящей, электропроводной и дегазированной жидкостью, что обеспечивает повышение чувствительности прибора примерно в 10 раз. При этом разность давлений в 1 мм рт. ст. будет соответствовать разнице уровней жидкости в 10—13 мм. Наименьшее давление, измеряемое с помощью прибора, в этом случае определяется давлением паров заполняющей жидкости. В жидкостном манометре Дубровина, который основан на фотоэлектрическом методе измерения, на фотоэлемент направляют тонкий световой пучок. При увеличении давления в аппаратуре поплавок, всплывая, перекрывает луч света, и неосвещаемый фотоэлемент включает через реле вакуумный насос [42].
444
Другой метод регулирования давления заключается в том, что к манометрической жидкости, например бутилгликолю, добавляют небольшое количество соли, например нитрита натрия, для повышения ее электропроводности. При этом сила тока увеличивается до 10"6 А (напряжение 6—8 В) и с помощью электронного реле (см. разд. 8.2.2) ее можно усилить до 10—15 А при напряжении 220 В. Для регулирования давления в правом колене манометра создают заданное давление, причем это колено термостатируют для исключения влияния изменений температуры (см. рис. 374,6). При равенстве давлений в правом колене и в ректификационной установке уровень манометрической жидкости в обоих коленах манометра одинаков. При повышении давления в установке уровень жидкости в правом колене понижается. В результате повышения уровня жидкости в левом колене электрический контакт замыкается и через электронное реле включается вакуумный насос, который вакуумирует систему до выравнивания давлений.
С помощью автоматического стенда с вакуумным насосом (рис. 377) во время испытаний с чистыми веществами в интервале давлений от 300 до 1 мм рт. ст. была достигнута точность регулирования ±0,1 мм рт. ст. [431. Такая точность для перегонки фенола при давлении 20 мм рт. ст. соответствует разнице температур кипения ±0,1 °C. Если, например, при разделении изомеров ксилола при давлении 70 мм рт. ст. необходимо определять темпера-
Регулятор давления Джильмоита, основанный на принципе поплавка с переменной глубиной погружения.
Рис. 377.
Автоматический стенд с вакуумным насосом, разработанный Крелем. Изготовитель: народное предприятие «Комбинат технического стекла», Ильменау.
445
Рис. 378.
Автоматический регулятор давления Штаге:
1 — поплавок; 2 — масляный манометр; 3 — контакт; 4,6 — штуцеры для присоединения к аппаратуре и вакуумному насосу; 5 — электромагнитный клапан.
Рис. 379.
Прибор для поддержания постоянного вакуума VK1:
1 — «автоматика»; 2 — «установка»; 3 — штуцер для залива запорной жидкости (уровень жидкости должен быть ниже конца контакта примерно на 2 мм).
Изготовитель: фирма Фишера «Лабораторная и исследовательская техника», Бони —курорт Годесберг.
туру кипения с точностью 0,1 °C, то для этого потребуется регулировать давление с точностью, превышающей ±0,15 мм рт. ст. [44 ]. Регулятор давления Штаге (рис. 378) имеет поплавок 1, который размещается во внутреннем колене масляного манометра 2, причем верхний конец контакта 3 лежит непосредственно на весах. Штуцер 4 служит для присоединения регулятора к колонне. При возрастании давления в колонне уровень масла вместе с поплавком
1 в манометре 2 опускается и реле открывает электромагнитный
клапан 5, соединенный с вакуумным насосом с помощью штуцера
6. В приборе Фишера [27], служащем для поддержания в системе
постоянного вакуума, регулирование осуществляется посредством
манометра с наклонной трубкой; манометр заключен в покрытый
слоем серебра вакуумированный кожух (рис. 379).
8.3.2. ИЗМЕРЕНИЕ И РЕГУЛИРОВАНИЕ ДАВЛЕНИЯ
В ИНТЕРВАЛЕ ОТ 1 ДО 10 * 1 * * * * * * 8 мм рт. ст.
Измерение вакуума в области очень низких давлений связано со значительными трудностями, поскольку при очень малых плотностях газа непосредственное измерение его давления затруднено. В этих случаях применяют косвенные методы измерения. Методом, основанным на применении тлеющего газового разряда,
446
можно лишь приближённо определять область измеряемых давлений. Флуоресцентный вакуускоп Бюргера позволяет определять остаточные давления, равные 10'1, 10'3 и 10'5ммрт. ст., а промежуточные значения давлений можно только оценивать. Для обеспече-
ния воспроизводимости результатов при пленочной и молекулярной дистилляции рекомендуется применять методы измерения давления, приведенные в табл. 68.
Методы измерения сверхвысокого вакуума, соответствующего остаточным давлениям от 2-10'10 до 10'12 мм рт. ст., описаны Хохом [46]. Сведения о преимуществах и недостатках манометров, приведенных в табл. 68, можно найти в работе Штекельмахера [47 ].
Альфатрон представляет собой ионизационный манометр, в котором в результате бомбардировки молекул остаточного газа «-частицами образуются ионы [45]. В молекулярном вакуумметре (абсолютном манометре Кнудсена) для измерения вакуума используют эффект радиации. Широкое применение находят молекулярные вакуумметры Геде [48] и вакуумметры Лангмюра с кварцевой нитью [49]. Работа мембранного микроманометра, так же как и
молекулярного вакуумметра, не зависит от химической природы исследуемого газа. Этот прибор позволяет измерять давление в интервале от КГ4 до 1,5-10'1 мм рт. ст. Методика измерения остаточных давлений с помощью диффузионных ртутных насосов описана Мюллером [50].
В настоящей монографии рассмотрены только некоторые модели вакуумметров, выпускаемых промышленностью. Подробное и полное описание этих приборов можно найти в справочнике по
Таблица 68.
Область применения манометров и интервал измеряемых давлений (по Мёнху [45])
Тип манометра или вакуумметра Вид измеряемого давления Зависимость показаний от вида газа-наполиителя Интервал измеряемых давлений, мм рт. ст.
Спиральный и мембран- Общее давление Не зависят 760-Ю-3
ный манометры Газоразрядный манометр Общее давление Зависят 10-2—Ю-»
(вакуумметр Филипса) Демпферный и вязкост- Общее давление Зависят ю-1—10'4
ный манометры Манометр Пирани, ос- Общее давление Зависят ю-1—10'4
нованный на измерении теплопроводности -
Компрессионный мано- Парциальное дав- Не зависят (толь- 760—10"1
метр Мак-Леода ление не конденсирующихся паров ко для газа) Ю-1— ю-s
Ионизационный манометр Общее давление Зависят IO-3— ю-6
Радиометр (молекулярный вакуумметр) Общее давление Не зависят 10"1—IO'8
447
технике высокого вакуума [49]. Для измерения давлений от 10"1 до 10"4 мм рт. ст. используются манометры Пирани, основанные на принципе измерения теплопроводности, и в интервале от 10-3 до 10~6 мм рт. ст. применяются ионизационные манометры. В настоящее время промышленностью выпускаются приборы, являющиеся комбинацией двух последних манометров.
Преимущество электрических вакуумметров состоит в том, что их показывающее устройство непрерывно фиксирует измеряемое давление, благодаря чему появляются возможности для его регулирования и записи показаний. Как вакуумметр, основанный на цринципе измерения теплопроводности, так и ионизационный вакуумметр, измеряют общее давление газов и паров, причем для определения остаточного давления газа их необходимо подключать к системе через охлаждаемую ловушку.
Калибруют эти приборы известным методом с помощью вакуумметра Мак-Леода при использовании предварительно осушенного воздуха. Соответствующие калибровочные кривые представлены в технической документации на прибор. Точность измерения манометра Пирани составляет 5% в интервале давления от 1 до 5-10~3 мм рт. ст., а ионизационного вакуумметра —3% в интервале давления от 1СГ3 до 10'6 мм рт. ст. Для подсоединения этих приборов к вакуумированной ректификационной установке они снабжаются штуцером со стандартным шлифом NS 14,5/23 или небольшим фланцем.
8.3.2.1. Компрессионный манометр Мак-Леода
Компрессионные манометры точно измеряют давление только идеальных газов. Они основаны на принципе сжатия определенного объема газа при измеряемом давлении в капиллярной трубке до давления, которое можно легко измерить. Измеряемое давление с достаточной точностью можно рассчитать по формуле:
p=A/tV2/V1 (197)
где р — измеряемое давление, мм рт. ст.; Д/г — разность уровней ртути в капиллярах, мм; Vi — объем стеклянного шараЗ (рис. 380) с капилляром 5 (считая от места присоединения 2 трубы 7), мл; У2 — объем сжатого газа в капилляре 5.
На рис. 380 показана одна из выпускаемых промышленностью моделей манометра Мак-Леода, в которой уровень ртути поднимается не с помощью груши со ртутью, а под действием атмосферного давления. Сосуд 12 заполняют тщательно очищенной ртутью и к штуцеру 6 подключают аппарат, в котором требуется измерить давление. Штуцер 10 соединяют с водоструйным насосом. При вакуумировании аппарата необходимо одновременно вакуумировать и пространство над ртутью в сосуде 12. С помощью крана 9 регулируют давление таким образом, чтобы не возникало слишком больших перепадов давления между объемом манометра и свободным объемом сосуда 12, так как иначе либо ртуть может подняться 448
очень высоко по манометрической трубке 1, либо воздух по этой трубке проникнет в сосуд с ртутью 12.
После установления в аппарате заданного давления с помощью крана 9 через штуцер 11 в сосуд 12 подают воздух, что приводит к подъему ртути в манометрической трубке 1. Для предотвращения попадания частиц пыли и капелек влаги в сосуд 12 целесообразно снабдить штуцер 11 специальным фильтром. Скорость подачи воздуха регулируют таким образом, чтобы ртуть медленно поднималась до отметки 8, указанной на верхнем конце капилляра 5. Измеряют разницу уровней ртути Ай и по формуле (197) рассчитывают давление р. Значение объема указано на стеклянном шаре 3, а объема V2 —на шкале капилляра 5.
Большинство Манометров данного типа откалиброваны таким образом, что измеряемое давление можно непосредственно регистрировать по шкале. При установке уровня ртути на отметке 4 по шкале, размещенной у трубы 7, определяют давление в интервале до КГ2 мм рт. ст. Перед каждым новым измерением с помощью подключения водоструйного вакуумного насоса нужно опустить уровень ртути в приборе ниже места присоединения 2 трубы 7. Объем стеклянного шара 3 и количество ртути, необходимое для заполнения манометров Мак-Леода, выпускаемых промышленностью, приведены в табл. 69.
Многие модели манометров Мак-Леода рассмотрены в книге Эберта [51 ]. Пневматически регулируемый и самоустанавливаю-щийся компрессионный манометр Мак-Леода, показания которого не зависят от давления окружающего воздуха, подробно описан Пехе [52]. В этой работе приведены также рекомендации по обеспечению воспроизводимости результатов измерений.,
На рис. 381 показана одна из моделей вакуускопа, являющаяся комбинацией укороченного U-образного манометра и компрессионного манометра. Данным прибором можно измерять давление
Рис. 380.
Компрессионный манометр Мак-Леода:
/ — манометрическая трубка; 2 — место присоединения трубы 7; 3 — стеклянный шар; 4, 8 — верхние отметки шара и капилляра; 5 — капилляр; 6, 10 — штуцеры для присоединения аппарата и водоструйного насоса; 9 — край; 11 — штуцер для подачи воздуха в сосуд со ртутью; 12 — сосуд со ртутью.
Рис. 381.
Вакуускоп.
Интервалы измеряемых давлений от 80 до 1 мм рт. ст. (по U-образному манометру) и от 4 до 10“2 мм рт. ст. (по компрессионному манометру).
V215 Э. Крель
449
Таблица 69.
Объем стеклянного шара и количество ртути, необходимое для заполнения манометров Мак-Леода
Интервал измеряемых давлений» мм рт. ст. Объем стеклянного шара, мл Объем ртути, мл
2—10-» 300 450
2—IO"4 100 250
15—IO'3 15 50
в интервале от 80 до 1 мм рт. ст. с помощью U-образного манометра и от 4 до 10"2 мм рт. ст. с помощью компрессионного манометра.
Для лабораторных ректификационных установок очень удобны поворотные манометры Мозера (рис. 382). Они рассчитаны на три интервала измеряемых давлений 700—1, 1—10-2 и 10"2—10"4 мм рт. ст.; для их заполнения требуется всего лишь 6—7 мл ртути. Переход от одного интервала к другому может быть осуществлен поворотом манометра на шлифе против часовой стрелки. Прибор заполняют ртутью настолько, чтобы в положении «г» ртуть, находящаяся при разряжении, только на половину заполняла расширение А. Манометр поворачивают до тех пор, пока столбик ртути не достигнет отметки Мг (интервал измеряемых давлений от 700 до 1 мм рт. ст.) или М2 (интервал от 1 до 10-2 мм рт. ст.). В каждом интервале измерений давление определяют по шкалам пъ п2 и п3. Как и большие манометры Мак-Леода, этот прибор перед каждым новым измерением необходимо возвращать в исходное положение. Манометр Мозера можно проверять и калибровать с помощью большого манометра Мак-Леода.
Компрессионный манометр Каммерера [53] также отличается весьма малым количеством используемой ртути (рис. 383). Его
Рис. 382.
Поворотный манометр Мозера:
а — вид сбоку; б — исходное положение; в — положение при измерении интервале от 700 до 1 мм рт. ст.; г — то же в интервале от 1 до 10“*; д —
то же в интервале от 10“2 до КГ* мм рт. ст.
450
Рис. 383.
Компрессионный манометр Каммерера.
При измерении давлений от 10 до 10‘а мм рт. ст. требуется 16 мл ртути, а в интервале* от 1 до 10“* мм рт. ст. — 17 мл.
преимуществом является также весьма простой метод заполнения ртутью с по-мощью насоса новой модели. Такие приборы выпускаются для интервалов измеряемых давлений от 10 до 10-8 мм рт. ст. и от 1 до 10-4 мм рт. ст.; для измерения остаточных давлений до 80 мм рт. ст. их снабжают U-образными трубчатыми манометрами.
В заключение еще раз отметим, что все компрессионные манометры можно применять для измерения парциального давления газов, неконденсирующихся при за-
данных степени сжатия и температуре. Пары веществ, имеющие при комнатной температуре давление выше 4 мм рт. ст., можно легко обнаружить по расхождению результатов измерений, воз-
никающему при переходе от одного интервала измерений к другому [51 ].
8.3.2.2. Регулирование давления в интервале от 100 до 10-8 мм рт. ст.
Об автоматическом регулировании остаточного давления в областях среднего и высокого вакуума в литературе имеется сравнительно мало сведений. При использовании вакуумметров, основанных на принципе измерения теплопроводности газа, Лапорт [49] рекомендует подключить к мостовой схеме Пирани сигнальное устройство, которое дает звуковой сигнал при увеличении давления выше заданного предела. Нисбет [54 ] описал прибор, позволяющий поддерживать в сосуде, продуваемом воздухом, постоянное давление 10-4 мм рт. ст. Мельпольдер [55] описал регулятор давления, обеспечивающий в интервале от 10-8 до 10-8 мм рт. ст. точность регулирования, равную ;±10'4 мм рт. ст. Схема данного регулятора приведена на рис. 384. Принцип его работы заключается во введении в манометр Мак-Леода четырех впаянных контактов 9—12. С помощью устройства 13 в манометре Мак-Леода каждую минуту поднимают уровень ртути. Регулирование давления осуществляется с помощью контактов 9 и 11. При уменьшении давления в системе ниже заданного контакт 10 замыкается, при этом он через реле 3 и 2 закрывает электромагнитный клапан 5. Этот клапан размещен на штуцере 4, соединяющем систему с вакуумным насосом. Вакуумированный аппарат подсоединяют к шту-
451
4
5
Рис. 384.
Схема регулятора давления Мельпольдера, работающего в интервале от 10"* до 10"® мм рт. ст.:
1 — переключатель; 2,3 — реле; 4,6 — штуцеры для присоединения вакуумного иасоса и аппарата; 5 — электромагнитный клапан; 7 — буферная емкость; 8 — охлаждаемая ловушка; 9—12 — впаянные контакты;
13 — устройство для подъема уровня ртути в манометрической трубке манометра Мак-Леода; 14 — манометр Мак-Леода.
церу 6, буферная емкость 7 служит для уменьшения колебания давления. В ловушке 8, охлаждаемой жидким азотом, задерживаются пары конденсирующихся веществ. При вакуумировании системы в период подготовки регулятора к работе переключатель 1 устанавливают в положение «автоматическое регулирование», как только остаточное давление в системе достигнет 10 мм рт. ст.
Электронный прибор Фишера [27] VKH для поддержания постоянного вакуума, основанный на принципе измерения теплопроводности газов, работает в интервале остаточных давлений от 100 до 10-3 мм рт. ст. (рис. 385). Датчик прибора подключается к вакуумируемой системе с помощью стандартного шлифа. Прибор
имеет переключатель, служащий для переключения регулятора с одного интервала
Рис. 385.
Электронный прибор для поддержания постоянного вакуума VKH
Изготовитель; фирма Фишера, «Лабораторная и исследовательская техника», Бонн— курорт Годесберг.
452
йЗМёрёния на другой. К выходному концу компенсационного усилителя непосредственно подсоединен магнитный клапан, размещенный на трубопроводе, который связывает систему с вакуумным насосом.
8.4. РЕГУЛИРОВАНИЕ ФЛЕГМОВОГО ЧИСЛА
И СКОРОСТИ ВЫКИПАНИЯ
8.4.1. РЕЛЕ ВРЕМЕНИ ДЛЯ РЕГУЛИРОВАНИЯ ФЛЕГМОВОГО ЧИСЛА
Автоматические головки ректификационных колонн, как правило (см. разд. 7.5), работают по принципу регулирования по времени. Флегмовое число при этом определяется отношением промежутка времени выключения реле (подача флегмы в колонну) к промежутку времени его выключения (отбор дистиллята). Устанавливаемые на головках колонн механические или электронные реле времени должны обеспечивать любую продолжительность отбора дистиллята. При малых флегмовых числах от 1 до 5 можно, например, устанавливать продолжительность отбора дистиллята, равной 1 с, что соответствует продолжительности подачи флегмы в колонну от 1 до 5 с. При работе с высокими флегмовыми числами в интервале от 10 до 50 продолжительность включения реле должна быть настолько уменьшена, чтобы скорость отбора дистиллята соответствовала скорости обогащения жидкости, орошающей колонну, высококипящим компонентом. Это означает, что особенно в период отбора промежуточных фракций промежуток времени включения реле (отбор дистиллята) необходимо постепенно сокращать, а промежуток времени выключения реле (подача флегмы в колонну) — соответственно увеличивать.
Эти условия соблюдены в механическом реле времени, разработанном автором [11]. Прибор имеет контактную шайбу, приводимую во вращение синхронным электромотором. Необходимое флегмовое число устанавливается с помощью шибера. Клеммы служат для подключения электронного реле, приводящего в действие электромагнит, установленный в головке колонны. Выходное напряжение электронного реле составляет 220 В. С увеличением флегмового числа одновременно сокращается период включения реле для отбора дистиллята:
Флегмовое число.....................
Период включения реле, с ...........
5 10 20 50
2 1 0,5 0,2
Если электронное реле снабдить переключателем для работы в нормальнозамкнутом и нормальноразомкнутом положениях, то
15 Э. крель 453
можно устанавливать обратные величины флегмовых чисел, т. е. получать флегмовые числа меньше единицы:
При нормальнозамкнутом положении .......................... При нормальноразомкнутом положении ........................
1 2 3 5 10
1 1/2 1/3 1/5 1/10
Длительность и частоту включений реле можно произвольно варьировать путем замены контактных шайб. Этот метод обеспечивает повышенную точность регулирования флегмового числа в интервале от 20 до 100 при аналитической ректификации. При опытно-промышленных и промышленных разгонках период включения реле при отборе дистиллята увеличивают до 30 с.
В настоящее время более предпочтительными являются электронные реле, не имеющие каких-либо механических устройств. Их продолжительность включения и выключения изменяется в интервале от 0,1 с до 20 мин с точностью до 1% 156]. Для задания определенного флегмового числа с помощью этих реле необходимо устанавливать две величины — продолжительность включения и продолжительность выключения. Промышленностью выпускается ряд моделей таких реле, которые обычно снабжены устройством для контроля за температурой кипения. При переключении головки колонны на работу с бесконечным флегмовым числом в верхней части колонны температура повышается до значения, установленного на контактном термометре, который размещен в головке колонны. При понижении температуры в головке колонны ниже установленной прибор автоматически восстанавливает предварительно заданное флегмовое число.
На рис. 386 показан ректификационный регулирующий прибор «Минитрон 5», который одновременно с регулированием продолжительности включения и выключения реле регулирует также температуры куба и обогревающего кожуха ректификационной
Рис. 386.
Регулирующий прибор для ректификационных установок «Мииитрои 5»: 1 — регулятор температуры куба; 2 — стабилизатор температуры обогревающего кожуха; 3 — регулятор температуры обогревающего кожуха; 4 — переключатель периода времени подачи флегмы; 5 — переключатель периода времени отбора дистиллята^ 6 — амперметр; 7 — выключатель контактного термометра; 8 — розетка для подключения к электросети; 9 — функциональный переключатель.
Изготовитель: фирма Фишера «Лабораторная и исследовательская техника», Бонн —курорт Годесберг.
454
колонны (см. разд. 8.2.2). Реле времени имеет 11 ступеней для установки продолжительности отбора дистиллята в интервале от 0,5 до 10 с и 11 ступеней для установки продолжительности подачи флегмы в колонну в интервале от 1 до 100 с. При этом для каждого выбранного периода времени включения реле можно устанавливать любой из одиннадцати промежутков времени подачи флегмы, что позволяет изменять флегмовое число в интервале от 1 до 200. Преимуществом прибора является также возможность подключения к транзисторному реле устройства, обеспечивающего прекращение отбора дистиллята или общее отключение ректификационной установки, что сопровождается звуковым и оптическим сигналами.
Техника регулирования подачи флегмы с помощью автоматических головок ректификационных колонн и электронных реле времени подробно рассмотрена в обзорной работе Рёка с сотр. [13 ]. Геммекер и Штаге [57] показали, что постоянный расход флегмы, а также его воспроизводимость и независимость от нагрузки колонны, можно обеспечить, только применяя регулируемые электромагнитные делительные устройства. Возникающие при этом погрешности регулирования могут быть обусловлены следующими причинами: а) флегмовое число превышает отношение периодов выключения и включения реле: при конденсации пара в нижней части делителя флегмы и непопадании этого конденсата в делительную воронку; при стекании части конденсата, образовавшегося в дефлегматоре, мимо делительной воронки; при попадании части паров в дефлегматор в момент отбора дистиллята, когда регулирование флегмового числа производится делением парового потока; б) флегмовое число меньше, чем отношение периодов выключения и включения реле: при наличии небольшого остаточного количества жидкости в «мертвом» объеме электромагнитного клапана; при конденсации пара в системе отбора дистиллята; при образовании в дефлегматоре капелек жидкости, не стекающих в колонну при отборе дистиллята, когда регулирование флегмового числа производится делением парового потока.
8.4.2. РЕГУЛИРОВАНИЕ СКОРОСТИ ВЫКИПАНИЯ
Необходимым условием регулирования флегмового числа по заданным периодам включения и выключения реле времени является постоянство нагрузки колонны по пару. При установке флегмового числа вручную регулирование скорости выкипания, необходимо для обеспечения постоянной разделяющей способности колонны и воспроизводимости результатов. Необходимую скорость выкипания можно поддерживать двумя методами: стабилизацией мощности электронагревателя куба (преимущественно в процессах дистилляции) и регулированием этой мощности по перепаду давления потока пара в колонне (в процессах ректификации).
Метод стабилизации мощности электронагревателя куба может быть, конечно, использован и в процессах ректификации, однако 15* 455
Рис. 387.
Устройство для регулирования нагрузки по перепаду давления в колонне: 1 — расходомер; 2 — холодильник; 3 — контактный манометр; 4 — подвижной контакт; 5 — реле.
Рис. 388.
Контактный манометр Креля-
Изготовитель: народное предприятие «Комбинат технического стекла», Ильменау.
этот метод требует частого контроля нагрузки колонны по пару, особенно при значительной разнице удельных теплот испарения компонентов кубовой жидкости. Регулирование мощности обогрева куба по перепаду давления потока пара в колонне более предпочтительно, поскольку перепад давления непосредственно связан со скоростью движения пара в колонне (см. разд. 4.11). Преимущество этого метода регулирования состоит также в том, что колебания напряжения в сети при использовании электронагревательных устройств или колебания давления и теплотворной способности газа при обогреве газовым пламенем не оказывают влияния на точность регулирования [13].
На рис. 387 показано устройство для регулирования нагрузки по перепаду давления, выполненное по нормалям «Дестинорм» [11 ]. Расходомер 1, служащий для измерения количества жидкости, стекающей из колонны в куб, имеет боковой патрубок с холодильником 2, предохраняющим от проникновения пара в контактный манометр 3. Чтобы предотвратить образование капелек конден-456
Рис. 389.
Контактный манометр с подвижным коленом:
/ — отметка Для заполнения жидкости —наполнителя;
2 — фильтр G1.
Изготовитель: фирма Шотт, Майнц,
сата в боковом патрубке или в холодильнике 2, можно продувать их азотом через счетчик пузырьков или, что еще проще, обогревать боковой патрубок с помощью электроспирали. Холодильник соединен с контактным манометром 3, снабженным подвижным контактом
4, который можно перемещать даже в процессе работы устройства. Одно колено контактного манометра при ректификации под атмосферным давлением остается открытым, а при вакуумной ректификации соединяется с вакуумированным трубопроводом, идущим к верхней части колонны.
Контактный манометр (рис. 388) в комбинации с реле, описанным в разд. 8.2.2, заполняют ртутью, а в комбинации с электронным реле — органической токопроводящей жидкостью. Эта жидкость по сравнению с ртутью повышает чувствительность прибора примерно в 10 раз. Сначала с помощью реостата устанавливают необходимую нагрузку колонны по пару, соответствующую определенному перепаду давления. Затем опускают контакт 4 (см. рис. 387) до соприкосновения его нижнего конца с мениском жидкости-наполнителя и несколько увеличивают силу тока. Реле 5 связано с электронагревателем куба и выключает его при замыкании контакта. Целесообразно использовать такую электроизмерительную схему, в которой регулируется только 1/3 общей мощности. Рекомендуется применять реле с запаздыванием включения, чтобы случайные скачки давления в кубе, возникающие при кипении, не приводили к включению электронагревателя. Можно пользоваться также контактным манометром с подвижным коленом (рис. 389), который имеет миллиметровую шкалу на колене и угловую
шкалу.
Изящный метод регулирования описал Штаге [58]. Регулирование осуществляется с помощью электронного устройства по температуре или по расходу флегмы, выраженному числом капель или миллилитров в 1 мин. Число капель флегмы определяется оптическим путем и пропорционально этому числу непрерывно регулируется мощность электрообогрева куба. Температура куба регулируется по температуре кипения кубовой жидкости в различные моменты перегонки. Нагрузка колонны по пару остается постоянной даже в период, когда имеет место смена фракций в кубовой жидкости,
457
8.5. ИЗМЕРЕНИЕ ФИЗИКО-ХИМИЧЕСКИХ
СВОЙСТВ ДИСТИЛЛЯТА
В ПРОЦЕССЕ РЕКТИФИКАЦИИ
Вследствие того что температура в верхней части колонны не определяет однозначно степень чистоты дистиллята, в процессе ректификации целесообразно определять и другие физико-химические свойства дистиллята без его отбора. Для этого необходимо проводить измерения на линии подачи флегмы в колонну, что дает возможность своевременно изменять режим процесса ректификации соответственно результатам измерений с целью получения требуемого состава дистиллята.
При перегонке на лабораторных и пилотных ректификационных установках можно непрерывно измерять следующие физико-химические свойства [59]: температуру затвердевания, показатель преломления и диэлектрическую проницаемость *. Во всех случаях, кроме определения температуры затвердевания, основная трудность состоит в обеспечении термостатирования исследуемой жидкости, температура которой при ректификации обычно непрерывно повышается.
8.5.1. ИЗМЕРЕНИЕ ТЕМПЕРАТУРЫ ЗАТВЕРДЕВАНИЯ
Применительно к ректификации высококипящих жирных кислот Янцен и Тидке предложили весьма удобное устройство для определения температуры затвердевания веществ с высокой температурой плавления (см. рис. 390). В этом устройстве, так же как и в приборе Тиле для определения температуры плавления, дистиллят (или флегма) сначала попадает через капельницу 3 в капилляр 5, омываемый термостатирующей жидкостью. Понижая температуру этой жидкости, определяют температуру затвердевания дистиллята по показаниям термометра 4. По байпасной трубке 2 в приемник 1 поступают следующие порции исследуемой жидкости. Повышая затем температуру термостатирующей жидкости, определяют таким же образом температуру плавления исследуемой жидкости.
* Наряду с указанными физико-химическими свойствами при ректификации на лабораторных и пилотных установках часто измеряют также вязкость, плотность, поверхностное натяжение и т. д. (см. разд. 8.5.4). — Прим. ред.
Рис. 390.
Устройстко Янцена—Тидке для определения температуры затвердевания веществ с высокой температурой плавления:
/ — приемник исследуемой жидкости; 2 — байпасная трубка;
3 — капельница; 4 — термометр; 5 — капилляр.
458
Мюллер и Ценшельский [60 ] разработали автоматический прибор, регистрирующий температуру плавления в течение любого периода времени перегонки с точностью ±0,25 °C.
8.5.2. ИЗМЕРЕНИЕ ПОКАЗАТЕЛЯ ПРЕЛОМЛЕНИЯ
Показатель преломления наряду с температурой кипения широко применяется для характеристики органических веществ [59]. При разделении близкокипящих веществ непрерывное определение
Рис. 391.
Рефрактометр «Ремат 10».
а — передняя панель прибора с кюветами К51 и двумя линейными показывающими шкалами; б — задняя панель прибора с кабелями для подключения к электросети.
Изготовитель: народное предприятие «Карл Цейс Иена».
459
Рис. 392.
Устройство Штаге с проточными рефрактометрами для измерения равновесных данных в системе пар—жидкость.
показателя преломления дистиллята позволяет своевременно изме-нять режим процесса ректификации, чтобы по возможности сократить объем промежуточных фракций и тем самым увеличить выход целевого продукта.
Фотоэлектрический рефрактометр позволяет фиксировать показания по шкале электрического прибора и одновременно записывать результаты измерений. Автоматические проточные рефрактометры с непрерывной регистрацией показаний разработаны Томасом с сотр. [61] и Лэтчумом [62]. Рефрактометр «Ремат 10», выпускаемый народным предприятием «Карл Цейс Иена» [63 ], также представляет собой проточный рефрактометр. Этот прибор позволяет определять показатель преломления движущейся пробы жидкости по разности между показателями преломления данной пробы и неподвижных сравнительных проб жидкостей. На рис. 391 показаны передняя и задняя панели данного прибора, работающего в интервале температур от —10 до +120 °C. Рефрактометр имеет четыре кюветы для различных пределов измерения показателя преломления.
Для определения показателя преломления удобен прибор Штаге, служащий для измерения равновесных данных в системе пар—жидкость (рис. 392). Два выпускаемых промышленностью рефрактометра устанавливают в приборе Штаге таким образом, что они позволяют определять в проточной системе концентрации жидкости и пара по показателям преломления. Для автоматической регистрации скорости изменения показателя преломления во времени можно использовать интерферометры. Прибор Кегелеса и Собера [64 ] с фотоэлектрической ячейкой диаметром 25 мм обеспе-
460
чивает точность показаний, соответствующую двум единицам в шестом знаке после запятой при использовании светового потока соответствующего зеленой линии Hg.
8.5.3. ИЗМЕРЕНИЕ ДИЭЛЕКТРИЧЕСКОЙ ПРОНИЦАЕМОСТИ
Тенденция к увеличению применения физических методов измерения привела к дальнейшей разработке техники измерения диэлектрической проницаемости. Измерение диэлектрической проницаемости (ДП) имеет особое преимущество при ректификации смесей, содержащих воду (ДП-80), а также смесей веществ с резко отличающимися значениями этого параметра. В качестве примера можно назвать смеси: уксусная кислота (ДП-6,13)--уксусный ангидрид (ДП-22,2) и метанол—толуол. Азеотропная смесь метанол—толуол, образующаяся при ректификации, имеет ДП-26,8, которая в значительной мере отличается от значений диэлектрической проницаемости исходных компонентов, равных соответственно 33,8 и 2,37 [65]. При определении концентрации толуола в бензоле данный метод измерения также оказывается наилучшим, хотя разница в значениях диэлектрической проницаемости у компонентов смеси равна всего 0,08.
На рис. 393 показано устройство Эме, служащее для измерения диэлектрической проницаемости в условиях ректификации [65]. Измерительную ячейку устройства можно помещать в трубу, подающую флегму в колонну, что обеспечивает возможность при необходимости своевременно изменять режим процесса ректификации. Монография Эме содержит ценный обзор полученных до настоящего времени значений диэлектрической проницаемости
указанием длины волны, температуры, при которой проводили измерение, и температурного коэффициента этой величины.
Преимущество мультидекаметра Сле-фогта [66 ] состоит в том, что измеряемая частота не является строго фиксированной и может изменяться в интервале от 10 кГц до 12 МГц. Грант [67] описал прибор для регистрации значений диэлектрической проницаемости, который весьма пригоден для использования в процессах ректифи-
для различных веществ с
Рис. 393.
Устройство Эме для измерения диэлектрической проницаемости в процессе ректификации:
/ — холодильник; 2 — штуцер для ввода эталонной жидкости; 3 — измерительная ячейка; 4 — экранированный кабель; 5 — регистрирующий прибор; 6 — подстарка.
4$|
кации. Области применения методов измерения диэлектрической проницаемости органических веществ исследованы Феллони и Тантилло [68].
8.5.4. ИЗМЕРЕНИЕ ДРУГИХ ФИЗИКО-ХИМИЧЕСКИХ СВОЙСТВ
При перегонке на лабораторных и пилотных ректификационных установках для непрерывного определения физико-химических свойств дистиллята и кубовой жидкости можно использовать методы измерения, применяемые в промышленности [2]. Однако во многих случаях измерительные приборы и устройства должны быть соответствующим образом модифицированы.
При аналитической разгонке можно использовать весовой плотномер Клазена [69 ], требующий 8 мл исследуемого вещества для каждого измерения. Точность измерения 10"3 г/см3. Для непрерывного измерения плотности жидкости применяют устройство, предложенное Линфордом [70].
В промышленности, особенно нефтеперерабатывающей, широкое применение нашли методы непрерывного измерения вязкости. Эти методы могут быть использованы и в условиях ректификации на лабораторных и пилотных установках. Обзор различных вискозиметров непрерывного действия, применяемых в процессах очистки нефти, представлен в работе Фрицше [71].
Проточные измерительные приборы, такие как инфракрасный спектрометр, ультрафиолетовый спектрометр и масс-спектрометр, все шире использующиеся в химической промышленности, могут найти применение и при ректификации на лабораторных и пилотных установках, особенно для аналитических разгонок [72]. Сиггиа [73 ] дал подробный обзор современных непрерывных методов анализа, применяемых при перегонке. В работе [74] можно найти сведения об аналитических анализаторах, предназначенных для исследования отдельных фракций.
В разд. 4.2 указывалось о том, какое значительное влияние оказывает на процесс разделения в ректификационной колонне поверхностное натяжение жидкости. Тензиометр «Лауда», по DIN 53914 (изготовитель: завод измерительных приборов «Лауда») позволяет автоматически измерять и регистрировать поверхностное натяжение жидкостей.
Все большее значение приобретают радиохимические методы контроля процессов перегонки. Благодаря экономии времени применение этих методов особенно целесообразно при анализе фракций в процессах ректификации многокомпонентных смесей углеводородов, спиртов или кислот с близкими физико-химическими свойствами. При радиохимическом анализе в исходную смесь вводят некоторое количество меченого вещества и затем определяют его концентрацию в выделяемых фракциях. Хьюгс и Мэлокой с помощью меченого вещества 14СН3ОН определяли
462
давление и состав в системе СН3ОН—-С2Н5ОН—Н2О и следили за режимом процесса ректификации. Очевидно, что аналогичным путем можно проводить испытания эффективности ректификационных колонн различных типов.
8.6. ИЗМЕРЕНИЕ РАСХОДА
И ДОЗИРОВАНИЕ ГАЗОВ И ЖИДКОСТЕЙ
Техника работы с газами подробно рассмотрена в монографиях Бернгауэра [76], Виттенбергера [77], а также Мюллера и Гнаука [78]. Поэтому ниже мы не будем детально обсуждать этот вопрос. Дозировать и измерять количества газа или пара в условиях перегонки на лабораторных и пилотных установках необходимо при низкотемпературной ректификации (см. разд. 5.3.1), перегонке с паром (см. разд. 6.1), а также при адсорбционной ректификации (см. разд. 6.3). Кроме того, для разделения легко воспламеняющихся или сильно гигроскопичных веществ может оказаться необходимым проводить ректификацию в атмосфере инертного газа, чаще всего азота, что также требует измерения расхода инертного газа.
Применительно к перегонке с насыщенным и перегретым водяным паром в разд. 6.1 были рассмотрены косвенные методы измерения расхода пара. Для измерения расхода газов и жидкостей при повышенных давлениях используют ротаметры с поплавками, вращающимися в потоке исследуемой среды. Расходомеры, основанные на счете пузырьков, и капиллярные реометры требуют предварительной калибровки по газовому счетчику, в то время как ротаметры поставляются заводами-изготовителями с калибровочными кривыми для определенных газов и жидкостей со шкалами соответственно в м3/ч и л/ч (О °C, 760 мм рт. ст.).
На рис. 394 показаны модели ротаметров, обычно используемые в лабораторных и промышленных установках. Применительно к пилотным ректификационным установкам подобные ротаметры выпускаются с пневматической или электроиндукционной. передачей показаний.
Для контроля за расходом охлаждающей воды при ректификации наиболее пригодны лабораторные ротаметры типа SW (рис. 395). В конические стеклянные измерительные трубки ротаметров вставляются поплавки, изготовляемые из различных материалов в зависимости от физико-химических свойств исследуемой среды. Методы автоматического регулирования расхода охлаждающей воды описаны в работе Ницпетера [79]. Промышленностью выпускаются ротаметры-регуляторы для жидкостей или газов, которые прекращают подачу исходной смеси в ректификационную колонну при выходе из строя водяного холодильника или при уменьшении расхода охлаждающей воды по сравнению с заданным значением. При восстановлении расхода охлаждающей воды регуляторы
463
Рис. 394.
Ротаметры со штуцерами для присоединения гибких шлангов (а), со штуцерами для присоединения гибких шлангов и кожухом для термостатиро-вания (б), со штуцерами в виде стандартных шлифов (в) и в специальной арматуре (г).
Изготовитель: фирма Хенига по изготовлению ротаметров, аппаратуры и машин, Аахен.
Рис. 395.
Лабораторный ротаметр типа SW16.1.
Измеряемый расход от 0,5 до 3 л/мин (интервал измерения I) и от 3 до 12 л/мин (интервал измерения II).
Изготовитель: народное предприятие «Завод исследовательских приборов», Знтц Фрайталь.
обеспечивают подачу исходной смеси в ректификационную установку [80].
При непрерывной ректификации сосуд с исходной смесью размещают на такой высоте, чтобы гидростатическое давление столба жидкости превышало давление в ректификационной колонне. Это обеспечивает непрерывную подачу разделяемой смеси в колонну. Если на линии подачи исходной смеси в колонну установлен обычный стеклянный кран, то приходится часто регулировать расход, так как по мере снижения уровня жидкости в напорном сосуде количество протекающей исходной смеси все время уменьшается. При использовании капельницы и многоходового крана можно непрерывно контролировать расход жидкости, однако практически равномерная подача жидкости обеспечивается лишь из напорного бака, работающего на принципе сосуда Мариотта. Из большого сосуда жидкость можно подавать под действием
464
сжатого воздуха с помощью резиновой ручной груши. В пленочной дистилляционной установке Креля сосуд для исходной смеси (см. рис. 196) снабжен стеклянным краном, который обеспечивает очень точное капельное дозирование жидкости. В капельнице Даммера и Крамера [81 ] (рис. 396) использованы оба указанные выше принципа: выравнивание давления по типу сосуда Мариотта и регулировка с помощью крана. Сосуд Мариотта с шестью сменными капиллярами, усовершенствованный Хенелем [82], позволяет точно дозировать по каплям малые количества жидкости.
Штаге применил в автоматизированной ректификационной установке «Лабодест» устройство для подачи исходной смеси с магнитным краном, регулируемым по времени (рис. 397). Время, в течение которого клапан закрыт, можно устанавливать в интервале от 1 до 120 с, а время, когда он открыт — от 1 до 2 с. При каждом открывании магнитного клапана определенное количество жидкости поступает в ниже расположенное устройство, измеряющее расход подаваемой исходной смеси. Клапан данного устройства устанавливается таким образом, чтобы поступившая жидкость полностью могла стечь за промежуток времени, когда магнитный
Устройство Штаге для подачи исходной смеси.
Рис. 398.
Йенский циркуляционный насос типа 100.
Изготовитель: народное предприятие «Йенский завод стекла Шотт», Иена.
Капельница [81, 88 ]:
1 — пробка клапана; 2 — отверстие; 3 — кожух для термостатироваиия;
4 — клапан с шаровым шлифом; 5 — капилляр для подачи капель; 6 — шпиндель клапана; 7 — запорная пробка с накаткой.
465
Рис. 399.
Зависимость иапора от производительности (по воде), при различных числах оборотов мотора п Йенского циркуляционного иасоса типа 100.
Рис. 400.
Электрический сильфоииый дозировочный насос для жидкостей и газов. Изготовитель: фирма Эрнст Хааге, Мюльхайм (Рур).
клапан закрыт. Бак для исходной смеси, соединенный с трубчатой вставкой, работает как сосуд Мариотта до тех пор, пока уровень жидкости в баке не превысит уровень жидкости в трубчатой вставке.
С помощью пневматического стеклянного крана Пинкафа и Вихтерле [83] можно, например, дозировать воду в интервале от 0,02 до 3500 мл/ч с точностью ±1,5 %. Вопросы дозирования веществ в лаборатории подробно обсуждаются в монографии Пинкафа [84 ].
Весьма удобен, особенно для пилотных установок, изготовляемый в Иене циркуляционный насос типа 100 (рис. 398). Он представляет собой простейшую модель центробежного насоса, выполненную из стекла. Напор и производительность этого насоса зависят от числа оборотов мотора; их можно определить по диаграмме, приведенной на рис. 399. Электрический сильфонный насос Хааге предназначен для ступенчатого дозирования газов и жидкостей при напоре до 20 м вод. ст. Стандартные модели без сальникового уплотнения имеют производительность от 15 мл/ч до 1500 л/ч. Их изготавливают из стали V4A, томпака и стеклопластика (рис. 400).
Особую группу составляют насосы перистальтического действия с гибкими шлангами из различных материалов. Их преимуще-466
ственно применяют для дозирования низкокипящих жидкостей, эмульсий, суспензий, газов и шламов различных видов [86]. Изменяя диаметр шланга (от 3 до 10 мм) и число оборотов вращения вала, можно дозировать вещества с расходом от нескольких мл/ч до 20 л/ч в интервале температур от —90 до +150 °C. На рис. 401 показан несоздающий пульсаций напора шланговый насос типа DP2-2, который можно использовать как для создания избыточного давления, так и вакуума. Этот насос имеет 16 отдельных независимых друг от друга каналов.
Для больших ректификационных установок непрерывного действия целесообразно применять поршневые насосы. В разд.
Рис. 401.
Шланговый насос типа DP2-2 с тремя различными скоростями подачи дозируемой среды.
Производительность от 3 до 2400 мл/ч.
Изготовитель: народное предприятие «Лабораторная техника», Ильменау.
Рис. 402.
Стеклянный центробежный насос с тефлоновым уплотняющим кольцом (прокладкой).
Изготовитель: фирма «Стеклянная техника», Висбаден — Ширштайн.
467
Рис. 403.
Насос для точного дозирования модели «Нормадос Р31».
Изготовитель: фирма Брана —Люббе. Гамбург.
производительность до 20 м3/ч Специализированная фирма Бран
5.1.3.1 были рассмотрены стеклянные дозирующие насосные головки с сильфонами из тефлона (см. рис. 143)188]. Для подачи различных сред можно применять также стеклянные центробежные насосы (рис. 402), имеющие и напор до 15 м. вод. ст. и Люббе (Гамбург) выпускает
для лабораторных и пилотных ректификационных установок ряд дозировочных насосов (рис. 403), которые можно использовать и для автоматического перемешивания различных жидкостей. Насосы модели «Нормадос» типов К и J с производительностью от 0,6 до 1121 л/ч применяют^ лабораторных и промышленных установках. Регулирование производительности осуществляется вручную, электрически или пневматически. Благодаря тому, что детали и
узлы насосов, имеющие одинаковое назначение, выполняют взаимозаменяемыми, из них можно собирать любые насосы данной модели. Насосы модели «Гномодос» рассчитаны на небольшую производительность (от 0,6 до 4,4 л/ч). Они имеют установочную гайку с накаткой, служащую для точной установки длины хода поршня.
Модель «Гномодос SO2» имеет несколько дозирующих насосных головок. Дозирующая насосная головка может выполняться в виде плунжерного или мембранного насоса. Передаточный механизм насоса позволяет изменять длину хода плунжера в интервале от 0 до 15 мм. К насосу можно подключать одновременно до пяти насосных головок.
9
Оборудование лаборатории. Ввод в эксплуатацию ректификационных установок
9.1. ПЛАНИРОВКА ПОМЕЩЕНИЯ
Основные нормы и правила проектирования новых лабораторий с учетом специфики техники перегонки рассмотрены в книге Бере [1], а также в работах Колемана [2] и Шрамма [3]. Планировка специальных лабораторий для аналитической дистилляции подробно описана Кинканноном и Вакером [4]. Для лаборатории, в которой проводится перегонка, необходимо помещение с протяженными и высокими стенами, удобными для крепления стендов, служащих для монтажа ректификационной аппаратуры. Примеры рациональной планировки и оборудования различных лабораторных помещений показаны на рис. 404—407.
Способы размещения пилотных ректификационных установок в промышленности демонстрируются на рис. 140, 141, 146 и 160.
Планировка помещения удлиненной формы (рис. 404). Для уменьшения шума вакуумные насосы размещают в отдельном помещении лаборатории для перегонки. Насосы 3 располагают у стены, отделяющей помещение для насосов от основного помещения лаборатории, чтобы по возможности сократить длину вакуумных коммуникаций. У противоположной стороны данной стены в основном помещении размещают стенд 6, предназначенный для монтажа вакуумных ректификационных установок. Для удобства регуляторы вакуума и вакуумметры устанавливают непосредственно за стендом. Помещение для вакуумных насосов можно одновременно использовать в качестве небольшой слесарно-механической мастерской.
У стены с окнами размещают лабораторные столы 1, к которым подводится электроэнергия и вода. На этих столах в основном проводят работы по измерению различных физико-химических свойств получаемых фракций дистиллята: показателя преломления с помощью рефрактометра Аббе или интерферометра, температур затвердевания и плавления; диэлектрической проницаемости и оптического вращения с помощью поляриметра. Рабочий стол 4, установленный в средней части основного помещения лаборатории, предназначен преимущественно для химических работ. У большей стены, выходящей в вестибюль, также размещают стенд 6. Для перегонки ядовитых веществ, вызывающих головную боль и головокружение (таких, как диэтиловый эфир, бензол, хлорированные углеводороды или органические нитросоединения) в лаборато-
469
Стена с окнами
Рис. 404.
Схема размещения оборудования лаборатории для перегонки, расположенной в комнате удлиненной формы, с помещением для вакуумных насосов (А) и боксом для работ с вредными веществами (£)<
/ — лабораторные столы; 2 — письменные столы; 3 — вакуумные насосы;
4 — рабочий стол; 5 — раковины; 6 — стенды для монтажа ректификационных установок; 7 — вытяжной шкаф.
Рис. 405.
Схема размещения оборудования лаборатории для перегонки, расположенной в укороченной комнате, с помещением для вакуумных насосов (Л) и боксом для работ с вредными веществами (Б)»
Обозначения те же, что иа рис. 404.
Рис. 406.
Лабораторный стол со штативами (в торцевой части).
Изготовитель: фирма Лейбольда, «Оборудование лаборатории», Кёльн— Байенталь.
рии выделяют специальное помещение — бокс. Это помещение должно быть снабжено хорошей вентиляционной установкой, обеспечивающей 40-кратный обмен воздуха в 1 ч. Бокс отделяют от основного помещения лаборатории прозрачной перегородкой из армированного стекла, через которую можно непрерывно наблюдать из лаборатории за работой установок в боксе. Пульты автоматизированных ректификационных установок с регистрирующую
Рис. 407.
Схема размещения оборудования лаборатории для перегонки, расположен* ной в укороченной комнате, с помещением для вакуумных насосов (Л) и высокими вытяжными шкафами.
Обозначения те же, что на рнс. 404.
щими приборами располагают в боксе у стены, обращенной к основному помещению лаборатории.
Планировка помещения укороченной формы (рис. 405, 407). Принцип планировки лабораторного помещения тот же, что и в варианте, изображенном на рис. 404. Стенды располагают вдоль свободных поперечных достаточно протяженных стен. Хорошо вентилируемый бокс, отделенный перегородкой от основного помещения лаборатории, предназначен для работы с вредными веществами. Рабочий стол 4, расположенный поперек комнаты, может
иметь на торце штативы для крепления небольших аппаратов (рис. 406).
Другой вариант планировки помещения укороченной формы показан на рис. 407. Если для определения свойств дистиллята преимущественно применяют химические методы, то в средней части основного помещения лаборатории размещают в поперечном направлении два больших рабочих стола 4. Вместо бокса устанавливают два вытяжных шкафа 7, которые снабжены стендами 6 для монтажа аппаратуры. Один из шкафов, имеющий высоту, равную высоте комнаты, размещают на полу (рис. 408). Второй шкаф
Рис. 408.
Вытяжной шкаф со штативом.
471
равен по высоте рабочему столу, его снабжают штативами для крепления небольших дистилляционных приборов. Скорость воздуха в дверном проеме вытяжного шкафа должна составлять 0,25—0,5 м/с.
9.2. СТЕНДЫ И КОММУНИКАЦИИ
Стенды для монтажа ректификационной аппаратуры могут быть как сборными, так и стационарно смонтированными. На рис. 409 показан стенд, собираемый из отдельных деталей специально для конкретной лабораторной установки. Передвижной стенд (рис. 410) позволяет размещать установку в любом месте лаборатории. Для монтажа больших ректификационных установок предпочтительно использовать стенды, крепящиеся к стене помещения. Укрепленные в стене шины Иордаля (рис. 411а) снабжены держателями (рис. 4116), которые можно устанавливать на любой высоте [5]. С помощью поперечных перекладин можно подгонять данное устройство под конкретную аппаратуру (см. рис. 408).
Исходя из многолетнего опыта автор рекомендует конструкцию стенда, показанную на рис. 412. Этот стенд состоит из отдельных секций шириной 1 м, он зацементирован в стену, покрытую метлахской плиткой снизу на высоту 1,5 м. Внизу стенда имеется ванна глубиной 80 см, облицованная кафелем. Ванна служит для сбора кубовой жидкости, которая может вылиться при поломке стеклянного куба. Кроме того, в случае загорания этой жидкости в ванне огонь будет локализован непосредственно возле установки.
Стенд, собираемый нз отдельных взаимозаменяемых деталей. Изготовитель: фирма Герард Плейгер, Анвере.
Рис. 410.
Передвижной стенд.
472
Рис. 412.
Секция стенда с ванной, облицованной кафелем, коммуникациями и электрощитом:
1 — двух- и трехполюсные розетки; 2 — общий выключатель; 3 — выключатели; 4 — контрольные лампы; 5,6 — трубопроводы для подачи воды и газа соответственно.
Рис. 413.
Стальные уголки с удлиненными прорезями для монтажа стендов с рабочими площадками.
Коммуникации для воды, газа, сжатого воздуха, водяного пара, а также линии вакуума прокладываются по стене. Отработанная вода отводится по сточному каналу. Настольные приборы и инструменты располагают на горизонтально размещенных деревянных щитах. В качестве распределителей электропитания для аппаратуры используются электрощиты, укрепляемые между двумя секциями стенда (см. рис. 412). Каждый щит снабжен 8— 10 штепсельными розетками с контрольными лампами и выключателями. По контрольным лампам легко определять, какая часть приборов установки включена. Общий выключатель дает возможность при необходимости выключать одновременно все электроприборы.
Для обслуживания ректификационных колонн, часто достигающих потолка комнаты, целесообразно применять узкие лестницы, которые можно хранить в углу за стендом.
Вентиляция стенда должна быть выполнена таким образом, чтобы воздух подавался у пола, а отсасывался у потолка. Обычно предусматривают 10-кратный обмен воздуха. Если при перегонке могут выделяться большие количества отходящих газов, то прокла
473
дывают специальные газоотводные трубопроводы, соединяющие конденсатор дистиллята с всасывающим отверстием вытяжной вентиляции.
Для обслуживания полупромышленных ректификационных установок в каждом конкретном случае создают специальные стенды с рабочими площадками (см. ниже рис. 424), которые также можно собирать из отдельных узлов и деталей. Монтаж таких стендов значительно упрощается при использовании стальных уголков с удлиненными прорезями (рис. 413).
9.3. СБОРКА УСТАНОВОК
Перед сборкой ректификационной установки, особенно состоящей из большого числа аппаратов, целесообразно составить монтажную схему, в.соответствии с которой разложить на полу узлы и детали установки. Затем необходимо проверить плотность посадки конусов и муфт в шлифовых соединениях, вставляя их без смазки. Смочив конус или нанеся на него мелом кольцевую полоску, можно легко обнаружить неравномерно отшлифованные части конуса и муфты. Шлифы со свободной посадкой конуса следует сразу же заменить. Такая предварительная проверка шлифов экономит много времени при последующей герметизации вакуумных ректификационных установок.
Монтаж установки обычно начинают от пола, причем при сборке установки необходимо предусмотреть возможность последующего снятия куба без демонтажа других аппаратов. Для этого целесообразно использовать вертикально перемещающиеся нагреватели кубов (см. рис. 327), сферические нагревательные приборы или пружинные колбодержатели (рис. 414). Отметим, что обычные
Рис. 414.
Пружнииый колбодержатель.
Рис. 415.
Стяжки с пружинами для крепления шлифовых соединений.
Рис. 416.
Клеммовый держатель для сферических шлифовых соединений.
474
Рис. 417.
Стяжка для плоских шлифовых соединений.
Рис. 418.
Держатель шлифовых соединений типа «Шлиф-кламмер» .
Изготовитель: фирма Эдвардс, Лондон.
кольцевые держатели не обеспечивают надежного закрепления куба.
Колонну устанавливают строго по отвесу. После закрепления колонны в вертикальном положении и проверки ее герметизации монтируют различные смежные аппараты и детали установки с последующей проверкой герметизации каждого отдельного узла установки.
Сборные детали установки должны иметь специальные крючки, предназначенные для крепления пружин, стягивающих шлифовые соединения. Для соединения шлифов крупных аппаратов используют пружинные стяжки (рис. 415). Клеммовый держатель для сферических шлифовых соединений показан на рис. 416- (см. разд. 3.1), а стяжка для плоских шлифовых соединений — на рис. 417. Легко разбираемым является держатель для шлифовых соединений модели «Шлиф-кламмер» (фирма Эдвардс, Лондон), крепящийся за выступ на стеклянной трубе (рис. 418). Резиновые манжеты для крепления шлифовых соединений (рис. 419) изготавливают для стандартных шлифов NS12,5—NS29. Они применимы при температурах до 130 °C [6]. Преимущество данных манжетов заключается в том, что они обеспечивают равномерное стягивающее действие по всей окружности шлифового соединения и позволяют отказаться от шлифовой смазки.
При использовании двойных муфт необходимо следить за тем, чтобы держатели не смещались в них по горизонтали или вертикали при затяжке винтов. В противном случае могут возникнуть меха-
475
Рис. 419.
Резиновые манжеты для крепления шлифовых соединений.
Рис. 420.
Двойная муфта (максимальный диаметр детали 14 мм).
Изготовитель: фирма Герард Плейгер, Анвере.
нические напряжения, которые обычно приводят к поломке стеклянных аппаратов, вследствие их термического расширения в процессе перегонки. Двойная муфта удобной конструкции показана на рис. 420. Муфта с крючками (рис. 421) предназначена специально для сборки стендов. Зажимные крючки и двойной контакт в трех точках гарантируют надежность соединения. Стандартная каркасная система, собираемая из большого числа взаимозаменяемых деталей, применяется для монтажа больших пилотных установок [7].
Нагревательные элементы, сферические сосуды с жидкостью и другие приборы можно точно устанавливать с помощью регулируемого штатива Маннхена—Риделя (рис. 422). С помощью этого штатива можно устанавливать аппараты на высоте до 15 см с точностью до 1 мм. Для крепления колонн и сосудов большого диаметра вместо обычных щековых клемм лучше использовать бандажи (рис. 423). Систематический обзор штативов различных конструкций представлен в работе Шварца [5].
Для сборки пилотных и полупромышленных ректификационных установок из стекла необходимы специальные соединитель-
Рис. 421.
Муфта с крючками.
Изготовитель: фирма Шпек, Франкфурт на Майне.
476
Рис. 422.
Лабораторный регулируемый штатив Маннхеиа-Риделя.
Рис. 423.
Бандажи для крепления колони.
Рис. 424.
Трубчатый стенд для ректификационной установки (общий объем аппаратов 200 л), изготовленный из одинаковых труб с оцинкованными соединительными муфтами.
Изготовитель: фирма «Стеклянная техника», Висбаден—Ширштайи.
ные детали, опорные и поддерживающие устройства. В каталогах предприятий технического стекла указано очень большое число таких деталей и устройств, предназначенных для сборки установок; рассмотреть их в данной книге в полном объеме не представляется возможным. Предприятия стекла выпускают также стенды для монтажа ректификационных установок. На рис. 424 показан стенд, который составлен из одинаковых труб, соединяемых с помощью трубчатых муфт, снабженных резьбой.
9.4. ГЕРМЕТИЗАЦИЯ УСТАНОВОК
В большинстве случаев возникают трудности при уплотнении шлифовых соединений и кранов. Перегоняемые жидкости обладают весьма различной растворяющей способностью по отношению к жировым смазкам шлифовых соединений, поэтому наиболее подходящую смазку следует выбирать на основе результатов предварительных испытаний (табл. 70).
477
Таблица 70.
Жировые смазки для шлифовых соединений и кранов
Смазка Максимальная рабочая температура, °C Состав или предприятие-изготовитель Область применения
Смазка Рамсэя 25 6 ч. вазелина, 1 ч. парафина, 6ч. пара-каучука Для обычных работ
Высоковакуумная 25 Предприятие Е. Лей- Для работ под высо-
смазка типа Р Высоковакуумная смазка типа R Смазка «Лителен» 30 150 больда, Кёльн ким вакуумом
Смазки типа 1 ч. растворимого Для работ с вещества-
BZ6564 и BZ6563 крахмала, 1 ч. глицерина [5]; предприятие Е. Мерка, Дармштадт ми, хорошо растворяющими жиры, например, диэтиловым эфиром и хлорированными парафиновыми углеводородами
Графитовая смазка Порошок графита, смесь графита и парафина, коллоидный раствор графита в воде Высокотемпературная перегонка
О применимости смазок на основе силикона в настоящее время нельзя сделать окончательного вывода, из-за недостатка опытных данных. Однако можно утверждать, что всегда имеется опасность попадания некоторой части смазки в колонну, что приводит к ухудшению смачиваемости насадки. Смазка «Лителен» (см. табл. 70), состоящая из литиевого мыла, практически не изменяет свою консистенцию с повышением температуры. Ее рекомендуется применять для смазки кранов в интервале рабочих температур от 0 до 150 °C. Обзор о методиках приготовления специальных смазок в лаборатории дан в работе Брооке [8].
Для кранов в основном применяют более мягкие смазки, чем для шлифовых соединений. Систематический обзор различных смазок представил Вагнер [9]. В работах Бернхауэра [10], Виттенбергера [11], Вольфа [12] и Фридрихса [13] даны подробные указания по уходу за стеклянными шлифовыми соединениями и кранами, особенно при плотной посадке муфт шлифов и сердечников кранов. Для соединения деталей, используемых в процессах молекулярной дистилляции, наиболее подходящими являются дегазированные смазки с низким давлением паров, такие как высоковакуумные смазки типов Р и R. Стандартные шлифы с ртутным затвором (рис. 425) обеспечивают практически полную герметизацию, однако их следует применять только в исключительных случаях вследствие опасности пролива ртути.
478
Шлифовые соединения без смазки даже при самой высокой степени чистоты обработки поверхностей конуса и муфты не обеспечивают достаточной герметичности при работе под вакуумом, и в частности не предохраняют от проникновения паров из ректификационной колонны. Поэтому верхнюю половину конуса шлифо-вого соединения следует всегда равномерно покрывать слоем смазки. Для этого лучше всего использовать гладкий деревянный шпатель. Пальцем можно нанести более равномерный слой, однако в этом случае имеется опасность попадания в смазку грязи, пота или влаги. Перед смазыванием, особенно при использовании смазки Рамсэя или апиезоновой смазки, конус шлифа можно слегка прогреть (но не на открытом пламени горелки!), чтобы нанесенная смазка при вращении конуса растеклась и покрыла его поверхность равномерным слоем. После этого конус вставляют в муфту и при нажатии вращают его до тех пор, пока все шлифовое соединение не станет прозрачным. Если с первой попытки это не удастся сделать, то всю процедуру повторяют снова, начиная с прогревания конуса.
Если при перегонке герметичность шлифового соединения нарушается из-за растворения или разложения жировой смазки, то уплотнение можно восстановить при помощи различных замазок. Для этого пригоден прежде всего пицеин с температурой плавления 80 или 105 °C. Хорошим заменителем пицеина является сургуч, однако он хрупок. При рабочих температурах до 250 °C применяют замазку из глицерина и свинцового глета, которая сильно затвердевает. Пицеин и сургуч не следует расплавлять на открытом газовом пламени, целесообразно нагреть уплотняемую поверхность до температуры около 100 °C большим факелом пламени горелки Бунзена, а затем наносить замазку шпателем, нагретым до той же температуры. Покрытую замазкой поверхность снова прогревают небольшим факелом пламени. При этом замазка распределяется равномерным слоем и проникает в зазоры между конусом и муфтой шлифового соединения.
Уплотнение шлифовых соединений можно обеспечивать также с помощью конусообразного тефлонового манжета, надеваемого на конус. При этом обеспечивается надежная герметизация и одновременно легкость разборки шлифового соединения. Французская фирма Гашо изготавливает манжеты типа «Родетс» для различных
, типоразмеров шлифовых соединений. Прецизионные шлифовые соединения можно применять и без смазки (см. разд. 3.1). Герметизацию сферических и плоских S шлифовых соединений в пилотных и полупромышленных ректификационных установках легко обеспечить с помощью уплотняющих тефлоновых колец и рубашек.
Рис. 425.
Стандартный шлиф с ртутным затвором.
479
9.5. ВВОД РЕКТИФИКАЦИОННЫХ УСТАНОВОК В ЭКСПЛУАТАЦИЮ
Подготовку к пуску ректификационной установки обычно начинают с заполнения куба разделяемой смесью. При обычной перегонке куб следует заполнять не более чем на 2/3 его номинальной емкости, а при вакуумной перегонке — только до половины. Одновременно рекомендуется вводить с жидкостью в куб небольшие твердые частицы, для обеспечения более равномерного кипения. Объем загружаемой жидкости следует определять с учетом повы-
шения температуры до ее ожидаемого значения в начале процесса перегонки. Для этого применяют нагреваемые мерные сосуды, которые особенно удобны при работе с веществами, находящимися при комнатной температуре в твердом состоянии *.
Токсичные и очень летучие вещества лучше подавать в куб из закрытого сосуда, создавая в последнем избыточное давление; по окончании заполнения куба его необходимо сразу же закрыть. Для обеспечения предварительного смачивания содержимого ректификационной колонны (насадки, неподвижные или движущиеся контактные устройства) рекомендуется по возможности загружать разделяемую смесь в куб сверху через дефлегматор. В тех случаях, когда необходимо предотвратить увлажнение разделяемой смеси, колонну перед загрузкой продувают теплым воздухом (феном):
при включенном электронагревателе кожуха.
Как уже указывалось, насадку перед загрузкой ее в колонну следует очень тщательно очистить. Следует иметь в виду, что
многократная промывка четыреххлористым углеродом и трихлорэтиленом не обеспечивают необходимой степени чистоты. Для
устранения прилипших к элементам насадки слоев загрязнений?
целесообразно сначала несколько раз промыть насадку горячим? бензолом, а затем сполоснуть ее трихлорэтиленом [14].
Перед проведением вакуумной перегонки необходимо сначала’ проверить герметичность установки. Для этого после достижений; в аппаратах установки заданного остаточного давления вакуумный насос отключают и в течение нескольких часов наблюдают з
изменением давления (см. разд. 5.4.1). Повторно герметичност] установки проверяют после разогрева установки, в условиях тепло вого расширения ее деталей. Электронагреватель куба включаю" по достижению заданного давления, а нагреватель кожуха ко лонны — после того, как начнется кипение жидкости. Охлажда!
щую воду подают в конденсатор во всех случаях до включени электронагревательных приборов, регулируя вручную ее расхо, по показаниям ротаметра. Следует отметить, что в пусковой перио, приходится неоднократно устанавливать расход воды. Поел
* При перегонке веществ, имеющих температуру плавления выше комиа' ной температуры, целесообразнее загружать разделяемую смесь по массе, опр< делив предварительно ее плотность при ожидаемой рабочей температуре nepi гонки и учитывая степень заполнения куба. — Прим. ред.
480
того, как пары достигнут верхней части колонны, устанавливают требуемую нагрузку (см. разд. 4.10.7) или предварительно создают режим работы, соответствующий «захлебыванию» колонны (см. разд. 4.10.8), для обеспечения лучшей смачиваемости насадки. Перед установкой заданного флегмового числа в ректификационной колонне не менее 0,5 ч проводят рабочий процесс с бесконечным флегмовым числом, что позволяет более точно определить начальную температуру отгонки (см. разд. 4.10.4).
Как уже указывалось выше, в куб необходимо вводить небольшие твердые частицы для обеспечения равномерного кипения жидкости. Обеспечивать равномерность кипения путем подачи воздуха через капилляр без необходимости не рекомендуется, так как это затрудняет точное определение рабочего давления при перегонке и в большинстве случаев вызывает частичную полимеризацию кубовой жидкости *. В качестве твердых частиц, обеспечивающих равномерность кипения, используют кусочки обожженной глины (от цветочных горшков), пемзы и тетраидальной насадки из платиновой проволоки. При перегонке веществ, обладающих сильной щелочной реакцией, в куб вводят оловянный порошок. Следует иметь в виду, что действие твердых частиц при вакуумной разгонке прекращается, если впустить в установку воздух. Облегчение парообразования в кубе при использовании твердых частиц обусловлено наличием в их порах маленьких пузырьков воздуха. Поэтому при проведении процесса вакуумной ректификации с перерывами необходимо каждый раз вводить в куб новые твердые частицы [15].
Если в начале перегонки жидкость в кубе очень сильно вспенивается, то можно уменьшить пенообразование путем введения небольшого количества противовспенивателей [16], из которых наиболее подходящими являются силиконовые масла.
При перегонке очень вязких жидкостей целесообразно непрерывно перемешивать содержимое куба с помощью магнитной мешалки или мешалки Хюбнера [17].
9.6. ТЕХНИКА БЕЗОПАСНОСТИ
Вопросам техники безопасности при лабораторных перегонках необходимо уделять особое внимание, поскольку зачастую приходится работать с токсичными, легковоспламеняющимися и взрывоопасными веществами, а при вакуумной перегонке всегда возможна опасность разрыва стеклянных сосудов [18]. Пределы взрываемости и температуры вспышки горючих газов и паров приведены в докладе Диттмара [19].
* При необходимости рекомендуется вводить в куб вместо воздуха азот, чтобы избежать вредного воздействия кислорода воздуха. — Прим. ред.
481
Электрические установки во взрывобезопасном исполнении необходимо в каждом конкретном случае предварительно испытывать.
Перегонку токсичных веществ в основном проводят в описанных выше отдельных боксах (см. разд. 9.1) или в обычных вытяжных шкафах. Взрывоопасные вещества, такие как диэтиловый эфир, лучше перегонять на открытом воздухе или при отсутствии данной возможности, в специально предназначенных помещениях. Если в разделяемой жидкой смеси растворены ядовитые газы, необходимо соединить конденсатор с всасывающим отверстием вытяжной вентиляции трубопроводом для отвода выделяющихся газов. При ректификации едких веществ следует работать в резиновых перчатках и защитных очках, поскольку всегда возможно появление капель жидкости на любых участках установки. Еще лучше пользоваться при работе специальным защитным костюмом.
Особые предосторожности необходимо соблюдать при вакуумной перегонке. Для предотвращения несчастных случаев следует пользоваться защитными очками, а еще лучше — специальными защитными приспособлениями для лица. Если ректификационная установка уже в значительной мере защищена колбонагревателем и обогревающим кожухом колонны, то надежное предохранение от осколков при разрыве стеклянных аппаратов и коммуникаций ^ожно обеспечить, закрывая неизолированные части аппаратуры полуцилиндрами из стекловолокна. В тех случаях, когда это по условиям опытов невозможно, например, при необходимости визуального наблюдения за процессом разделения, следует устанавливать защитные приспособления из проволочной сетки или листов органического стекла. Буферные сосуды целесообразно помещать в выложенные стекловолокном деревянные ящики или в корзину из проволочной сетки. Вакуумные насосы с ременным приводом обязательно закрывают деревянными ящиками.
Оборудование специальных лабораторий, в которых проводятся работы в условиях высокого давления, а также перегоняют токсичные вещества, подробно описано в работе Крейга и Дау [20].
1. ЛИТЕРАТУРА
1. RECHENBERG С. V., Einfache und fraktionierte Destination in Theorie und Praxis, Schimmer u. Co. AG, Miltitz 1923.
2. YOUNG S., Theorie und Praxis der Destination, Springer-Verlag, Berlin 1932 (Ubers, aus d. Engl.).
3. THORMANN K., Destillieren und Rektifizieren, Verlag Spamer, Leipzig 1928 (TOPMAH R. Перегонка и ректификация. Химтеоретиздат. М., 1935).
4. BADGER W. L., und McCABE W. L., Elemente der Chemie-Ingenieur-Technik, Springer-Verlag, Berlin 1932 (Ubers, aus d. Engl.). (БЕДЖЕР B., МАК-КЭБ В. Основные процессы и аппараты химических производств. Госхимтехиздат. М.—Л., 1933).
5. McCABE W. L„ und THIELE Е. L. Ind. Engng. Chem., int. Edit. 17 (1925), 605.
6. JANTZEN E., Dechema Monogr. 5 (1932), 48; Angew. Chem. 36 (1923), 592.
7. RIRSCHBAUM E., Destillier- und BektifiZiertechnik, 4. Aufl., Springer-Verlag, Berlin/Heidelberg/New York, ,1969.
8. GYULA N., Leparlas (Destination), Muszaki Konyvkiado, Budapest 1955.
9. JACOBS J., Destillier- und Rektifizieranlagen, R. Oldenbourg-Verlag, Munchen 1950.
10. ROBINSON C. S., und GILLILAND E. R-, Elements of Fraction al Distillation, McGraw-Hill Bo'k Company, New York 1950.
10a. PERRy J. H., Chemical Engineers Handbook, 3. Aufl., McGraw-Hill Book Company, New York 1953.
11. SCHULTZE Gg. R., und STAGE H. Die Kolonnendestillation im Laboratorium, VDI-Forschungsheft 424, VDI-Verlag, Berlin 1944.
12. THORMANN K, Dechema—Erfahrungsaustausch, Laboratoriumstechnik, Arbeitsmethoden und Gerate, Destillieren und Rektifizieren, Dechema, Frankfurt/Main, September 1950.
13. THORMANN K., Dechema—Erfahrungsaustausch, Techn. Apparate und Anlagen, Trennen fliissiger Mischungen, Destillieren, Betriebstechnik, Dechema, Frankfurt/Main, August 1952.
14. STAGE H., und SCHULTZE Gg. R., Die grundlegenden Arbeiten fiber Theorie, Apparate sowie Verfahren der Destination und Rektifikation. VDI-Verlag, Berlin 1944.
15. ROSE A. und E., Distillation Literature Index und Abstracts 1941—1945, 1946—1952, Applied Science Laboratories, State College, Pennsylvania 1953.
16. ZIC Referatedienst Chemische Verfahrenstechnik, hrsg. von der Zentralstelle fur Information der chemischen Industrie, Berlin 1972 ff.
Forschritte der Verfahrenstechnik, hrsg. von der Ingenieurwissenschaftlichen Abteilung der Farbenfabriken Bayer AG, Leverkusen, Verlag Chemie GmbH, Weinheim/Bergstr. 1952 ff.; ab Band 9 (1971 ff.) hrsg. von der Verfahrenstechnischen Gesellschaft im VDI, VDI-Verlag, Dusseldorf.
Verfahrenstechnische Berichte (Vorankfindigungsblatter, Karteiblatter und halbjahrliche Sach-, Autoren- und Firmenregister), hrsg. von der Ingenieurwissenschaftlichen Abteilung der Farbenfabriken Bayer AG, Leverkusen, Verlag Chemie GmbH, Weinheim/Bergstr.
17. WALSH T. J., Ind. Engng. Chem., seit 1956.
483
16. ORLlGEK A. 6., und POLL H., Hilfsbuch fiir Mineraloitechniker, fid. 1: Die Eigenschaften von Kohlenwasserstofien, Mineralolprodukten und Hilfsstoffen, Springer-Verlag, Wien 1951.
19. SIGWART K-, Destination und Rektifikation, in: Ullmanns Encyklopadie der technischen Chemie, Bd. 1, Urban & Schwarzenberg, Miinchen/Berlin 1951, S. 429 bis 470.
20. KORTUM G„ und BUCHHOLZ-MEISEN HEIMER H., Die Theorie der Destination und Extraktion von Fliissigkeiten, Springer-Verlag, Berlin/Gottingen/Heidelberg 1952.
21. BOSNJAKOVIC Fr., Technische Thermodynamik, Bd. 1 (6. Aufl.) und Bd. 2 (5. Aufl.), Theodor Steinkopff Verlag, Dresden 1972/1971. {БОШНЯКОВИЧ Ф. Техническая термодинамика. T. 1,2. Госэнергоиздат. М.—Л., 1955, 1956).
22. GATTERMANN L., und WIELAND Н., Die Praxis des organischen Chemikers, 41. Aufl., Walter de Gruyter & Co., Berlin 1962. {ГАТТЕРМАН Л., ВИЛАНД Г. Практические работы по органической химии. Госхимиздат. М.—Л., 1948. Перевод с 31-го немецкого издания).
23. WITTENBERGER W., Chemische Laboratoriumstechnik, 6. Aufl., Springer-Verlag, Wien/New York 1963.
24. WEYGAND C., und HILGETAG G., Organisch— chemische Experimentierkunst, 4. Aufl., Johann Ambrosius Barth, Leipzig 1970. {ВЕЙГАНД K-, ХИЛЬГЕТАГ Г. Методы эксперимента в органической химии. Химия. М., 1968).
25. BERNHAUER К., Einfiihrung in die organisch—chemische Laboratoriumstechnik, Springer-Verlag, Wien 1947.
26. CARNEY Th. P., Laboratory Fractional Distillation, Mac-Millan, New York 1949.
27. ROSENGART M. J., Die Technik der Destination und Rektifikation im Laboratorium, VEB Verlag Technik, Berlin 1954 (Obers, aus d. Russ.). (Р03ЕНГАРТ M. И. Техника лабораторной перегонки и ректификации. Госхимиздат. М.—Л., 1951).
28. ROSE A., und Е., und MITARB., Technique of Organic Chemistry, Vol. IV, Distillation, Interscience Publishers, New York, 1951.
29. WEBER U. von, Gewinnung atherischer Ole durch Destination, in: GILDEMEISTER E., und HORFFMANN Fr., Die atherischen Ole (bearb. von W. Treibs), Akademie-Verlag, Berlin 1966.
30. SIGWART K-, Destillieren und Rektifizieren, in: H0UBEN-WEYL, Methoden der organischen Chemie, (hrsg. von E. Muller), Bd. 1/1, Georg Thieme Verlag, Stuttgart 1958.
31. ZUIDERWEG F. J., Laboratory Manual of Batch Distillation, Interscience Publishers, New York 1957.
32. COULSON E. A., und HERINGTON E. F. G., Laboratory Distillation Practice, George Newnes, London 1958.
33. BUKALA M„ MAJEWSKI J., und RODZINSKI W., Fraktionierte Destination im Laboratorium, Panstwowe Wydawnictwo Naukowe, Warschau 1958.
34. Industrial and Engineering Chemistry, Chemical and Process Engineering, VDI-Zeitschrift, Chemische Technik.
35. HOPPE K-, KUNNE H., und BENDIX H., Fortschritte in der Destination, Rektifikation, Sorption und Extraktion, VEB Deutscher Verlag fiir Grundstoffindustrie, Leipzig 1970.
36. TREYBAL E. R., Masstransfer Operations (International Student Edition), 2. Aufl., McGraw-Hill Book Company, New York 1968.
GEISELER G., und MITARB., Ausgewahlte physikalische Methoden der organischen Chemie, Akademie-Verlag, Berlin 1963.
KEIL B., und MITARB., Laboratoriumstechnik der organischen Chemie (hrsg. von H. Furst),- Akademie-Verlag, Berlin 1961.
WIBERG К. B., Laboratory Technique in Organic Chemistry, McGraw-Hill Book Company, New York 1960.
484
37. Organikum—Organisch—Chemisches Grundpraktikum, 13. Aufl., VEB Deutscher Verlag der Wissenschaften, Berlin 1974.
Anorganikum—Lehr- und Praktikumsbuch der anorganischen Chemie (hrsg. von L. Kolditz), 7. Aufl., VEB Deutscher Verlag der Wissenschaften, Berlin 1975.
38. ROCK H., Destination im Laboratorium—Extraktive und azeotrope Destination, Dr. Dietrich Steinkopff Verlag, Darmstadt 1960.
39. STAGE H., und BOSE K-, Die Belastungsverhaltnisse in Fflllkorpersaulen imter Destillationsbedingungen, Springer-Verlag, Berlin/Gottingen/Heidel-berg 1962.
MULLER E., und STAGE H., Experimented Vermessung von Dampf—Fliissigkeits—Phasengleichgewichten, Springer-Verlag, Berlin/Gottingen/Heidelberg 1961.
40. HALA E„ PICK J., FRIED V., und VILIM 0., Gleichgewicht— Fliissigkeit—Dampf (hrsg. von W. Mannchen), Akademie-Verlag, Berlin 1960; englische Ausgabe: Vapour—Liquid Equilibrium, 2. ed., Pergamon Press Ltd., London 1968.
41. HALA E., WICHTERLE I., POLAK J. und BOUBLIK T„ Vapour— "Liquid Equilibrium Data at Normal Pressures, Pergamon Press Ltd., London 1969.
42. КОГАН В. Б., ФРИДМАН В. М., КАФАРОВ В. В. Равновесие между жидкостью и паром. Справочное пособие. Наука. М.—Л., 1966.
ОГОРОДНИКОВ С. К., ЛЕСТЕВА Т. М., КОГАН В. Б. Азеотропные • смеси. Справочник. Химия. Л., 1971.
43. BILLET R., Grundlagen der thermischen Fliissigkeitszerlegung, Bibliographisches Institut, Mannheim 1961; BILLET R., Verdampfer-Technik, Bibliographisches Institut, Mannheim 1965.
43a. BILLET R., Industrielle Destination, Verlag Chemie GmbH, Weinhe, Bergstr. 1973.
44. BILLET R., Optimierung in der Rektifiziertechnik, unter besonderer Berflcksichtigung der Vakuumrektifikation, Bibliographisches Institut, Mannheim 1967.
45. OLIVER E. D., Diffusional Separation Processes: Theory, Design and Evaluation, John Wiley & Sons, New York 1966.
46. PRATT H. R. C., Countercurrent Separation Processes, Elsevier Publishing Company, Amsterdam 1967.
47. KRELL E., FREY H.-Р., GAW A LEK G., und WERNER G., Einfiihrung in die Trennverfahren, VEB Deutscher Verlag fiir Grundstoffindustrie, Leipzig 1975.
48. HOPPE K-, und MITTELSTRASS M., Grundlagen der Dimensionierung von Kolorinenboden, Theodor Steinkopff Verlag, Dresden 1967.
49. JUNGNICKEL H-, und OTTO W., Tiefe Temperaturen in der Verfahrenstechnik (Bd. 11 der Reihe «Beitrage zur Verfahrenstechnik»), VEB Deutscher Verlag fiir Grundstoffindustrie, Leipzig 1972.
50. FRANK W., und KUTSCHE D., Die schonende Destination, Otto Krausskopf-Verlag GmbH, Mainz 1969.
51. ОЛЕВСКИЙ В. M., РУЧИНСКИЙ В. P. Ректификация термически нестойких продуктов. Химия. М., 1972.
52* HOLLO J., KURUCZ Е„ und BORODI A., The Applications of Molecular Distillation, Akademiai Kiadd, Budapest 1971.
53. SCHUBERTH H., Thermodynamische Grundlagen der Destination und . Extraktion, Bd. I, VEB Deutscher Verlag der Wissenschaften, [Berlin 1972.
54. BRAUER H., und MEWES D., Stoffaustausch, einschl. chemischer Reaktionen, Verlag Sauerlander AG, Aarau und Frankfurt/Main 1971. BRAUER H., Grundlagen der Einphasen- und Mehrphasenstromung, Verlag Sauerlander AG, Aarau und Frankfurt/Main 1971.
485
55. HOLLAND C. D., Multicomponent Distillation, Prentice-Hall International, Englewood Cliffs 1963.
56. Chemie-Ing.-Techn. 42 (1970), 14, 945—957.
57. BILLET R., CZ-Chemie-Techn. 3 (1974) 10, 353—361.
2. ЛИТЕРАТУРА
1. SC HELEN Z_ H., Zur Geschichte der pharmazeutisch—chemischen Destilliergerate. Beilage zum Bericht April 1911, Verlag Schimmel und Co. AG, Miltitz.
2. GILDEMEISTER E., Die atherischen Ole, Verlag Schimmel und Co. AG, Miltitz 1928.
3. RECHENBERG C. von, Einfache und fraktionierte Destination in Theorie und Praxis, Verlag Schimmel und Co. AG, Miltitz 1923.
4. UNDERWOOD A. J. V., Trans. Instn. chem. Engr. 13 (1935), 34.
5. FORBES R. J., Short history of the art of destination, Verlag E. J. Bull, Leiden 1948.
6. FORBES R. J., und DIJKSTERHUIS E. J., »A History of Science and Technology, Bd. I: Ancient Times of the Seventeenth Century; Bd. 11: The Eighteenth and Nineteenth Centuries. Pelican Books Nr. A 498 und A 499, Verlag Penguin Books Ltd., Harmondsworth/Middlessex 1963.
7. SCHNEIDER IV., Lexikon alchemistisch—pharmazeutischer Symbole, Verlag Chemie GmbH, Weinheim/Bergstr. 1962.
8. LIPPMANN E. 0. von, Entstehung und Ausbreitung der Alchemie, Bd. 1 u. 2, Verlag Julius Springer, Berlin 1919, 1931; Bd. 3, Verlag Chemie GmbH, Weinheim/Bergstr. 1953.
9. LIPPMANN E. 0. von, Abhandlungen und Vortrage zur Geschichte der Naturwissenschaften 1 (1906), 2 (1913), Verlag Chemie GmbH, Weinheim/Bergstr.
10. LIPPMANN E. 0. von, Beitrage zur Geschichte der Naturwissenschaften und der Technik 1 (1923), 2 (1953), Verlag Chemie GmbH, Weinheim/Bergstr.
11. STAGE F., Angew. Chem., Ausg. B. 19 (1947), 181.
12. KREIS H., Liebigs Ann. Chem. 224 (1884), 259; YOUNG S. J. Chem. Soc. [London] 75 (1889), 679; FRIEDRICHS J., Z. angew. Chem. 32 (1919), 340.
13. Glas-Instrumenten-Techn. 14 (1970), 196.
14. KRELL E., Glas-Instrumenten-Techn. 11 (1967) 1—5, Beilage 1—20.
3. ЛИТЕРАТУРА
1. DDR-Standards und Fachbereichsstandards der DDR, Hrsg: Amt fiir Standardisierung der Deutschen Demokratischen Republik, Berlin 1968.
2. Verzeichnis der DIN-Normblatter, Stand vom 28.2.72, Beuth-Vertrieb GmbH, Berlin (West).
3. TELLE W., Chemische Laboratoriumsgerate, 2. Aufl., VEB Deutscher Verlag fiir Grunds toff Industrie, Leipzig 1969.
4. FRIEDRICHS F., Die Glashfltte 76 (1949), 3—6.
5. FLIEDNER K., Glasapparatetechnik 5 (1958), 33—37; Beilage zur Chem, Techn. 10 (1958).
6. KUEHNHANSS G., und MIT ARB., Chem. Techn. 9 (1957), 407—409.
7. TGL 10192—10200, 11641—11644, 12023—12032, 20277 DIN—проект 28800—28808.
7a. HAMPEL B., Chemiker-Ztg. 95 (1971) 7, 316—325.
8. KRELL E., Chem. Techn. 6 (1954), 452—455.
9. PADELT E., und LAPORTE H., Einheiten und Grofienarten der Naturwissenschaften, 2. Aufl., VEB Fachbuchverlag, Leipzig 1967.
10. FORSTER H., Einheiten, Grofien, Gleichungen und ihre praktische Anwendung, VEB Fachbuchverlag, Leipzig 1968.
486
11. HAEDER W.,und GARTNER E., Die gesetzlichen Einheiten in der Technik Verlag Beuth—Vertrieb, Berlin 1970.
12. KRELL E„ Chem. Techn. 7 (1955), 30—32.
13. LESSEES A. G., Chem. Engng. 63 (1956), 208—210.
14. ORLICEK A. F., P6LL H„ und WALENDA H., Hilfsbuch fflr Mineralotechniker, Bd. 1: Die Eigenschaften von Kohlenwasserstoffen, Mineralolprodukten und Hilfsstoffen, Springer-Verlag; Wien 1951.
15. BAEHR H., Chemie-Ing.-Techn. 23 (1953), 676.
16. KRAMER M. T., Glas-Instrumenten-Techn. 15 (1971), 251—254; 17 (1973), 43—48, 137—139.
4. ЛИТЕРАТУРА
1. SIGWART K-, Destination und Rektifikation, in: Ullmanns Encyklopadie der technischen Chemie, Bd. 1, Urban Schwarzenberg, Mflnchen/Berlin 1951, S. 429/470.
2. TGL 21 120.
з’. KUHN W.', Helv. chim. Acta 25 (1942), 252—295; KUHN W., und RYFFEL K., Helv. chim. Acta 26 (1943), 1693—1721.
За. НИСЕЛЬСОН Л. А., ТРЕТЬЯКОВА К- В., ЯТКО М. Е. ЖПХ, 1973, т. 46, № 7, с. 1613—1614.
3b. GRACZYK J., Nukleonika 1 (1973), 1, 25—33.
4. MEIER W., und HUBER M., Chemie-Ing.-Techn. 39 (1969), 797—800.
5. HUBER M„ und HILTENBRUNNER R., Chem. Engng. Sci. 21 (1966), gjg_____
6. KAFAROV V. V., D0R0KH0V J. N., und SHESTOPALVOV V. V., Hydrodynamische Parameter von Ftillkorperkolonnen, Tagungsheft 4, Distillation 1969, Brighton/England, Referat: Chemie-Ing.-Techn. 42 (1970) 14 953
7. JONAS, Chr., Chem. Techn. 26 (1974) 12, 87—91.
8. ZUIDERWEG F. J., und HARMENS A. Chem. Engng. Sci. 9 (1958) 2/3, 89—103.
9. KRELL E„ Chem. Techn. 16 (1964) 10, 591—600.
10. ТИТОВ А. А., ЗЕЛЬВЕНСКИЙ Я- Д- Хим. пром., 1974, Ns 9, с. 50—53.
11. KRELL Е., Plaste und Kautschuk 14 (1967) 4, 251—254.
Ila. KWASNIAK J., Verfahrenstechnik 8 (1974) 11, 315-322.
lib. YILMAZ T.,und BRAUER H., Chemie-Ing.-Techn. 45 (1973) 14, 928—934.
11c. KUNNE H.—J., Chem. Techn. 25 (1973) 9, 533—538.
lid. DIETZi H., REINEMANN G., und COMEL M„ Chem. Techn. 25 (1973)
3, 143—148.
lie. GODAU H.—J., Chem. Techn. 26 (1974) 9, 561—564.
Ilf. BILLET R., CZ-Chem. Techn. 3 (1974), 9—12.
llg. ADOLPHI H.—V., Chem. Techn. 26 (1974), 10, 643—645.
lift. АНТОНОВ Ю. E„ САФИН P. III., PENT В. И. TOXT, 1975, t. 9, № 1, c. 140—142.
12. STAUDINGER H., Anleitung zur organlschen qualitativen Analyse, 6. Aufl., Springer-Verlag, Berlin/Gottingen/Heidelberg 1955, S. 12—13.
13. STAGE H., Erdol und Kohle 3 (1950), 478—490.
14. HILDEBRAND J. H., und ROTARIU S. J., Analytic. Chem. 24 (1952), 770—773.
15. MAUSER H., und KORTUM G., Z. Naturforsch. 10 a/4 (1955), 317—322.
16. MANNCHEN W., Einftihrung in die Thermodynamik der Mischphasen, VEB Deutscher Verlag fiir Grundstoffindustrie, Leipzig 1965.
17. SCHUBERTH H., Thermodynamische Grundlagen der Destination und Extraktion, Bd. I, VEB Deutscher Verlag der Wissenschaften, Berlin 1972.
18. DREISBACH R. R., Pressure—Volume—Temperature Relationship of Organic Compounds, 3. Aufl., Handbook Publ., Inc., Sandursky, Ohio 1952.
19. DREISBACH R. R., Physical Properties of Chemical Substances, The Dow Chemical Company, Midland, Michigan 1951—1953.
487
20. STULL D. R., Ind. Engng. Chem. 39 (1947), 517/550 u. 1684.
21. STAGE H., Fette u. Seifen 53 (1951), 677—682.
22. STAGE H., Fette u. Seifen 55 (1953), 217—224.
23. STAGE H., MULLER E., und FALDIX P., Erdol u. Kohle 6 (1953), 375—380.
24. STAGE H-, MULLER E., und FALDIX P., Chem. Techn. 6 (1954), 31/36.
25. JORDAN T. E., Vapor Pressure of Organic Compounds, 2. Aufl., Interscience Publishers, New York 1954.
26. RIDDICK J. A., und TOOPS JR., E. E., Organic Solvents, Physical Properties and Methods of Purification, Interscience Publischers, New York 1955.
27. SAGE B. H., und LACEY W. N., Some Properties of the Lighter Hydrocarbons, Hydrogen Sulfide and Carbondioxide American Petroleum Institute, New York 1955.
28. GLASER F., und RU.LAND H., Chemie-Ing.-Techn. 29 (1957), 772—775.
29. JAECKEL R., Destination und Sublimation in Fein- und Hochvakuum, in: HOUBEN—WEYL, Methoden der organischen Chemie (hrsg. von Eugen Muller), Bd. 1/1, Georg Thieme Verlag, Stuttgart 1958.
30. KIENITZ H„ Bestimmung des Dampfdruckes, in: HOUBEN—WEYL, Methoden der organischen Chemie (hrsg. von Eugen Muller), Bd. III/l, Georg Thieme Verlag, Stuttgart 1958, S. 255—324.
31. MILAZZO G., Chemie-Ing.-Techn. 28 (1956), 646—654.
32. SWIETOSLAWSKI IF., Ebulliotrie Measurements, Reinhold, New York 1945. SWIETOSLAWSKI W., und ANDERSON J. R„ in: A. WEISSBERGER; Physical Methods of Organic Chemistry, Vol. I, Interscience Publishers, New York 1994, S. 107.
33. WEBER U. von, J. prakt. Chem. 1/4 (1955), 318—323.
34. EBLIN P.„ J. chem. Educat. 27 (1950), 67.
35. HERRINGTON E. F., und MARTIN J. F., Trans. Faraday Soc. 49/2 (1953), 154—162. t '
36. NATELSON S„ und ZUCKERMANN J. L., Ind. Engng. Chem., analyt. Edit. 17 (1945) 739 bis 740.
37. GOULD C., JR., HOLZMANN G., und NIEMANN C., Analytic Chem. 19 (1947), 204—206.
38. WIBERLEY F. J., SIEGERIEDT R. K-, und BENEDETTI-PICHLER A. A., Mikrochem. verein. Mikrochim. Acta 38 (1951), 417—475.
39. НЕСМЕЯНОВ A. H. Атомная энергии. 1957, т. 3, вып. 9, с. 227—237.
40. СКЛЯРЕНКО С. И., МАРКИН Б. И., БЕЛЯЕВА Л. Б. ЖФХ, 1958, т. 32, № 8, с. 1916—1921.
41. HENNING F., und STOCK A., Z. Physik 5 (1921), 226.
42. Smith und Menzies, Z. physik. Chem. 75 (1911), 501.
43. EUCKEN A., und SUHRMANN R., Physikalisch—chemische Praktikumsaufgaben, 7. Aufl., Akademische Verlagsgesellschaft Geest & Portig KG, Leipzig 1968, S. 100—106.
44. KORTUM G., MOEGLING D„ WOERNER F.; Chemie-Ing.-Techn. 22 (1950), 453—457.
45. SCHLUNDER E. U., Chemie-Ing.-Techn. 35 (1963) 7, 482—487.
46. ROCK H., Z. physik. Chem., N. F. 4 (1955), 242—245.
47. PETER M., Magyar Kemiai Folyoirat 10 (1953), 295—300, Referat in: Chem. Techn.. 6 (1954), 307.
47a. KOLLAR G., und NAGY E., Chem. Techn. 23 (1971), 1, 35—36.
48. PEARSON D. E., J. chem. Educat. 28 (1951), 60—62.
49. ENGLISH W. D., J. Amer. chem. Soc. 74 (1952), 2927—2928.
50. DUHRING U., Wiedem. Ann. 11 (1880), 163; 52 (1894), 556.
51. RECHENBERG G. von, Einfache und fraktionierte Destination in Theorie und Praxis, Verlag Schimmel & Co. AG, Miltitz 1923.
52. JANTZEN E., und ERDMANN W„ Fette u. Seifen 54 (1952), 197—20Г.
53. КИРЕЕВ В. А., ЖПХ, 1934, т. 7, № 1—2, с. 1—15.
488
54. ANTOINE С., C. R. hebd. Seances Acad. Sci. 107 (1888), 681, 863, 1143/45.
55. THOMSON G. W., Chem. Rev. 38 (1946), 1—39.
56. RIEDEL L„ Chemie-Ing.-Techn. 26 (1954), 83—89.
57. RECKHARD, H., Erdol u. Kohle 116 (1958), 234—241; Chemiker-Ztg., 84 (1960), 33—36.
58. WICHTERLE I., und LINER J., Die Antoinschen Dampfdruck-Konstanten reiner Verbindungen, Verlag Academia, Prag 1971 (in engl. Sprache).
59. Berechnung thermodynamischer Stoffwerte von Gasen und Flflssigkeiten, VEB Deutscher Verlag fur Grundstoffindustrie, Leipzig 1966, S. 77—89.
60. OTHMER D. F., und YU E.—S., Ind. Engng. Chem. 60 (1968), 1, 22—35. 60a. ZIA T., und THODOS G., Canad. J. chem. Engng. 52 (1974) 5, 630—635.
61. SCHWAB G. M„ Z. f. angew. Chem. 43 (1930), 155—156.
62. STANGE R., Chem. Techn. 5 (1953), 78—79.
63. OTHMER D. F., MAURER P. W., MOLINARY C. J., KOWALSKI R.C., Ind. Engng. Chem. 49 (1957), 125—137.
64. WITTENBERGER W., Chemiker-Ztg, 76 (1952), 371—373.
65. BERGHOLM A., Svensk kern. Tidskr. 60 (1948), 96.
66. HOFFMANN W„ und FLORIN F., Z. Ver. dtsch. Ing., Beih. Verfahfenstechn., (1943), 47—51.
67. COX E. R., Ind. Engng. Chem. 15 (1932), 592—593.
68. DAVIS D. S., Ind. Engng. Chem. 17 (1925), 435.
69. CALINGART G., und DAVIS D. S„ Ind. Engng. Chem. 17 (1925), 1287.
70. FALDIX P., und STAGE H., Chem. Techn. 5 (1953), 526—528, 718—719.
71. STAGE H„ und JUILFS J., Chemiker-Ztg. 77 (1953), 511, 538, 575; 78 (1954), 43, 112, 182, 217.
72. THORMANN K., Destillieren und Rektifizieren, Spamer-Verlag, Leipzig 1928. {TOPMAH К., Перегонка и ректификации, Химтеоретиздат, М., 1935).
73. MATZ W., Die Thermodynainik des Warme- und Stoffaustausches in der Verfahrenstechnik, Bd. 1 und 2, Dr Dietrich Steinkopff Verlag, Darmstadt. 1949/1953.
74. KORTUM G., und BUCHHOLZ-MEISENHEIMER H., Die Theorie der Destination und Extraktion von FlOssigkeiten, Springer-Verlag, Berl in/Go tt ingen/Heidelberg 1952.
75. BOSNJAKOVI^ Fr., Technische Thermodynamik, Teil II, 5. Aufl., Verlag Theodor Steinkopff, Dresden 1971.
(БОШНЯКОВИЧ Ф., Техническая термодинамика, т. 2, Госэнергоиздат, М—Л., 1956).
76. FLORIN F., Brennstoff-Chem. 33 (1952), 161—166.
77. McCABE W. L., und THIELE E. W., Ind. Engng. Chem., ind. Ed. 17 (1925), 605.
78. HALA E., PICK J-, FRIED V., und VILIM 0., Rovnovaha kapalinapara Nakladetelstoi ceskoslovenska akademie red, Prag 1955; 2. engl. Ausgabe: Vapour-Liquid-Equilibrium, Pergamon-Press, London 1968; deutsche Ausgabe: Gleichgewicht—Flfissigkeit—Dampf, Akademie-Verlag, Berlin 1960.
79. Chemie-Ing.-Techn. 41 (1969) 12, 735—736.
80. WILSON G. M., J. Amer. chem. Soc. 86 (1964), 127.
81. DONTH E. J., und WEICHMANN R., Chem. Techn. 20 (1968) 2, 93—95.
82. KRUG K., HABERLAND D., und BITTRICH H.—J., Chem. Techn. 23 (1971) 7, 410—415.
83. RENON H., und PRAUSNITZ J. M„ American Institute of Chemical Engineers Journal 14 (1968), 135.
84. Р03ЕНГАРТ M. И., Техника лабораторной перегонки и ректификации. Госхимиздат, М.—Л., 1951.
85. ORLICEK A. F., und POLL Н., Hilfsbuch ffir Mineraloltechniker, Bd. I u. Il, Springer-Verlag, Wien 1951/1955.
86. VOORWIJK J. F„ Chem. Engng. 63 (1956), 201—202.
87. HOLLO J., und LENGYEL T., Ind. Engng. Chem. 51 (1959), 957—960; Collect czechoslov. chem. Commun. 23 (1958), 1735—1743.
16 э. Крель 489
HOLLO J., Periodica polytechn. [Budapest]; chem. Ingenieurwes., 2 (1958), 113—128; Fette, Seifen, Anstrichmittel 62 (1960) 10, 913—918.
88. UMSTATTER H., und FLASCHKA H„ Erdol u. Kohle 1 (1948), 151—157.
89. WILLIAMS R. A., und HENLEY E. J., Chem. Engng. J. 1 (1970) 2, 145—151.
90. MELPOLDER F. W., und HEADINGTON С. E., Ind. Engng. Chem. 39 (1947), 763—766; ROSE A., und BILES R., Chem. Engng. Progr. 51 (1955), 138—140.
91. ROSE E„ Ind. Engng. Chem. 33 (1941), 596.
92. HAWKINS J. E., und BRENT J. A., Ind. Engng. Chem. 43 (1951), 2611—2621.
93. ЦЫПКИНА 0. ЖПХ, 1958, т. 28, № 2, с. 185—192.
94. KORTUM G„ Chemiker-Ztg. 74 (1950), 151—154.
95. CARLSON H. C., und COLBURN A. P., Ind. Engng. Chem. 34 (1942), 581.
96. ORLICEK A. F., Osterr. Chemiker-Ztg. 50 (1949), 86.
97. LAAR J. J. VAN, und LORENZ R., Z. anorg. Chem. 145 (1925), 239; Z. phys. Chem. 72 (1910), 723; 83 (1913), 599.
98. MARGULES M„ Wiener Sitz. Ber. 104 (1895), 1243.
98a. GREWER Th., und SCHMIDT A., Chemie-Ing.-Techn. 45 (1973) 17, 1063-1066.
99. LANDOLT H., und BORNSTEIN R., Physikalisch — Chemische Tabellen, 6. Aufl., Springer-Verlag, Berlin/Gottingen/Heidelberg 1950/55.
100. PERRY J. H„ Chemical Engineers' Handbook, 4. Aufl., McGraw-Hill, Book Company, New York 1963, S. 1942ff.
101. YU CHIN CHU, Distillation Equilibrium Data, 2. Aufl., Reinhold, New York 1956.
102. CHU WANG, LEVY und PAUL, Vapour Liquid Equilibrium Data, Edwards T. W., Ann. Arbor Mich., 1956.
103. KIRSCHBAUM E., Destillier- und Rektifiziertechnik, 4. Aufl., Springer-Verlag, Berlin/Heidelberg/New York 1969.
103a. HUBNER W., und SCHLUNDER E. U., Chemie-Ing.-Techn. 45 (1973) 5, 247—253.
103b. VOGELPOHL A., und CERETTO R., Chemie-Ing.-Techn. 44 (1972) 15, 936—938.
104. JACOBS J., Destillier — Rektifizier — Anlagen., R. Oldenbourg Verlag, Mflnchen 1950.
105. КОГАН В. Б., ФРИДМАН В. М., КАФАРОВ В. В., Равновесие между жидкостью и паром. Справочное пособие. Наука, М.—Л., 1966.
106. HALA Е., WICHTERLE I., POLAK J-, und BOUBLIK Т„ Vapour-Liquid Equilibrium Data at Normal Pressure, Pergamon Press, London 1969.
106a. The Institution of Chemical Engineers, London, fiihrt einen Daten-Service (PPDS-Physical Property Data Service) durch, der auch die Phasengleichgewichtsdaten Fliissig/Dampf erfa₽t.
107. KORTUM G„ und FREIER H.—J., Chemie-Ing.-Techn., 26 (1954), 670—675.
108. WEBER U. von, Z. phys. Chem., N. F. 38 (1963) 3/4, 129—139.
109. GELBIN D., Chem. Techn., 15 (1963) 1, 6—9.
110. STAGE H., MULLER E., und GEMMEKER L., Chemiker-Ztg./Chem. Apparatur 85 (1961), 11.
111. OTHMER D. F„ Ind. Engng. Chem., ind. Edit. 20 (1928), 743.
112. STAGE H., und FISCHER W. G., Glas-Instrumenten-Techn. 12 (1968), 11, 1167—1173.
113. GILLESPIE D. C„ Ind. Engng. Chem., analyt. Edit. 18 (1946), 575.
114. ROCK H., und SIEG L., Z. physik. Chem., N. F. 3 (1955), 355—364; Chemie-Ing.-Techn. 28 (1956), 133.
115. KORTUM G., und BIEDERSEE H., von, Chemie-Ing.-Techn. 42 (1970), 8, 552—560.
490
116. SCHMIDT R., WERNER G„ und SCHUBERTH H., Z. phys. Chem. 242 (1969). 381—390.
117. WICHTERLE I., und HALA E., ind. Engng. Chem. Fundamentals 2 (1963) 2, 155—157.
118. NEUMANN A., und WALCH W„ Chemie-Ing.-Techn. 40 (1968) 5, 241—244.
118a. STAGE H„ und FISCHER W-. G., verfahrenstechnik 7 (1973) 6, 1—3.
119. KUMARKRISHNA RAO, V. N„ SWAMI D. R., und
NARASINGARAO M., Amer. Ind. chem. Eng. 3 (1957), 191—197.
120. JOST W., ROCK H., SCHRODER W., SIEG W., und WAGNER H. G., Z. physik. Chem., N. F. 10 (1957), 133—136;
SCHRODER Ц7., Chemie-Ing.-Techn. 30 (1958), 523—525.
121. LYDERSEN A. L., und HAMMER E. A., Chem. Engng. Sci. 7 (1958), 241—245.
122. JUNGHANS W., und WEBER U. von, J. prakt. Chem. 4/2 (1955), 2g5______273
123. MAI K. L., und BABB A. L., Ind. Engng. Chem. 47 (1955), 1749—1757.
124. ЗЕЛЬВЕНСКИЙ И- Д-, ШАЛЫГИН В. А., ЖФХ, 1957, т. 31, № 7, с. 1501—1509.
125. REDLICH О., und KISTER А. Т., Ind. Engng. Chem. 40 (1948), 341—348.
126. HERINGTON E. F. G., Nature [London] 161 (1947), 610—611.
HAMER D. F. 0., und MITARB., Ind. Engng. Chem. 42 (1950), 120.
HAASE R., und JOST W., Z. phys. Chem., N. F. 9 (1956), 300—301.
TIERNEY J. W., Ind. Engng. Chem. 50 (1958), 707—710.
127. HERINGTON E. F. G„ J. appl. Chem. 2 (1952), 11—23.
128. BITTRICH H.—J., Chem. Techn. 14 (1962) 9, 527—533.
129. TAO L. C., Ind. Engng. Chem., Fundamentals 1 (1962) 2, 119—123.
130. ROLLING H., Chemie-Ing.-Techn. 24 (1952), 405—411.
131. BOWMAN J. R„ und CICHELLI M. T., Ind. Engng. Chem. 41 (1949), 1985.
132. JUNGE C., Chem. Techn. 8 (1956), 579—588.
133. ANSCHUTZ R„ Chem. Techn. 9 (1957), 516—519.
134. FISHER G. T., Ind. Engng. Chem., Process Design and Development 2 (1963), 284—288.
135. BILLET R., Chemie-Ing.-Techn. 30 (1958), 513—515.
136. THUM 0., Chemie-Ing.-Techn. 29 (1957), 675—678.
137. NERETNICKS I., ERICSON I., und ERIKSSON S., Brit. chem. Engng.
14 (1969) 12, 1711 bis 1712.
138. WEBER U. VON, Chem. Techn. 2 (1950), 241—246.
139. VOGELPOHL A., Chemie-Ing.-Techn. 43 (1971) 20, 1116—1121.
139a. DOMENECH S., GUICLION C., und ENJALBERT M., Chem. Engng. Sci. 29 (1974) 7, 1519—1528, 1529—1535.
140. BILLET R., Chemie-Ing.-Techn. 30 (1958), 407—416.
140a. WILCOX W. R., Ind. Engng. Chem. Fundamentals 3 (1964) 1, 81—83.
141. THORMANN K., Chem. Techn. 2 (1950), 255—256.
142. CAVERS S. D., Ind. Engng. Chem., Fundamentals 4 (1965) 2 , 229—230.
143. POHL K-, Chemie-Ing.-Techn. 28 (1956), 562—564.
144. HORVATH P. J.t und SCHUBERT R. F., Chem. Engng. 10 (1958).
145. FLATT R., Chimia [Zurich] 9 (1955), 232—237.
146. STAGE H., und SCHULTZE Gg. R., Oel u. Kohle 5/6 (1944), 90—95.
147. ZUIDERWEG F. J., Laboratory of Batch Distillation, Interscience Publishers, New York 1957.
148. SCHAFER W., Angew. Chem., Teil В 19 (1947), 251—253.
149. FISCHER W., Chemie-Ing.-Techn. 23 (1951), 116.
150. MATZ W., Angew. Chem., Teil В 19 (1947), 131—134.
151. HILBERATH F., Oel u. Kohle 39 (1943), 875—886.
152. BRAGG L. B., und LEWIS J. W„ Wld. Petroleum Rep. 14 (1943), 61.
152a. STRANGIO V. A., und TREYBAL R. E„ Ind. Engng. Chem. Process Design and Development 13 (1974) 3, 279—285.
16*
491
153. ROSE A. und E., und Mitarb., Distillation, Interscience Publishers, New York 1951.
154. ROSE A., Ind. Engng. Chem. 33 (1941), 594.
155. FENSKE M. R., TONGBERG S. 0., und QUIGGLE D., Ind. Engng. Chem. 26 (1934), 1169.
156. RICHTER H., Oel u. Kohle 40 (1944), 282—288.
157. POHL H., Erdol u. Kohle 5 (1952), 291—294.
158. ZUIDERWEG F. I., Chemie-Ing.-Techn. 25 (1953), 297—308.
159. SIZMANN R., Chemie-Ing.-Techn. 33 (1961), 659—668.
160. MANNING R. E., und CANNON M. R., Ind. Engng. Chem. 49 (1957), 346—349.
161. STUKE B., Chemie-Ing.-Techn. 25 (1953), 677—682.
162. HEISE F., HILLER G., und WAGNER H. Gg., Chemie-Ing.-Techn.
41 (1969) 20, 1100 bis 1105.
162a. KAUTER A., und WAGNER H. Gg., Chemie-Ing.-Techn. 42 (1970), 1127; 46 (1974), 997.
162b. BORCHARDT E., und WAGNER H. Gg., Chemie-Ing.-Techn. 43 (1971), 95g_______gg2
163. CHILTON T. H., und COLBURN A. P., Ind. Engng. Chem., ind. Edit. 27 (1935), 255 bis 260, 904.
164. CARNEY Th.. P., Laboratory Fractional Distillation, Mac-Millan, New York 1949.
165. DANATOS S., und OSBURN J. 0., Chem. Engng. 65 (1958), 147—150.
166. ARKENBOUT G. J., und SMIT W. M„ Separation Sci. 2 (5) (1967), 575—596.
167. MERKEL F., Arch. Warmewirtsch. Dampfkesselwes. 10 (1929), 13—17.
168. WEBER U. von, in: GILDEMEISTER E„ und [HOFMANN Fr., Die atherischen Ole (bearb. v. W. Treibs), Bd. I, Akademie-Verlag, Berlin 1966.
169. ROBINSON C. S., und GILLILAND E. R., Elements of Fractional Distillation, 6. Aufl., McGraw-Hill Book Company, New York 1950, 476—478.
170. WEBER U. von, Chem. Techn. 2 (1950), 241—246.
171. ROLLING H., Chemie-Ing.-Techn. 22 (1952), 405—411.
172. KOHRT H. U., Angew. Chem., Teil В 20 (1948), 117—124.
173. HAUSEN H„ Z. angew. Physik 4 (1952), 41—51.
174. WIJK W. R. van, und THISSEN H. A. C., Chem. Engng. Sci. 1 (1952), 121/123; 3 (1954), 145—152.
175. ЛЬВОВ С. В. — ДАН СССР, 1947, т. 57, № 4, с. 375—378.
175а. MOSTAFA Н. A., Chemie-Ing.-Techn. 47 (1975), 2 , 63.
175b. CHIEN Н. Н. J., Chem. Engng. Sci. 8 (1973) 11, 1967—1974.
175c. SEROV V. V., ABRAMENKO V. P„ und ZYKOV D. D„ Int. chem. Engng. 13 (1973) 3, 514—516.
176. KRELL E„ Chem. Techn. 4 (1952), 200—207.
176a. FALDIX P.,und STAGE H., Chemie-Ing.-Techn. 41 (1969) 23, 1265—1269.
176b. MULLER W„ Chem. Techn. 26 (1974) 1, 16—20.
176c. BILLET R., Verfahrenstechnik 8 (1974) 3, 65—72.
177. THORMANN K-, Dechema-Erfahrungsaustausch, Techn. Apparate und Anlagen, Trennen flussiger Mischungen, Destillieren, Betriebstechnik, Dechema, Frankfurt/Main, August 1952.
178. ELLIS S. R. M., und FRESHWATER D. C., Perfum. essent. Oil. Rec.
45 (1954), 380 bis 386.
179. COLBURN A. P., Trans. Amer. Inst. chem. Engr. 37 (1941), 805.
180. UNDERWOOD A. J. V., Chem. Engng. Progr. 44 (1948), 603.
181. FENSKE M. R„ Ind. Engng. Chem. 24 (1932), 482.
182. HARBERT W. D., Ind. Engng. Chem. 37 (1945), 1162.
183. BRUUN P. J., Meded. Landbouwhogeschool, Wagningen 61 (9), (1961), 1—94.
184. UNDERWOOD A. J. V., Trans. Instn. chem. Engr. [London] 10 (1932), 112—152.
185. MURCH D. P., Ind. Engng. Chem. 45 (1953), 2616—2621.
492
186. MULLIN J. W., Ind. Chemist, 33 (1957), 390, 408—417.
187. KIRSCHBAUM E., Angew. Chem., Teil В 19 (1947), 13—14; Chemie-Ing.-Techn. 28 (1956), 639—644.
188. КАЗАНСКИЙ Б. А. и др., ЖОХ, 1942, t. 12, № 1—2, c. 112—123.
189. БУШМАКИН И. H., ЛЫЗЛОВА P. В. — ЖПХ, 1952, t. 25, l№ 3, c. 303—312.
190. PERKTOLD F„ Angew. Chem., Teil В 19 (1947), 184—185.
191. DAVID A., Dechema-Monogr., 23 (1954), 126—175.
192. MYLES M., und MITARB., Ind. Engng. Chem. 43 (1951), 1452—1456.
193. ALLENBY О. C. W., und L'HEUREUX C„ Analytic. Chem. 22 (1950), 1340.
194. H ALDEN W ANGER K-, Chemie-Ing.-Techn. 23 (1951), 437—440.
195. ZUIDERWEG F. J. (edit.), Recommended Test Mixtures for Distillation Columns, The Institution of Chemical Engineers, London 1969.
196. BRANDT H., und ROCK H., Chemie-Ing.-Techn. 29 (1957), 397—402.
197. FELDMANN J., und MITARB., Ind. Engng. Chem. 45 (1953), 214.
198. ЗЕЛЬВЕНСКИЙ Я- Д., ШАЛЫГИН В. A. — Хим. пром., 1962, № 6, с. 38—41.
199. SCHULTZE Gf. R., und STAGE H., Oel u. Kohle 40 (1944), 68.
200. COLLINS F. C., und LANTZ V., Ind. Engng. Chem., analyt. Edit. 18, (1946), 673—677.
201. NAUMANN K., und LEIBNITZ E., Chem. Techn. 8 (1956), 458—471.
202. ОБОЛЕНЦЕВ P. Д„ ФРОСТ A. B. — Ytefyr. хозяйство, 1947, т. 25, № 8, с. 35—45.
203. STRUCK R. T., und KINNEY C. R., Ind. Engng. Chem. 42 (1950), 77—82.
204. WEBER U. von, Personliche Mitteilung.
205. ZUIDERWEG F. H„ Chem. Engng. Sci. 1 (1952), 174—193.
206. KIRSCHBAUM E., BUSCH W., und BILLET B„ Chemie-Ing.-Techn. 28 (1956), 475—480.
207. SZAPIRO S., Zeszyty naukowe Politechn. Lodzkiej, Chem. 2 (1955), 33—37.
208. BRAUER H„ Chemie-Ing.-Techn. 29 (1957), 520-530, 785—790.
209. ЗЕЛЬВЕНСКИЙ fl. Д„ ТИТОВ А. А., ШАЛЫГИН В. A. — Хим. пром., 1963, № 2, с. 36—43.
210. WEYGAND С., und HILGETAG G., Organisch-chemische Experimentierkunst, 4 Aufl., Johann Ambrosius Barth, Leipzig 1970. (ВЕЙГАНД К., ХИЛЬГЕТАГ Г. Методы эксперимента в органической химии, Химия. М., 1968).
211. BERNHAUER К., Einfiihrung in die organische-chemische Laboratoriumstechnik, Springer-Verlag, Wien 1947.
212. PESTEMER M., Angew. Chem. 63 (1951), 118—122.
213. BRANDT P. L., PERKINS R. B., und HALVERSON L. K„ Oil Gas J. 7 (1946).
214. LENTZ H., Chemie-Ing.-Techn. 41 (1969) 15 , 855—861.
214a. LEUNG L. S., HUTTON В. E. T., und NICKLIN D. J., Ind. Engng. Chem. Fundamentals 14 (1975) 1, 63—67.
215. STAGE F„ Angew. Chem., Teil В 19 (1947), 182.
216. SCHULTZE Gg. R., und STAGE fH., Uber die Wirksamkeit der neuen Normalkolonne des «Ausschusses fiir Laboratoriumsdestillation», Fiat-Report Nr. 1 133 vom 23.12.1947.
217. SIGWART K., Destination und Rektifikation, in: Ullmanns Encyklopadie der technischen Chemie, Bd. 1, Urban und Schwarzenberg, Miinchen/Berlin 1951, S. 429—470.
218. THORMANN K., Dechema-Erfahrungsaustausch, Laboratoriumstechnik, Arbeitsmethoden und Gerate, Destillieren und Rektifizieren, Dechema, Frankfurt/Main, September 1950.
219. MACH E., VDI-Forschungsheft 375 Nov./Dez. 1935.
220. BARTH W., Chemie-Ing.-Techn. 23 (1951), 289—293; 26 (1954), 29—34.'
221. LEVA M., Flow through Packings and Beds, Part I, New York 1957.
493
222. KAST W., Chemie-Ing.-Techn. 36 (1964) 5, 464—468.
223. TEUTSCH T„ Chemie-Ing.-Techn. 36 (1964), 5, 496—523.
224. GELBE H., Chemie-Ing.-Techn. 40 (1968), 528—530.
225. GLASER H., Chemie-Ing.-Techn. 41 (1969) 7, 444—450
226. HECKMANN C. J., und KRELL E., Chem. Techn. 5 (1953), 699—704.
227. NEUMANN F., Chemie-Ing.-Techn. 33 (1961), 439—442.
228. REED T. M., und FENSKE M. R., Ind. Engng. Chem. 42 (1950), 654—660.
229. STAGE H., Aus einem Prospekt liber Labodest-Baukasten-Destillationsgerate, Zeichnung Nr. 741, 98, 156. STAGE H., und BOSE K., Chemie-Ing.-Techn. 27 (1955), 439.
230. U MHOLTZ C. L., und WINKLE M. van, Petroleum Refiner 34 (1955), 114—117.
231. SCHUMACHER R., Erdol u. Kohle 2 (1949), 501—508.
232. BORCHERT R., und JUBITZ W., Infrarotstrahler zur Erwarmung, Trocknung und Aushartung, SVT Band 5, VEB Verlag Technik, Berlin 1951.
233. JUNGE C., Chem. Techn. 6 (1954), 37—38.
234. GROSSE-OETRINGHAUS H., Erdol u. Kohle 1 (1948), 273—280.
235. REID R. C., und SHERWOOD T. K-, The Properties of Gases and Liquids, 2. Aufl., McGraw-Hill Book Company, New York 1966.
236. HECHT G., und MIT ARB., Berechnung thermodynamischer Stoffwerte von Gasen und Fliissigkeiten, VEB Deutscher Verlag fiir Grundstoffindustrie, Leipzig 1966.
237. STAGE H., Fette u. Seifen 55 (1953), 284—290.
238. KHARBANDA 0. M. P., Nomograms for Chemical Engineers, Heywood & Company, London 1958.
239. ORLICEK A. F., und POLL. H., Hilfsbuch fiir Mineraloltechniker, Bd. I u. II, Springer-Verlag, Wien 1951/55.
240. HAUSSLER W„ Technik 9 (1954), 51—52.
241. COLE P. J., Analytic, chem. 22 (1950), 946—947.
242. KRELL E., Chem. Techn. 4 (1952), 200—207.
243. MEIER-GROLMAN F. W., und WESOLOFSKY F., Oel u. Kohle 39 (1941), 297—300.
244. BECK R., Chemie-Ing.-Techn. 44 (1972), 8, 596—598.
245. GEMMEKER L., und STAGE H-, Glas-Intrumenten-Techn. 7 (1963) 1, 18—23.
246. KAPLICK K., und SCHUBERT M., Datenverarbeitung in der Verfahrenstechnik—Digitale Rechenautomaten, Bd. 6 der Reihe: Beitrage zur Verfahrenstechnik, VEB Deutscher Verlag fiir Grundstoffindustrie, Leipzig 1969.
247. WAGNER M., Analogrechner in der Verfahrenstechnik, Bd. 10 der Beihe: Beitrage zur Verfahrenstechnik, VEB Deutscher Verlag fiir Grundstoffindustrie, Leipzig, 1972.
248. ADLER H., Elektronische Analogrechner, 3. Aufl., VEB Deutscher Verlag der Wissenschaften, Berlin 1970.
249. SAVAS E. S., Computer Control of Industrial Processes, McGraw-Hill Book Company, New York/Toronto/London 1965.
250. ROTH H., Chem. Techn. 20 (1968), 2, 77—82.
251. SCHUBRING A., Chemie-Ing.-Techn. 32 (1960) 3 , 223—227.
252. SEALEY C. J., Chem. Engng. Sci. 25 (1970), 561—568.
253. LEIBNITZ E„ und SCHUHLER C., Z. physik. Chem. 217 (1961), 217—229.
254. NAGEL 0., SMITS H., WOLF D., und GUNZLER H., Chemie-Ing.-Techn. 41 (1969) 17, 983—987.
255. PILHOFFER Th., Chemie-Ing.-Techn. 42 (1970), 7J509—515. JOHNSON M. L„ und LUPFER D. E., Chem. Engng. Progr. 62 (1966) 6, 75—79.
256. WANG I. C., und HENKE G. E., Hydrocarb. Process, 45 (1966), 8, 155.
257. SARGENT R. W. H., und MURTAGH B. A., Trans. Instn. chem. Engr. 47 (1969), 85.
258. STAINTHORP F. P., und M1TARB. in: P1RIC J. M.: Efficient
494
Computer Methods for the Practising Chemical Engineer, London 1967, S. 181—189.
259. BLATT E., und FLEISSNER H., Glas-Instrumenten-Techn. 15 (1971), Sonderheft 1333 bis 1337.
260. BRAUER H., und MEWES D„ Chemie-Ing.-Techn. 44 (1972), 1/2, 93—96.
261. BITTER R. C., Chemie-Ing.-Techn. 40 (1968) 13, 651—656.
262. WAGNER H., und BLASS E„ Chemie-Ing.-Techn. 45 (1973), 63—67.
263. REICHELT W., und BLASS E., Chemie-Ing.-Techn. 43 (1971) 17, 949________956.
REICHELT W., Chemie-Ing.-Techn. 43 (1971) 22, 1196—1203; 44 (1972) 18, 1068—1071.
264. KOHLER R., und SCHOBER G., Chem. Techn. 25 (1973) 12, 718—721.
265. KIRBACH V., FRENZEL L„ und ST RANKMULLER H., Chem. Techn.
25 (1973), 12, 721 bis 722.
5. ЛИТЕРАТУРА
1. STAGE H.,und GEMMEKER L., Chem. Labor Betrieb 15 (1964) 5, 177—188;
6, 232 bis 242; 7, 290—298; 8, 325—338.
la. STAGE H., CZ-Chemie-Technik 1 (1972), 263—272.
2. BODENHEIMER W., Nature [London] 173 (1954), 124.
3. JANTZEN E„ und WITGERT H., Fette u. Seifen 46 (1939), 563.
3a. FISCHER W. G., GIT Fachz. Lab. 18 (1974) 7, 689—691.
3b. BERTELE E„ und HUMBEL F. A., GIT Fachz. Lab. 17 (1973) 6, 667—674.
4. BABCOCK M. J., Analytic. Chem. 21 (1949), 632—633.
5. SHRADER S. A., und RITZER J. E., Chem. Fabrik 12 (1935), 173.
6. KLENK E., Hoppe-Seyler’s Z. physiol. Chem. 242 (1936), 250.
7. FISCHER W. S., Chemiker-Ztg. 94 (1970) 5, 157—161; Glas-Instrumenten-Techn. 13 (1969), 535—540.
8. WINTERS J. C., und DINERSTEIN R. A., Analytic. Chem. 27 (1953), 546—550.
NERHEIM A. G., und DINERSTEIN R. A., Analytic. Chem. 28 (1956), Ю29__1033.
9. SPlEGELBERG C., Dissertation, F. U., Berlin 1956.
10. GOULD JR., C. W., HOLZMANN G., und NIEMANN C., Analytic. Chem. 20 (1948), 361.
11. BREGER J. A., Analytic. Chem. 20 (1948), 980.
12. PASCHKE R. F., und MITARB., J. Amer. Oil Chemists’Soc. 31 (1954), 5—7.
13. PFEIL E„ Angew. Chem. 54 (1941), 161—184.
14. EIGENBERGER E., Mikrochemie 26 (1939) 204.
15. CLASEN H., Chemie-Ing.-Techn. 28 (1956), 45—48.
16. STAGE H., Fette u. Seifen 55 (1953), 217—224.
17. GUNTHER G., KUHNHANSS K., und HUTTIG E., Chem. Techn. 7 (1955), 656—662.
18. PROFFT E„ und BUCHMANN G„ Chem. Techn. 8 (1956), 417—418. BUCHMANN G.; Chem. Techn. 14 (1962) 10, 617—620; 12, 741—744.
19. FURST H., und SCHWETLICK K-, Chem. Techn. 8 (1956), >458—471.
20. OLECHNOWITZ A. F., Dissertation, Halle-Wittenberg 1956.
21. FI EBACH K., Dissertation, Leipzig 1957.
22. GUTWASSER H.,und MULLER A., Chem. Techn. 21 (1969) 11,756—759. GUTWASSER H., Chem. Techn. 22 (1970) 8, 467—471.
23. MAIR B. J., und MITARB., Analytic. Chem. 30 (1958), 1814—1817.
24. NAUMANN K., und LEIBNITZ E., Chem. Techn. 8 (1956), 458—471.
25. OLDROYD D. M., und GOLDBLATT L. A., Ind. Engng. Chem., analyt. Edit. 18 (1946), 761—763.
495
26. PETROCELLI J. A„ PUZNIAK T. J., und CLARK R. 0., Analytic. Chem. 36 (1964), 1008—1011.
27. Prospekt «Aerograph Research Notes 1965». Wilkens Instrument u. Research fAG, Basel/Schweiz.
28. STAGE H., CZ-Chemie-Technik 2 (1973) 1, 35—40.
29. HAMPEL B„ Chemiker-Ztg. 95 (1971), 7 , 316—325.
30. RIEGER H., Chem. Techn. 22 (1970) 1, 36—40.
31. CARPENTER J. K-, und HELWIG R. W., Ind. Engng. Chem. 42 (1950), 571—578.
32. JORDAN D. G., Chemical Pilot Plant Practice, Interscience Publishers, New York 1955.
33. DIETZ H., und KARDOS J., Chem. Techn. 19 (1967) 12, 713—719.
34. BILLET R., Chemie-Ing.-Techn. 42 (1970) 14, 910—917.
35. KULLMANN W. W., Pharmaz. Ind. 15 (1953), 159—162.
36, JOB C., und ZELLNER H., Pharmaz. Ind. 17 (1955), 161—163. Hersteller: Schott u. Gen., Mainz.
36a. Prospektblatt «Quickfit, Einstufige Wasserdestillationsapparatur».
Leistung 3,5 bis 4 1/h dest. Wasser.
37. ШПОЛЬСКИЙ Э. ВЛ Атомная физика т. 1, 2. Наука. М., 1974.
38. БРОДСКИЙ А. И. Химия изотопов, Изд. АН СССР, М., 1957.
39. BRUNNEE К., und V0SHAGE И., Massenspektrometrie—Physikalische und apparative Grundlagen sowie Anwendungen, Verlag Karl Thi'emig, Mflnchen 1964.
BIRKENFELD H., HAASE G., und ZAHN H., Massenspektrometrische Isotopenanalyse, 2. Aufl., VEB Deutscher Verlag der Wissenschaften, Berlin 1969.
WEITZEL K., HUBNER H., HUBNER G., und MAASS /., Anwendung stabiler Isotope in Forschung und Technik, VEB Deutscher Verlag fiir Grundstoff Industrie, Leipzig 1963.
40. CONN A. L., und WOLF J. E., Ind. Engng. Chem. 50 (1958), 1231—1234, DBP Nr. 944848, ausgegeben am 28.6.1956.
41. MERCEA V., Isotopenpraxis 2 (1966), 8, 313—322.
42. MULLER G„ MAUERSBERGER K., und SPRINZ H„ Analyse stabiler Isotope durch spezielle Methoden, Akademie-Verlag, Berlin 1969.
43. BAERTSCHI P., KUHN W., und KUHN H., Nature London 171 (1953), 1018.
44. SELLERS E. S., und AUGOOD D. R., Trans. Instn. chem. Engr. 34 (1956), 53—78. •
45. TIMMERHAUS K. D., WEITZEL D. H., und FLYNN T. M., Chem. Engng. Progr. 54 (1958), 35—46.
46. WEISSER R. H„ Techn. Rundschau Sulzer 40 (1958), 71—73.
47. SCHAVORONKOV N. M., und SAKODYNSKIJ K- L, Kernenergie 4 (1961) 12, 893—904.
48. CLUSIUS K., und MEYER H„ Helv. chim. Acta 36/7 (1953), 2045—2055.
49. GROTH W., IHLE H., und MURRENHOFF A., Angew. Chem, 68 (1956), 644—648.
50. GLUSIUS K., und SCHLEICH K-, Helv. chim. Acta 42 (1959), 232—239. McINTEER В. B., und POTTER R. M., Ind. Engng. Chem. Proc. 4 (1965) 1, 35—42.
51. ДЕВЯТЫХ Г. Г., ЗОРИН А. Д., НИКОЛАЕВ Н. И. — ЖПХ, 1958, т. 31, № 3, с. 368—375.
52. СЕВР ЮГОВА Н. Н., УВАРОВ О. В., ЖАВОРОНКОВ Н. М. Атомная энергия, 1956, т. 4, с. 113—117.
53. KISTEMAKER J., BIGELEISEN J. В., und NIER А. О., Proceedings of the International Symposium on Isotope Separation (Amsterdam, April 23—27, 1967), North-Holland Publishing Comp., Amsterdam 1958.,
54. ЯГОДИН Г. А., УВАРОВ О. В., ЖАВОРОНКОВ Н. М. — ДАН СССР, 1956, т. 111, № 2, с. 384—387.
55. CLUSIUS К., und SCHLEICH К., Helv. chim. Acta 41 (1958), 1342—1358.
496
56. KIRSHENBAUM J., Physical Properties and Analysis of Heavy Water, McGraw-Hill Book Company, New York 1951.
57. UREY H. C., J. chem. Soc. [London] 1947, 562.
58. BENEDICT M., Survey of Heavy Water Production Processes, A/Conf., 8/P/819, Schweiz, 30.6.1955.
{БЕНЕДИКТ M., Обзор методов получения тяжелой воды. Мирное использование атомной энергии. Материалы Международной конференции в Женеве, август 1955, том. 8, стр. 819. Гос. Научно-технич. изд. химии. литературы. М., 1955).
59. KVHN W., Chemie-Ing.-Techn. 25 (1953), 12—18, 29 (1957), 6—16.
KVHN W., BAERTSCHI Р., und THURKAUF М., Chimia [Zurich] 8 (1954), 109—122, 145—156.
KUHN W„ Helv. chim. Acta 35 (1952), 1684—1736.
KUHN W., Helv. chim. Acta 37 (1954), 1407—1422.
60. BAERTSCHI P., und KUHN tW., Final^Concentration of Heavy Water by Rectification, A/Conf. 8/P/927, Schweiz, 8.7.1955.
61. BECKER E. №., Angew. Chem. 68 (1956), 6—13.
HUBER M., Die Rektifikation als Speziaifall der Theorie der Isotopentrennung, Sulzer-Verfahrenstechnik, Achema 1961.
62. HANNY J., HARTMANN F., und HILTBRUNNER R., Techn. Rundschau Sulzer 4 (1961), 21—26.
63. DIXON 0. G., J. Soc. chem. Ind. 68 (1949), 88—91.
64. KRELL E., Chemie-Ing.-Techn. 32 (1960), 233—240.
65. УВАРОВ) О. В., СОКОЛЬСКИЙ BaA., ЖАВОРОНКОВ H. М. — Хим. пром., 1956, № 7, с. 20—21.
66. DOSTROVSKY J., LLEWELLYN D. R„ und VR0MEN В. Н., J. chem. Soc. 4 (1952), 3509—3525.
67. JACKSON R. F., und PIGFORD R. L., Ind. Engng. Chem. 48 (1956), 1020—1026.
68. РОЗЕН A. M. — ДАН СССР, 1956, т. 107, № 2, с. 295—298.
69. LONDON Н., Separation of Isotopes, Verlag George Newnes Ltd., London 1961.
COHEN K., Theory of Isotopic Separations, McGraw-Hill Book Company, New York 1951.
70. STASCHEWSKI D., Chemie-Ing.-Techn. 40 (1968) 748; 43 (1971) 14, 810—816.
71. SUROWIEC A. J., Ind. Engng. Chem. 52 (1960) 9, 798.
GUPTA D., und RAY S. N., Ind. Engng. Chem. ProC. 1 (1962), 255—262. ПЕТЛЮК Ф. Б., ПЛАТОНОВ В. M., КИРСАНОВ И. В. — Хим. пром., 1964, № 6, с. 45—53.
72. BAERTSCHI Р„ und KUHN W., Helv. chim. Acta 40 (1957), 1084—1103.
73. STAGE H., Chemische Arbeitsgemeinschaft Verfahrenstechnik, Jan./Febr. 1970, 44—54.
73a. SCHWAB H„ Chemie-Ing.-Techn. 44 (1972) 11, 704—707.
74. GELBIN D„ Chem. Techn. 21 (1969) 6, 329—334.
75. CANNON N. R., Ind. Engng. Chem. 53 (1961), 629.
76. MCWHIRTER I. R., und LLOYD W., Chem. Engng. Progr. 59 (1963), 7, 58.
77. BURSTIN H., und WINKLER J., Chemiker-Ztg. 22 (1931), 212.
78. ROLLING H., und TRAMM H., Chemie-Ing.-Techn. 21 (1949), 9.
79. CAPITANI C., und MILANI E„ Ind. chimica 36 (1954), 671—684.
80. STAGE H., Dechema-Monogr. 15 (1950), 156—170. -4*
81. SIGWART K., Destillieren und Rektifizieren, iny HOUBEN-WEYL., Methoden der organischen Chemie, hrsg. von E. Muller, Bd. 1/1, Georg Thieme Verlag, Stuttgart 1957. ч Л- «Ф
82. HAUSEN H., und SCHLATTERER R., Chemie-Ing.-Techn.’21 (1949), 453—460.
KIRSCHBAUM E., und LIPPHARDT G., Chemie-Ing.-Techn. 29 (1957), 393—397.
497
83. OTTO IF., Chem. Techn. 14 (1962) 4, 228—231.
84. KIRSCHBAUM E., Destillier- und Rektifiziertechnik, 4. Aufl., Springer-Verlag, Berlin/Heidelberg/New York 1969.
85. FABUSS B., Z. Ver. ung. Chemiker 8 (1953), 323—324.
86. FISCHER V., Ann. Physik 6 (1947), 139—152.
87. HERRMANN E., Chemiker-Ztg. 80 (1956), 102—104, 131—134, 169—173.
88. TROSTER E., Chemie-Ing.-Techn. 32 (1960) 8, 525—531.
89. JUNGE C., Chem. Techn. 6 (1954), 37—38.
90. TRENNE H., Erdol u. Kohle 2{(1951), 63—66.
91. KUHN W., Helv. chim. Acta 25 (1952), 252—295.
92. WEBER U. von, Chem. Techn. 2 (1950), 241—246.
93. VOIGT G., Kernenergie 2 (1959), 14—39.
93a. BLASS E., Chemie-Ing.-Techn. 45 (1973) 13 , 865—872.
93b. WONDRAK A. M., und KOHLER F., Chemie-Ing.-Techn. 46 (1974), 1, 37.
94. PODBIELNIAK W. J., Ind. Engng. Chem., analyt. Edit. 3 (1931), 177;
5 (1933), 119.
95. GROSSE-OETRINGHAUS H., Erdol u. Kohle 2 (1949), 286—299.
96. WUSTROW W., Z. analyt. Chem. 108 (1937), 305.
97. STOKES Ch. S., und HAUPTSCHEIN M„ Analytic. Chem. 24 (1952), 1526.
DRAW IN H. W., Vakuum-Techn. (1961), 2.
98. LERER E., und KUSS E., Z. physik. Chem. Abt. A, Bd. 163 (1933), 73.
99. MILLER G. H., Operations with Gases in Technique of Organic Chemistry, Vol. Ill, Part. II, Chapter V, Interscience Publishers, New York 1957.
100. MULLER G., und GNAuCK G., Reinste Gase, VEB Deutscher Verlag der Wissenschaften, Berlin 1965.
101. KOCH H., und HILBERATH F„ Brennstoff-Chem. 21 (1940), 197—203.
102. GEMMEKER W., und STAGE H., Glas-Instrumenten-Techn. 6 (1962), 10, 372—376.
103. KLEIN E.,und WAIZ H., Glas-Instrumenten-Techn. 11 (1967), 5, 405—411.
104. PODBIELNIAK W. J., und PRESTON S. T., Petroleum Engr. 5 (1955), 205—211.
105. Hersteller: Ernst Haage, Miihlheim—Ruhr.
106. SIMONS G., Ind. Engng. Chem., analyt. Edit. 10 (1938), 648.
107. PODBIELNIAK W. J., Apparatus und Methods for Precise Fractional Distillation Analyses, American Chemical Society, Atlantic City Meeting 1941.
108. ROSE A. und E., und MITARB., Distillation, Interscience Publishers, New York 1951.
109. PRESTING W., und MITARB., Freiberger Forschungshefte 5, Akademie-Verlag, Berlin 1953, S. 231—238.
110. STAGE H., Destillieren und Rektifizieren, Abschnitt 19 in: Fortschritte der Verfahrenstechnik 1952/53, Verlag Chemie GmbH, [Weinheim/Bergstr. 1954.
111. BERNHAUER K., Einfiihrung in die organisch—chemische Laboratoriumstechnik, Springer-Verlag, Wien 1947.
112. GEMMEKER L., und STAGE H., Glas-Instrumenten-Techn. 6 (1962) 11, 455—458.'
113. HOLLAND-MERTEN E. L., Tabellenbuch der Vakuumverfahrenstechnik in der Grundstoffindustrie, VEB Deutscher Verlag fiir Grundstoffindustrie, Leipzig 1968.
114. JAECKEL R., Kleinste Driicke, ihre Messung und Erzeugung, in: Technische Physik in Einzeldarstellungen, Springer-Verlag, Berlin/Gottingen/ Heidelberg 1950.
LEYBOLD, Vakuum-Taschenbuch, hrsg. von K- DIELS und R. JAECKEL, Springer-Verlag, Berlin/Gottingen/Heidelberg 1962.
KEUNECKE E., Chemiker-Ztg. 81 (1957), 239—244.
YARWOOD J., Hochvakuumtechnik, 3. Aufl., Rudolf A. Lang Verlag, Berlin 1955 (Obers, aus d. Engl.).
498
PUPP W., Vakuumtechnik, Grundlagen und Anwendungen, Thiemig-Taschenbuch Bd. 43, Verlag Karl Thiemig, Munchen 1973.
115. MULLER L., Freiberger Forschungshefte, Ausg. В 6, Akademie-Verlag, Berlin 1954, S. 81—115; Freiberger Forschungshefte 11, Akademie-Verlag, Berlin 1957, S. 575.
116. SPENDLOVE M. J., Vakuum-Techn. 6 (1957), 15—18 u. 36—44.
117. HORSLEY W., J., appl. Chem. 8 (1958), 13; Chemie-Ing.-Techn. 30 (1958), 736.
118. PARKER S. G., und WILSON 0. W„ Ind. Engng. Chem. Proc. 4 (1965) 4, 365—368.
118a. БЕЗОБРАЗОВ E. Г. и др.. Хим. пром., 1974, № 4, с. 61—62.
119. LAPORTE H., Hochvakuum, VEB Wilhelm Knapp Verlag, Halle 1951.
120. ECHOLS Jr., L. S„ und GELUS E., Analytic. Chem. 19 (1947), 668.
121. ULUSOY E., und SAKALOZ T., Theoretische Grundlagen der Isotherm— Destination (Mitt. I—III), Revue de la faculte des sciences de I’universite d’Istanbul, Serie C, Tome XXHI/XXIV, Fasc. 3—4 (1958/59).
122. FRANK W., und KUTSCHE D., Die schonende Destination, Otto Krausskopf-Verlag GmbH, Mainz 1969.
123. BILLET R„ und RAICHLE L., Chemie-Ing.-Techn. 39 (1967) 3, 133—137.
124. GELBE H„ Chemie-Ing.-Techn. 40 (1968) 11, 528—530.
125. HARRIES W., Chemie-Ing.-Techn. 21 (1949), 139—142.
126. MON CH G. C., Hochvakuumtechnik, Rudolf A. Lang Verlag, Popneck 1950; Neues und Bewahrtes aus der Hochvakuumtechnik, 2. ber. Nachdruck, VEB Verlag Technik, Berlin 1961.
127. HOLLAND-MERTEN E. L., Die Vakuum-Pumpen in der Verfahrenstechnik, VEB Wilhelm Knapp Verlag, Halle 1950.
128. ARDENNE M„ von, Technik 13 (1958), 275—285.
129. KIENEL G., Glas-Instrumenten-Techn. 10 (1966) 9, 761—770.
130. KRELL E„ Chem. Techn. 2 (1950), 252—255; 4 (1952), 443—450.
131. WITTKA F., Angew. Chem. 53 (1940), 557.
132. GEMMEKER L„ und STAGE H., Glas-Instrumenten-Techn. 8 (1964) 6, 413—428; 7, 503—508.
STAGE H., Chemieanlagen + Verfahren (1972), Heft 2.
133. UTZINGER G. E., Chem. Techn. 7/8, (1943), 61.
134. JANOSFIA P., Dechema—Monogr. 55 (1965).
135. FURST H„ und SCHIMKE D., Chem. Techn. 22 (1970) 9, 553—555.
136. DIETER K., Chemiker-Ztg. 94 (1970) 12, 445—451.
137. BILLET R„ Chemie-Ing.-Techn. 29 (1957), 733—742.
138. EGLI R., Glas-Instrumenten-Techn. 11 (1967) 6, 509—515.
139a. KRAMER M. T., GIT Fachz. Lab. 18 (1974) 7, 862—868.
139. BURROWS G., Molecular—Distillation, Oxford—Clarendon—Press, Oxford 1960.
HOLO J., und MITARB., The Applications of Molecular Distillation, Akademiai Kiado, Budapest 1971.
140. RIDGWAY-WATT P„ Chem. and Ind., 27, 5. 1961, 680—692.
140a. HABENDORFF R„ CZ-Chem.-Techn. 2 (1973), 21—24.
141. HICKMAN К. C. D., und TREVOY T. J., Chemie-Ing.-Techn. 25 (1953), 672—676.
142. LANGMUIR S„ Physic. Rev. 8 (1916), 176.
143. BURROWS G., J. appl. Chem. 7 (1957), 375—384.
144. JAECKEL R., Erdol u. Kohle 4 (1951), 1761.
145. PERRY E. S„ Methods in Enzymol 3 (1957), 383—391.
JAECKEL R„ und OETJEN G.—W., Chemie-Ing.-Techn. 21 (1949), 169—176.
JAECKEL R., und MITARB., Vakuumtechnik und Molekulardestillation, in: Fortschritte der Verfahrenstechnik, Bd. 2, 1954/55, S. 29—43; Bd. 3, 1956/57, S. 62—73, Verlag Chemie GmbH, Weinheim/Bergstr. 1954/57.
JAECKEL R., Destination und Sublimation im Fein- und Hochvakuum, in: HOUBEN-WEYL, Methoden der organischen Chemie, hrsg. von
499
E. MULLER, Bd. 1/1, Georg Thieme Verlag, Stuttgart 1958.
146. FRANK W., Glas-Instrumenten-Techn. 9 (1965) 11, 777—782.
147. UTZINGER G. E„ Chemie-Ing.-Techn. 26 (1954), 129—131.
148. MELPOLDER F. W„ und MITARB., Analytic. Chem. 27 (1955), 974—977.
149. МАЛЮСОВ В. А., ЖАВОРОНКОВ H. M„ Collect. Czechoslow. Chem. Commun, 23, № 9, 1720—1726 (1958).
150. МАЛЮСОВ В. А., УМНИК H. H., ЖАВОРОНКОВ Н. М., Хим. пром., 1958, № 5, с. 36—42.
151. HICKMAN К. С. D., Ind. Engng. Chem. 39 (1947) 686—694.
152. . HICKMAN К. C. D.; Ind. Engng. Chem., analyt. Edit. 29 (1937), 968.
153. HICKMAN К. C. D., Патенты США № 2117802, 1938; № 2210928, 1940 г.
154. EMBREE N. D., Ind. Engng. Chem. 29 (1937), 975.
155. GORRIZ A. M., und MITARB., An. Real Soc. espan. Fosica Quim., Ser. В 48 (1952), 825 bis 850; Ser. В 49 (1953), 19—22, 107—114, 579—586.
156. FAWCETT E. W., Kolloid-Z. 86 (1939), 34—91.
157. MASCH L.—W., Chemie-Ing.-Techn. 22 (1950), 141—146.
158. HICKMAN K., Ind. Engng. Chem. 48 (1956), 496—499.
159. CARNEY T. P., Laboratory Fractional Distillation, MacMillan, New York 1949.
160. МАЛЮСОВ В. А., МАЛАФЕЕВ H. А., ЖАВОРОНКОВ H. М., ЖФХ, 1958, т. 32, № 10, с. 2403—2409.
.161. SCHNEIDER К. W., Erdol u. Kohle 4 (1951), 4—9.
162. SIMONS J. Н., Ind. Engng. Chem. analyt. Edit. 10 (1938), 30.
163. UY EHA H.,und HAGIHARA Y„ J. chem. Engng. Japan 3 (1970) 1, 13—17.
164. BEWILOGUA L., VERGES P., und VINZELBERG H., Isotopenpraxis 9 (1973) 3, 97—100.
. 165. LI EDEL К. H., Gias in der Chemie—Laborgerate, Gro₽apparate, Rohrleitungen, Otto Krausskopf-Verlag GmbH, Mainz 1973.
6. ЛИТЕРАТУРА
1. FINDLAY A., Die Phasenregel und ihre Anwendungen, Verlag Chemie GmbH, Weinheim/Bergstr. 1958 (Ubers, aus d. Engl.).
2. BADGER W. L., und McCABE W. L., Elemente der Chemie-Ingenieur-Technik, Springer-Verlag, Berlin 1932 (Obers, aus d. Engl.).
3. STAGE H., Fette u. Seifen 55 (1953), 513—516.
4. STAGE H., Erdol u. Kohle 3 (1950), 478—490.
5. HOFFMANN E. J., Azeotropic and Extraktive Distillation, John Wiley & Sons, London 1965.
6. Патент Германии N 767 162 3.12.1951 от 27.11.1936.
7. SIZMANN R., und STUKE B., Chemie-Ing.-Techn. 27 (1955), 669—675.
8. ALGERMISSEN J., Chemie-Ing.-Techn. 30 (1958), 502—510.
9. MERKEL H„ Chemie-Ing.-Techn. 24 (1952), 511—512.
10. STAGE H.,BUNGER R.,und JOHAS A., Fette u. Seifen 55 (1953), 580—583.
11. POZZI und ESCOT, Bull. Soc. chim. France 31 (1904), 932.
12. PFEIL E., Angew. Chem. 54 (1941), 161—184.
13. BERNHAUER K., Einfuhrung in die organisch—chemische Laboratoriumstechnik, Springer-Verlag, Wien 1947.
14. THORMANN K., Chem. Fabrik 13 (1940), 3—9.
15. WEBER U. von, Gewinnung atherischer Ole durch Destination, in; GILDEMEISTER, E., und HOFFMANN, Fr., Die atherischen Ole (bearb. von W. Treibs), Akademie-Verlag, Berlin 1966.
16. RIGAMONTI R., Chemiker-Ztg. 80 (1956), 628—631.
17. ROCK H-, Destination im Laboratorium, kExtraktive und azeotrope Destination, Dr. Dietrich Steinkopff Verlag, Darmstadt 1960.
18. KUMMERLE K., Chemie-Ing.-Techn. 32 (1960), 513—517.
19. GERSTER J. A., Chem. Engng. Progr. 65 (1969) 9, 43.
19a. MULLER E., Verfahrenstechnik 8 (1974) 3, 88—93.
500
20. LECAT M., Tables azeotrophiques, Azeotropes binaires orthobares, 2. Aufl., Brussel 1949 (Eigenverlag).
21. ОГОРОДНИКОВ С. K-, ЛЕСТЕВА T. M., КОГАН В. Б., Азеотропные смеси. Справочник, Л., Химия, 1971.
22. STUKE В., Chemie-Ing.-Techn. 25 (1953), 133—136.
23. COULSON Е. A., und HERRINGTON Е. F., J. Chem. Soc. [London] 1947, 597—607.
24. KUHN W., und MASSIN I P., Helv. chim. Acta 33 (1950), 737.
KUHN W., und KUHN H. J., Helv. chim. Acta 40 (1957), 2433—2454.
25. ENiiSTUN В. V., Commun. Fac. Sci. Univ. Ankara, Ser. В 5 (1954) 1—21.
26. ЛИТВИНОВ H. Д. — ЖФХ, 1953, т. 27, № 4, с. 476—490.
27. SWIETOSLAWSKI W., Przemysl. chem. 7 (1951), 363—370; Bull. Soc. chim. beiges 62 (1953), 10—20; Bull. Acad, polon. Sci., Cl. Ill, Vol. I, 1—2, 1953.
SWIETOSLAWSKI W., Physikalische Chemie des Steinkohlenteers, N. J. Hoffmann Verlag, Koln 1954.
SWIETOSLAWSKI W., Azeotropie und Polyazeotropie, Bd. 1, Panstwowe Wydawnictwo Naukowe, Warshau 1957.
28. LECAT M., Azeotropisme et Distillation, in: Grignard, Traite^de Chimique Organique I, Paris 1935.
28a. OTHMER D. F., Verfahrenstechnik 8 (1974) 3, 94—99; 4, 118—123.
29. SCHUBERTH H., Thermodynamische Grundlagen * der Destination und Extraktion, Bd. I, VEB Deutscher Verlag der Wissenschaften, Berlin 1972.
30. ZIEBORAK K-, Prace Glownego Inst. Chem. przemosloweje, (1951), 1—44.
31. ORSZAGH A., Przemysl. chem. 9 (32) (1953), 333—336.
32. MALESINSKI I- W., Bull. Acad, polon. Sci., Ser. Sci. chim., geol., geogr. 6 (1958), 105 bis 109.
33. STCKI J-, Bull. Acad, polon Sci., Ser. Sci. chim., geol., geogr. 6 (1958), 47—53.
34. BERG L., und MITARB., Hydrocarbon Processing 45 (1966), 103; Ind.. Engng. Chem. 37 (1945), 585; 38 (1946), 1249.
BERG L., Chem. Engng. Progr. 65 (1969) 9, 52—57.
35. ROSE A. und E., und MITARB., Distillation, Interscience Publischers, New York 1951.
36. FRANCK H. G., Angew. Chem. 63 (1951), 260—263.
37. COULSON E. A., und JONES J. J., J. Soc. chem. Ind. 65 (1946), 163—175.
38. DUMMETT G. A., Petroleum [London] 18 (1955), 41—45.
39. HUNSMANN W., und SIMMROCK К. H„ Chemie-Ing.-Techn. 38 (1966) 10, 1053—1059.
39a. ПЕТЛЮК Ф. Б., АВЕТЬДН В. С. — TOXT, 1973, т. 7, № 3, с. 307—312.
40. LINO М„ und MITARB., J.rchem. Engng. Japan 4 (1071) 1, 33—36.
41. MAIR B. J., GLASGOW A. R., und ROSSINI F. D., J. Res. nat. Bur. Standards 27 (1941), 39.
42. MAIR B. J., und MITARB., Analytic. Chem. 30 (1958), 393—400.
43. FELDMAN J., und ORCHIN M. Патент США № 2583554, 1952.
44. SCHUBERTH H., Chem. Techn. 21 (1969) 1, 28—34, 51.
45. ШЕЙНКЕР Ю. H., ПЕРЕСЛЕНИ E. M. — ЖФХ, 1952, t. 26, № 8, c. 1103—1109.
46. RUETHER J. A., und LU В. C. Y., Canad. J. chem. Engng. 50 (1972) 2, 266—270.
47. NUTTING H. S., und HORSLEY L. H., Ind. Engng. Chem., analyt. Edit. 19 (1947), 602—603.
48. JOFFE J., Ind. Engng. Chem. 47 (1955), 2533—2535.
49. MALES I NSKI W„ Bull. Acad, polon. Sci., Cl. HI 4 (1956), 371—379.
50. KORTUM G„ und BUCHHOLZ-MEISENHEIMER H„ Die Theorie der Destination und Extraktion von Fliissigkeiten, Springer-Verlag, Berlin/Gottingen/Heidelberg 1952.
51. HORSLEY L. H., Azeotropic Data, Bd. I und II, American Chemical
501
Society, Washington 1952/1962; Advances in Chem., Series No. 6 and 35 (Хорсли Л. Таблицы азеотропных смесей, ИИЛ, М., 1951).
52. EWELL R. И., HARRISON J. М., und BERG L., Ind. Engng., Chem. 36 (1944), 871.
53. MARSCHNER R. F., und GROPPER W. P., Ind. Engng. Chem. 38 (1946), 262—268.
54. HORSLEY L. H„ Analytic. Chem. 19 (1947), 603.
55. MEISSNER H. P„ und GREENFELD S. H., Ind. Chemist 40 (1948), 438_____<12
56. НОВИКОВ К- E., НАТРАДЗЕ А. Г. Хим. пром., 1958, № 2, с. 38—41.
57. SKOLNIK H„ Ind. Engng. Chem. 40 (1948), 442.
58. NAKANISI K-, ICHINOSE S„ und SKIRAI H., Ind. Engng. Chem., Fundamentals 7 (1968), 3 , 381—387.
58a. BRANDANI V., Ind. Engng. Chem. Fundamentals 13 (1974) 2, 154—156.
59. KUHN W., Helv. chim. Acta 37 (1954), 1585—1605.
60. SZAPIROj S., Przemysl chem. 12 (35) (1956), 106—110.
61. KORTUM G., und BITTEL A., Chemie-Ing.-Techn. 28 (1956), 40—44, 282—284.
61a. HUNSMANN W„ Chemie-Ing.-Techn. 33 (1961) 8, 537—540.
61b. HEGENER B., HESSE D., und WOLF D., Chemie-Ing.-Techn. 45 (1973) 14, 942—945.
62. HOUDRY PROCESS CORP. Патент США Ns 2776936, 1957.
63. GREWER, Th., Chemie-Ing.-Techn. 43 (1971), 655—658.
63a. ПАВЛОВ С. Ю., СКОРИКОВА В. В., БОЙКОВА С. 5. — ТОХТ, 1972, т. 6, Ks 3, с. 474—477.
64. STAGE Н., Erdol u. Kohle 3 (1950), 478—490.
65. SCHEIBEL Е. G., Chem. Engng. Progr. 44 (1948) 927—931.
65а. ГАРБЕР Ю. Н., МИРОНЕНКО В. Ф. — ТОХТ, 1974, т. 8, № 6, с. 846—852.
66. КОГАН В. Б. — ЖФХ, 1955, т. 29, № 8, с. 1470—1476.
67. КАФАРОВ В. В., ГОРДИЕВСКИЙ Л. А. — ЖПХ, 1956, т. 29, № 2, с. 176—183.
68. КОГАН В. Б. —Хим. пром., 1957, № 6, с. 36—41.
69. ROCK Н., Chemie-Ing.-Techn. 28 (1956), 489—495.
HORSLEY L. Н., Analytic. Chem. 19 (1947), 508—600; 21 (1949), 831.
70. PORTER R. S„ und JOHNSON J. F., Ind. Engng. Chem. 52 (1960) 8, 691—694.
71. HILDEBRAND, Solubilities, 2. Ausg., New York 1936 (Гильдебранд Д. Г. Растворимость неэлектролитов, ГОНТИ НКТП, М., 1938).
72. Патент Великобритании № 613867, 1948.
73. Патент США № 2549586, 1951; GRISWOLD J., und MITARB., Ind. Engng. Chem. 38 (1946), 65—70.
74. Патенты США №№ 2610704, 1952; 2588063, 1952; 2371342, 1945.
75. GARNER F. H„ ELLIS S. R. M., und GRANVILLE W. H., J. Inst. Petroleum 42 (1956), 148—154.
76. NULL H. W., und PALMER D. A., Chem. Engng. Progr. 65 (1969) 9, 47.
77. GELBIN D., Chem. Techn. 11 (1959), 70—73.
78. NAGEL und SINN R„ Chemie-Ing.-Techn. 39 (1967) 6/e, 275—282.
79. KORTUM G., und FALTUSZ E., Chemie-Ing.-Techn. 33 (1961), 599—606.
80. GARWIN L., und HUCHINSON К- E., Ind. Engng. Chem. 42 (1950), 727—730.
81. DAUBACH E., Diplomarbeit, Marburg 1948.
82. JOST W., Chemie-Ing.-Techn. 23 (1951), 64.
83. TURSI R. R., und THOMPSON R., Chem. Engng. Progr. 47 (1951), 304—308.
84. NOVELLA E. C., und TARASSO J. M., An. Real. Soc. espan. Fisika Quim., Ser. В 48 (1952), 441—448.
85. SCHIER К. H., Dissertation, Martin-Luther-Universitat, Halle—Wittenberg 1959.
502
86. BELCK L., Chemie-Ing.-Techn. 23 (1951), 90—91.
87. BOGART M. J. P., und BRUNJES A. S., Chem. Engr. Progr. 44 (1948), 95.
88. RIEDER R. M., und THOMPSON A. R., Ind. Engng. Chem. 42 (1950), 379
89. GUYER A., GUYER JR. A., und KARTH JOHNSON B., Helv. chim. Acta 38 (1955), 946 bis 954.
90. MIRO A. R., de la GANDARA J. L. 0., und GONZALES J. R. A., An. Real Soc. espan. Fisika Quim., Ser. В 53 (1957), 171—184, 185—194.
91. FURTER W. F., Chem. Engng. (1958) Juni, CE 173—CE 177.
92. SCHREIBER E., SCHUTTAU E„ RANT D., und SCHUBERTH H., Z. physik. Chem. [Leipzig] 247 (1971) V2, 23—40.
SCHUBERTH H„ Z. physik. Chem. [Leipzig] im Druck.
93. FUCHS 0., und ROTH F. H., Chem. Fabrik 11 (1938), 401—406.
94. INO M., und MITARB., J. chem. Engng. Japan 4 (1971) 1, 22—26.
95. JAQUES D., und FURTER W. F„ Canad. J. chem. Engng. 50 (1972) 4, 502—505.
96. FURTER W. F., und MERANDA D„ GVC/AIChe-J., Joint Meet. 2 (1974), D 1—3, 1—10.
97. SADA E., KITO Sh„ YAMAJI H., und KIMURA M., J. appl. Chem. Biotechnol. 24 (1974) %, 229—238.
7. ЛИТЕРАТУРА
1. LIEDEL К- H., Gias in der Chemie-Laborgerate, Groflapparate, Rohrleitungen, Otto Krausskopf-Verlag GmbH, Mainz 1973.
2. MONCH G. CH., Hochvakuumtechnik, Rudolf A. Lang Verlag, Popneck 1950.
3. EITEL W., und MITARB., Glastechnische Tabellen, Springer-Verlag. Berlin 1932.
4. VVB Westglas, Ilmenau, Technik 8 (1953), 675—680.
5. REIGER H., Silikattechnik 4 (1953), 339—342; Chem. Techn. 22 (1970), 1, 36—40.
6. POMEROY H. H., Analytic. Chem. 24 (1952), 1076.
7. KAMMEL H., Silikattechnik 2 (1951), 227—231.
8. KRELL E., Chem. Techn. 4 (1952), 200—207.
9. Kataloge der Glasgerateindustrie
FRIEDRICHS F., Das Gias im chemischen Laboratorium, 3. Aufl., Springer-Verlag, Berlin/Gattingen/Heidelberg 1960.
TELLE W., ABC der Laboratoriumsgerate, VEB Fachbuchverlag Leipzig 1953.
TELLE W., Chemische Laboratoriumsgerate, 2. Aufl., VEB Deutscher Verlag fiir Grundstoffindustrie, Leipzig 1969.
10. STAGE H., Erdol u. Kohle 3 (1950), 377—383.
11. KRELL E„ Chem. Techn. 4 (1952), 443—450.
12. KINZA V., DDR-Gebrauchsmuster, 10175.
13. WOLF F. W. D., Glas-Instrumenten-Techn. 4 (1960), 43—52, 126—131, 343_____349.
14. HECKMANN C. J., Chem. Techn. 24 (1972) 9, 533—536; 25 (1973) 1, 20—23.
14a. REICHELT W., Chemie-Ing.-Techn. 45 (1973) 1, 25—29.
15. MALEWSKI W., Chemie-Ing.-Techn. 42 (1970) 6, 385—390.
16. SIGWART K-, Destination und Rektifikation in: Ullmanns Encyklopadie der technischen Chemie, Bd. 1, Urban & Schwarzenberg, Munchen/Berlin 1951.
17. RAY J. D„ Rev. sci. 28 (1957), 200—201.
18. GARNEY Th. P., Laboratory Fractional Distillation, MacMillan, New York 1949.
19. THORMANN K-, Dechema-Erfahrungsaustausch, Arbeitsmethoden und Gerate, Destillieren und Rektifizieren, Dechema, Frankfurt/Main, September 1950.
503
20. FENSKE M. R., A. P. 2 037 316 vom 4.4.1936.
21. ROSE A. und E., und MITARB., Technique of Organic Chemistry, Vol. IV Interscience Publishers, New York 1951.
22. KUHN W., Chemie-Ing.-Techn. 25 (1953), 12—18.
KUHN W„ und MITARB., Chimija [Zurich] 8 (1954), 109—122.
23. CRAIG L. C., Ind. Engng. Chem., analyt. Edit. 8 (1936), 219; 9 (1937), 441.
24. KUHN W., Helv. chim. Acta 25 (1942), 252—295.
25. KUHN W„ und RYFFEL K-, Helv. chim. Acta 26 (1943), 1693—1721.
JOST W., Angew. Chem., Teil В 20 (1948), 231.
COENEN A., Dissertation, T. H. Aachen 1941.
MAIR B. J., und WILLINGHAM Ch. B., J. Res. nat. Bur. Standards
22 (1937), 519—527.
26. WEST HAVER C. J., Ind. Engng. Chem. 34 (1942), 126.
27. DONNELL С. K-, und KENNEDY R. M., Ind. Engng. Chem. 42 (1950), 2327______2332
28. JANTZEN E., und WIECKHORST 0., Chemie-Ing.-Techn. 26 (1954), 392______396
29. KOCH R., und FEIND K-, Chemie-Ing.-Techn. 30 (1958), 577—584.
30. BECK F., Chemie-Ing.-Techn. 35 (1963), 837—844.
31. FISCHER W. G., Glas-Instrumenten-Techn. 13 (1969) 5, 535—540; 14 (1970)
8, 933 bis 936; 15 (1971) 7, 753—759; 15 (1971) 8, 832—838.
Chemiker-Ztg. 94 (1970) 5, 157—161.
Fette, Seifen, Anstrichmittel 72 (1970), 444—454.
Fette, Seifen, Anstrichmittel 75 (1973), 131—133.
32. MACURA H., und GROSSE-OETRINGHAUS H., Brennstoff-Chem. 19 (1938), 437—439.
33. MANNING R. E., und CANNON M. R., Ind. Engng. Chem. 49 (1957), 346—349.
34. "HOPPE K., und MITTELSTRASS M., Grundlagen der Dimensionierung
i . von Kolonnenboden (Technische Fortschrittsberichte, Bd. 61), Verlag Theodor iSteinkopff, Dresden 1967.
35. STAGE H., Glas-Instrumenten-Techn. 13 (1969) 12, 1285—1299.
STAGE H., Chemieanlagen und-verfahren (1970), Heft 1/2.
36. KEESOM W. H., und MITARB., Physica 1 (1934), 1109/14.
37. BRUUN J. G„ und SCHICKTANZ sS. T., J. Res. nat. Bur. Standards 7 (1931), 851—882.
38. STAGE F., Angew. Chem., Teil’B 19 (1947), 182.
39. Патент ФРГ № 862598, 1952; ~ KIRSCHBA UM E., Chemie-Ing.-Techn.
25 (1953), 73—80.
40. FRITZ P. W., Glas-Instrumenten-Techn. 11 (1967) 3, 161—162; Патент ФРГ на полезную модель 1957085.
41. OLDERSHAW С. F., Ind. Engng. Chem., analyt. Edit. 13 (1941), 265—268.
42. HILBERATH F., Oel u. Kohle 39/40 (1943), 875—880.
43. WIJK W. R. van, und THISSEN H. A. C„ Chem. Engng. Sci. 3 (1954), 153—160.
44. UMHOLTZ C. L., und WINKLE M. van. Petroleum Refiner 34 [(1955), 114—117.
45. JONES P. D., und WINKLE M. van, Ind. Engng. Chem. 49 (1957), 232—238.
46. Entwickelt in dem Forschungslabor von C. F. BRAM u. Co. Hersteller: the Emil Griner u. Co.
47. McGURE G. V., und'MADDOX R. N., Ind. Engng. Chem. Proc. 6 (1971), 1, 9.
48. WAGNER C. D., LUKENS JR., H. R., und HUGHES R..R., Nucleonics 14 (1956), 78—83.
49. NEWITT D. M., DOMBROWSKI N., und KHELMAN F. H., Trans. Instn. Chem. Engr. 32 (1954), 244—261.
50. МЕЛИКЯН P. А., ЖПХ, 1958, t. 31, № 4, c. 550—558.
51. WIDMER G., Helv. chim. Acta 7 (1924), 194—196.
52. GOLODETZ A., Chem. Industrie (niss.) 35 (1912), 102—108, 141—145.
504
53. LECKY H. S., und EWELL J?. H., Ind. Engng. Chem., analyt. Edit., 12 (1940), 544—547.
54. STALLCUP W. D., und MITARB., Ind. Engng. Chem., analyt. Edit. 14 (1942), 503—547.
55. BOWER JR., J. R., und COOKE L. M., Ind. Engng. Chem. 15 (1943), 290—293.
56. PODBIELNIAK W. J., Ind. Engng. Chem. 13 (1941), 639—645.
57. BREZINA V., Glas-Instrumenten-Techn. 14 (1970) 5, 493—494.
58. STEDMAN D. F., Trans. Amer. Inst. chem. Engr. 33 (1937), 153—161.
59. KOCH H„ und RAAY H., van, Chemie-Ing.-Techn. 8 (1950), 172.
60. BRAGG L. B., Ind. Engng. Chem. 45 (1953), 1676.
61. DRP nach Prospekt Ernst Haage, Miihlheim/Ruhr.
62. ELLIS S. R. M., und VARJAVANDI J., Chem. Process Engng. 39 (1958), 239.
63. JUCHHEIM G„ Glas-Instrumenten-Techn. 10 (1966) 10, 914—917.
64. SPERANDIO A., RICHARD M., und HUBER M., Chemie-Ing.-Techn.
37 (1965), 3, 322 bis 328.
ZOGG M., Chemie-Ing.-Techn. 45 (1973) 2 , 67.
65. ТИМОФЕЕВ А. В., АЭР0В M. E„ Хим. пром., 1971, Ns'5, c. 61—62.
65a. KWASNIAK J., Verfahrenstechnik 8 (1974) 11,315—318. '
66. SHAWORONKOW N. M., und MALJUSOW W.’A., Chem. Techn. 24 (1972) 7, 397—401.
67. ZUlDERWEGiF. T., ChemAEngng. Sci. 1 (1952), 164—174, 184—194.
68. PODBIELNIAK} W. J., Патент Германии № 595250, 1934.
69. BENNER F. C., DINARDO A., und TOBIN D. J., Ind. Engng. Chem. 43 (1951), 722 bis 727.
70. BYRON E. S„ BOWMAN J. R., und COULL J., Ind. Engng. Chem. 43 (1951), 1002—1010.
71. WILLINGHAM С. B., SEDLAK V. A., ROSSINI F. D., und ESTHAVER J. W., Ind. Engng. Chem., Ind. Edit. 39 (1947), 706—712.
72. JOST W., Chemie-Ing.-Techn. 25 (1953), 356.
JOST W., Rektifikation unter extremen Bedingungen, in: Dechema-Monogr., 22 (1954), 30—72.
73. JOST W., SIEG L„ und BRANDT H., Chemie-Ing.-Techn. 25 (1953), 291_____292.
74. ГЕЛЬПЕРИН Н.И.,ХАЦЕНКО M. С., ЖПХ, 1952, т. 25, № 6, с. 610—615.
75. MURRAY К. Е., J. Amer. chem. Soc. 28 (1951), 235.
76. ИРЛИН А. Л., БРУНС Б. П., ЖАХ, 1950, т. 5, № 1, с. 44—47.
77. HOHER G., Untersuchungen uber die Trennfahigkeit einer Labor-Rektifikationskolonne mit rotierender Wendel als Austauschflache, Ingenieur-Arbeit, Ingenieurschule fiir Chemie, Leipzig 1958.
78. NEUMANN F., Chemie-Ing.-Techn. 33 (1961) 7, 485—491.
78a. МАМИН В. H. u др. Химико-фармацевтический журнал, 1974, т. 8, № 3, с. 51—55.
79. TOELDTE W., Seifen—Ole—Fette—Wachse 74 (1948), 126.
80. N. N., Glas-Instrumenten-Techn. 10 (1966) 4, 259/260.
81. KRAMER M. T., Glas-Instrumenten-Techn. 15 (1971) 8, 856—860.
82. BREZINA V., Glas-Instrumenten-Techn. 10 (1966), 3, 162—172.
83. FRIEDRICHS F., Z. angew. Chem. 33 (1920), 29—32.
84. FRIEDRICHS J., und v. KRUSKA H., Chem. Fabrik 7 (1934), 284—287.
85. MACH F„ und HERRMANN R., Chem. Fabrik 6 (1933), 211—213.
86. EICHHORN E., Untersuchung der Wirkung von Laboratoriumskiihlern, Ihgenieur—Hauptarbeit, Ilmenau 1955.
87. GEMMEKER L., und STAGE H., Glas-Instrumenten-Techn. 6 (1962) 3, 69—78; 4, 114 bis 119; 5, 169—174; 6, 206—208; 9, 327—328; 10, 372—376; 12, 494—497.
88. KRELL E„ Chem. Techn. 5 (1953), 581—587.
89. SCHNEIDER K. W., und SCHMID Ch., Erdol u. Kohle 2 (1949), 508—510.
89a. KLOSS H.—J., CZ-Chem.-Techn. 3 (1974), 89—93.
505
90. KRAMER M. T., Glas-Instrumenten-Techn. 16 (1972) 4, 445—448; 5, 571—574; 8, 943—946.
91. Prospekt Normag, Hofheim/Ts., Liste 10; Chemie-Ing.-Techn. 26 (1954), 231.
92. SCHULZ E. 0., Glasinstrumentenkunde, Wagner u. Sohn. Weimar 1937.
93. HUBNER G., Chemiker-Ztg. 83 (1959), 581—583.
94. LLOYD L. E„ und HORNBACHER H. G., Analytic. Chem. 19 (1947), 120______122. ' '
95. COLLINS F. C., und LANTZ V., Ind. Engng. Chem., analyt. Edit. 18 (1946), 673—677.
96. K1ESELBACH K-, Analytic. Chem. 19 (1947), 815.
97. STAGE H., und GEMMEKER L., Glas-Instrumenten-Techn. 6 (1962) 12, 494—497.
98. N. N., Glas-Instrumenten-Techn. 10 (1966) 3, 178—179.
99. STAGE H., und GEMMEKER L., Glas-Instrumenten-Techn. 7 (1963)
11, 608—614; 12, 687—704; 8 (1964) 6, 413—428; 7 , 503—508.
STAGE H., Glas-Instrumenten-Techn. 14 (1970) 3, 213—220; 4, 337—349.
100. Hersteller: Glaswerk Schott und Gen., Mainz.
101. ROCK E. J., und JANZ G. F., Analytic. Chem. 22 (1950), 626.
102. GRASSMANN W., und DEFFNER C., Chemiker-Ztg. 76 (1952), 623.
103. SCHRAM E., Analytic. Chem. 25 (1953), 1424.
104. STAGE H., und GEMMEKER L., Glas-Instrumenten-Techn. 7 (1963)
11, 608—614; 12, 687—704 8 (1964) 6 , 413—428; 7, 503—508.
STAGE H., Glas-Instrumenten-Techn. 14 (1970) 3, 213—220; 4, 337—349.
105. HUBNER G„ Chemiker-Ztg. 76 (1952), 67.
106. Hersteller: Edmund Buhler, Tubingen.
107. Hersteller: Dr. Hofmann und Roth, Stuttgart-Hdedelfingen; s. auch KRELL E., Chem. Techn. 7 (1955), 599—608.
108. Hersteller: W. C. Heraeus GmbH, Hanau.
109. BORCHERT R., und JUB1TZ W., Infrarotstrahler zur Erwarmung, Trocknung und Ausbartung, VEB Verlag Technik, Berlin 1951.
110. KLEES K. F., Chem. Rdsch. [Solothurn] 7 (1954), 358—359.
111. SCHULZE H., Chemie-Ing.-Techn. 23 (1951), 565—569.
112. WEBER U. von, Chem. Techn. 1 (1949), 159—160.
113. ZAMPACHOVA L., Chem. Prumysl (tschech.) 7/32 (1957), 408—413.
114. K0LL1NG H., und TRAMM H., Chemie-Ing.-Techn. 21 (1949), 9—14.
115. Hersteller: Bronwill Scientific Ind., New York.
116. KORTUM G., und B1TTEL A., Chemie-Ing.-Techn. 28 (1952), 40—44.
117. ROSE A. und E., und MITARB., Technique of Organic Chemistry, Vol, IV, Interscience Publisners, New York 1951.
118. Hersteller: W. C. Heraeus GmbH, Hanau, Elektrothermal Engineering Ltd. London und VEB Asbestdraht, Berlin.
118a. SELLIN J., Chemie—anlagen + verfahren 4 (1975), 87—91.
119. Alleinvertrieb fiir «Calorex»: Heraeus—Wittmann GmbH, Heidelberg.
120. LEVA M., Tower Packings and Packed Tower Design, 2. Aufl., The United States Stoneware Company, Akron/Ohio 1953.
121. KERENYI E„ und SCHULTHE1SZ I., Proc, and Conf, on Appl. Phys.
Chem., Veszprem, Aug. 2—5 1971, Akademiai Kiado, Bp 1971, Vol. 2, S. 67—74.
121a. REICHELT W., Chemie-Ing.-Techn. 45 (1973) 2, 53—57.
122. Hersteller: Podbielniak Inc., Chicago 11.
123. BRAUN R., Chemie-Ing.-Techn. 33 (1961), 350—352.
124. DIXON O. G., J. Soc. chem. Ind. 68 (1949), 88, 119, 299.
125. HUBER M.,und SPERANDIO A., Chem.-Ing.-Techn. 36 (1964) 3,221—227.
126. ELLIS S. R. M., und VARJAVAND1 J., Chem. Process Engng. 7 (1958), 239—243.
127. Unveroffentlichte Mefiergebnisse des Institutes fiir Apparatebau und Verfahrenstechnik^der TH Karlsruhe (pers. Mitteihing).
128. MacMAHONl H. 0., Ind. Engng. Chem., ind. Edit. 39 (1947), 712—718.
129. ’Hersteller:;The United States Stoneware Company, Akron/Ohio.
130. Herstellen^Hydronil Ltd., Chemical Plant Division, Fenton/England.
506
131. KIRSCHBAUM E„ Z. Ver. Dtsch. Ing. 98 (1956), 1801—1802.
ALT C., MEIER A., und RICHTER G., Chemie-Ing.-Techn., 27 (1955), 502—506.
132. Hersteller: Vereinigte Fiillkorperfabrik GmbH u. Co., Baumbach/Westerwald.
133. FUCHS 0., Chemie-Ing.-Techn. 23 (1951), 537—540.
134. FORSYTHE W. L., und MITARB., Ind. Engng. Chem. 39 (1947), 714.
135. WOLF F., und GUNTHER W., Dechema-Monogr. 29 (1957), 364—377.
136. KRELL E., Chem. Techn. 16 (1964) 10, 591—600.
137. KRELL E., Plaste und Kautschuk 14 (1967) 4, 251—254.
138. TELLER A. J., Chem. Engng. Progr. 50 (1954) 2, 65—71; Ind. Engng. Chem.
50 (1958), 8, 1201—1206.
139. N. N., Chem. Engng. 80 (1973) 19, 88.
140. EGBERONGBE S. A., Process Engng. (1973) 2, 82—83, 85.
141. STURMANN H., Leistungssteigerung von Fiillkorperkolonnen durch neue Wandformen, Dissertation, Hamburg 1937.
142. KRELL E. Патент ГДР № 63761, 1968. «Kolonne mit verminderter Randgangigkeit».
8. ЛИТЕРАТУРА
1. ROLFSON F. B„ und MITARB., Analytic. Chem. 20 (1948), 1014—1019.
2. BAYERL V., und QUARG M., Taschenbuch des Chemietechnologen, 3. Aufl., VEB Deutscher Verlag fiir Grundstoffindustrie, Leipzig 1968.
3. KAHTEPMAH Л.Б., ДУХАН Б.С., ИВАНОВ 17. Г. Химия и технология топлив и масел. 1958, т. 3, № 5, с. 57—60.
4. SALZER F„ Chemie-Ing.-Techn. 31 (1959), 405—409.
5. J1RMANN R„ Glas-Instrumenten-Techn. 14 (1970) 1, 11—12.
6. STAGE H., CZ-Chemie-Techn. 1 (1972), 263—272.
7. ZAPPE D., HECKMANN M., GUTWASSER H., und DORNBUSCH E., Heckmann-Mitt. Nr. 5 (1968).
8. ULUSOY E., Chemiker-Ztg. 79 (1955), 46—48.
9. STAGE H., GEMMEKER L., FISCHER G., und RICHTER R., Glas-Instrumenten-Techn. 5 (1961), 138—145 und 204—211.
10. FISCHER W. G., und WEYAND H., Glas-Instrumenten-Techn. 14 (1970) 1, 23—26.
11. KRELL E., Chem. Techn. 9 (1957), 266—274.
12. KORTUM G., und BITTEL A., Chemie-Ing.-Techn. 28 (1956), 40—44.
12a. KOUKOL H., KUPKA H. J., und HENNING G., GIT Fachz. Lab. 19 (1975), Sonderheft Labormedizin, 493—497.
13. BRANDT H., und ROCK H., Chemie-Ing.-Techn. 25 (1953), 511—514. BRANDT H., ROCK H., und LANGERS F., Chemie-Ing.-Techn. 29 (1957), 86—91.
14. ABEGG H., Chimia [Zurich] 6 (1952), 258—267.
15. LAPORTE H., Messung, Erzeugung und Konstanthaltung hoher bis tiefer Temperaturen, VEB Fachbuchverlag Leipzig 1961.
16. K'OSSLER I., und VODEHNAL J., Chem. Prumysl (tschech.) 7 (32) (1957), 292______297.
17. KADLEC M., Chem. Listy (tschech.) 51 (1957), 987—990.
17a. STAGE H., CZ-Chemie-Technik 5 (1973), 203—208.
18. VANVOR H., Glas-Instrumenten-Techn. 8 (1964), 415.
19. TELLE W., Chemische Laboratoriumsgerate, 2. Aufl., VEB Fachbuchverlag, Leipzig 1969.
20. PIATTI L., und MARTI W., Chemie-Ing.-Techn. 28 (1956), 473—475.
21. Hersteller: Siemens AG, Erlangen — Dr. Neumann GmbH, Mtinchen 80.
22. LOFFLER H. J., und HENRICI H., Chemie-Ing.-Techn. 30 (1958), 718. SOBLE A. B., Chem. Engng. Progr. 56 (1957), 444—451.
23. Hersteller: VEB Thermometerwerk, Geraberg.
507
24. Hersteller: Juchheim KG, Solingen.
25. KLEINTEICH R., Glas-Instrumenten-Techn. 3 (1959), 89—93.
26. WALTER M., Relaisbuch, Franckh’sche Verlagshandlung, Stuttgart 1951.
27. FISCHER W., Glas-Instrumenten-Techn. 14 (1970), 5, 513—516.
28. THORMANN K-, Dechema-Erfahrungsaustausch, Arbeitsmethoden und Gerate, Destillieren und Rektifizieren, Dechema, Frankfurt/Main, September 1950.
29. HUTLA V., Chem. Listy (tschech.) 51 (1957), 1964—1965.
30. JAECKEL R., Dechema—Erfahrungsaustausch, Vakuumtechnik, Dechema, Frankfurt/Main 1956.
31. LECK J- H., Pressure Measurement in Vakuum Systems, The Institute of Physics, London 1957.
32. NICKEL E., Gas- und Wasserfach, Beilage Bau und Betrieb 6 (1954), Nr. 5 (Ausg. Gas).
33. FARQUARSON J., und KERMICLE H. A., Rev. sci. Instr. 28 (1957), 324 325
34. DOSCH R., Glas-Instrumenten-Techn. 4 (1960), 9—17.
35. HOLLAND-MERTEN E. L., Tabellenbuch der Vakuumverfahrenstechnik in der Grundstoffindustrie, VEB Deutscher Verlag fiir Grundstoffindustrie, Leipzig 1964.
36. HOHLSCHUTTER H. W., und KAMPF G„ Angew. Chem. 66 (1956), 355.
36a. Hersteller: Airflor—Lufttechnik GmbH, Rheinbach.
37. CROMPTON R. W., und ELFORD M. T., J. sci. Instruments 34 (1957), 405—407.
38. STORZBACH W., Glas-Instrumenten-Techn. 14 (1970) 1, 36—37.
39. KRELL E., Chem. Techn. 3 (1951), 333—336.
40. GILMONT R., Ind. Engng. Chem., analyt. Edit. 18 (1946), 633—636.
41. Hersteller: Dr. Friedrichs — Dr. Matschke KG Normschliff-Glasgerate, Wertheim/Main.
42. YOUNG C. G., und MITARB., Canad. J. Technol. 29 (1951), 447—450.
43. KRELL E., Chem. Techn. 10 (1958), 327.
44. COULSON E.A.,und WARNE R. J., J. sci. Instruments 21 (1944), 122—123.
45. MONCH G. C-, Hochvakuumtechnik, Rudolf A. Lang Verlag, PofJneck 1950; Neues und Bewahrtes aus der Hochvakuumtechnik, VEB Wilhelm Knapp Verlag, Halle 1959.
46. HOCH H., Vakuum-Techn. 16 (1966) 1/2, 8—13.
47. STECKELMACHER W., J. sci. Instruments 1 (1951), 10—19.
48. GAEDE W., Z. techn. Physik 15 (1934), 664.
49. JAECKEL R., Kleinste Driicke, ihre Messung und Erzeugung, in: Technische Physik in Einzeldarstellungen, Springer-Verlag, Berlin/Gottingen/Heidelberg 1950.
LAPORTE H., Hochvakuum, seine Erzeugung, Messung und Anwendung im Laboratorium, VEB Wilhelm Knapp Verlag, Halle 1951;
Vakuummessungen, VEB Verlag Technik, Berlin 1955.
Y ARWOOD J., Hochvakuumtechnik, Rudolf A. Lang Verlag, Berlin 1955. • LEYBOLD, Vakuum-Taschenbuch, hrsg. von K. Diels und R. Jaeckel, 2. Aufl., Springer-Verlag, Berlin/Gottingen/Heidelberg 1962.
FRANK W., und KUTSCHE D., Die schonende Destination, Otto Krausskopf-Verlag GmbH, Mainz 1969. .,
50. MULLER B., Glas-Instrumenten-Techn. 14 (1970) 7 , 798—807.
51. EBERT H., Kompressionsvakunmmeter, Friedr. Vieweg-J- Sohn GmbH, Braunschweig 1951.
52. PECHE G., Glas-Instrumenten-Techn. 7 (1963) 1, 4—7; 2, 48—49.
53. Hersteller: Rudolf Brand, Wertheim/M., Glashiitte.
54. NISBET J. S., J. sci. Instruments 26 (1949), 271—273.
55. MELPOLDER F. W., Ind. Eng. Chem. 39 (1957), 617.
56. FISCHER H. E„ Analytic. Chem. 20 (1948), 982.
57. GEMMEKER L., und STAGE H., Glas-Instrumenten-Techn. 6 (1962) 12, ; 494—497 ; 7 (1963) 1, 18—23.
508
58. STAGE H., Chemie-Ing.-Techn. 22 (1950), 374—375.
STAGE H., GIT Fachz. Lab. 19 (1975) 3, 182.
59. HOUBEN-WEYL, Methoden der organischen Chemie, hrsg. von E. Miiller, Physikalische Methoden, Teil 1 und 2, Georg Thieme Verlag, Stutgart 1955.
60. MULLER R. H., und ZENCHELSKY S. T., Analytic Chem. 24 (1952), 844—847.
61. THOMAS G. R., und MITARB., Analytic. Chem. 22 (1950), 1221—1223.
62. LATCHUM J. W., Патент США № 2529030, 1950.
63. NEBE W., RIEGLER H., und MENZEL W., Jenaer Rundschau 18 (1973), Messe—Sonderheft 87—89.
64. KEGELES G., und SOBER H. A., Analytic. Chem. 24 (1952), 654—660.
65. OEHME F., Chemische Analyse durch Messung von Dielektrizitats-Konstanten, Verlag VEB Laborchemie, Apolda 1953.
OEHME F., Dielektrische MefJmethoden, 2. Aufl., Verlag Chemie GmbH, Weinheim/Bergstr, 1962.
66. SLEVOGT К. E., Dechema-Monogr. 14 (1950), 97—120; 17 (1951), 95—107.
67. GRANT R. A., J. appl. Chem. 8 (1958), 136; Chem.-Ing.-Techn. 30 (1958), 738.
68. FELLONI L„ und TANTILLO E., Ann. Chimica 47 (1957), 751—758. 69. CLASEN H., Chemie-Ing.-Techn. 28 (1956), 45-48; 29 (1957), 110—111. 70. LINFORD A., Ind. Chemist. 34 (1958) 403, 481—488.
71. FRITSCHE R. W., Petroleum Processing 7 (1952), 1138—1143.
72. KOCH 0., Chemie-Ing.-Techn. 30 (1958), 181—187.
KIENITZ H., Chemie-Ing.-Techn. 32 (1960) 10, 641—650.
73. SIGGIA S., Continons Analysis of Chemical Process Systems, John Wiley &Sons, London 1959.
74. Chemie-Ing.-Techn. 36 (1964) 9 A, 1455—1457.
75. HERFORTH L.,und KOCH H., Praktikum der angewandten Radioaktivitat, 2. Aufl., VEB Deutscher Verlag der Wissenschaften, Berlin 1972.
76. BERN HAU ER K-, Einfiihrung in die organisch—chemische Laboratoriumstechnik, Springer-Verlag, Wien 1947.
77. WITTENBERGER W., Chemische Laboratoriumstechnik, 6. Aufl., Springer-Verlag, Berlin/Gottingen/Heidelberg 1963.
78. MULLER G., und GNAUCK G., Reinste Gase, VEB Deutscher Verlag der Wissenschaften, Berlin 1965.
79. HINZPETER G„ Vakuum-Techn. 11 (1962), 2.
80. Hersteller: Fischer, Labor- u. Verfahrenstechnik, Bonn-Bad Godesberg.
81. DAMMER H. J., Glas-Instrumenten-Techn. 15 (1971) 2, 115—116.
82. HANEL R., Chem. Techn. 15 (1963) 3, 176—177.
83. PINKAVA J., und WICHTERLE 0., Collect, czechoslov. chem. Commun. 20 (1955), 697 bis 707.
PINKAVA J., und KALAB V., Collect, czechoslov. chem. Commun. 20 (1955), 707—712.
84. PINKAVA J., Laboratoriumstechnik kontinuierlicher chemischer Prozesse, Nakladatelstvi technicke Literatury (SNTL), Prag 1958.
85. SCHMIDT W., Chem. Techn. 10 (1958), 131—134.
86. HEUSCH R., Fette, Seifen, Anstrichmittel 72 (1970), 11, 969—977.
87. POHLENZ W., Pumpen fiir Flfissigkeiten, VEB Verlag Technik, Berlin 1970.
88. LI EDEL К. H., Gias in der Chemie — Laborgerate, GrofJapparate, Rohrleitungen, Otto Krausskopf-Verlag GmbH, Mainz 1973.
9. ЛИТЕРАТУРА
1. BEHRE A., Chemisch-physiktalische Laboratorien und ihre neuzeitlichen Einrichtungen, Akademische Verlagsgesellschaft Geest & Portig,
’ Leipzig 1950.
2. COLEMANN H. S., und MITARB., Laboratory Design., Reinhold, New York 1952.
3. SCHRAMM W., Chemische und biologische Laboratorien, 3. Aufl., Verlag Chemie GmbH, Weinheim/Bergstr. 1969.
509
' 4. KINCANNON С. В., und BAKER M. 0., Analytic. Chem. 29 (1957), 1189—1193.
5. SCHWARZ H., Stative, Aufhangung von Apparaturen, Bn: Houben-Weyl, Methoden der organischen Chemie (hrsg. von E. Miiller), Bd. 1/1, Georg Thieme Verlag, Stuttgart 1958.
6. Hersteller: Ernst Haage, Miilheim-Ruhr.
7. Hersteller: Geru, Geroldwill/Schweiz.
8. BROOKE M„ J. Chem. Educat. 28 (1951), 602—603.
9. WAGNER G., Osterr. Chemiker-Ztg. 43 (1940), 229.
10. BERN HAUER K-, Einftihrung in die organised—chemische Laboratoriumstechnik, Springer-Verlag, Wien 1947.
11. WITTENBERGER W., Chemische Laboratoriumstechnik, 7. Aufl., Springer-Verlag, Berlin/Heidelberg/New York 1973.
12. WOLF F. W. D., Glas-Instrumenten-Techn. 4 (1960), 34—52, 126—131.
13. FRIEDRICHS F., Das Gias im chemischen Laboratorium, 3. Aufl., Springer-Verlag, Berlin/Gottingen/Heidelberg 1960.
14. BRAUER H., Chemie-Ing.-Techn. 29 (1957), 520—530.
15. GREINACHER H., Ausgewahlte Aufgaben und Probleme aus der Experimentalphysik (§ 28: Der Siedeverzug), Springer-Verlag, Wien 1953.
16. Патенты США № 2580880, 1952 ; 2580922, 1952.
17. HUBNER G., Chemiker-Ztg. 75 (1951), 229—230.
18. The General Safety Committee of the Manufacturing Chemist’s Association. Guide for Safety in the Chemical Laboratory, Van Nostrand, New York 1954.
19. DITTMAR P., Erdol u. Kohle 11 (1958), 25—30.
20. CRAIG L. E., und DEW J. E., Ind. Engng. Chem. 51 (1959), 1249—1252.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Азеотроп гетерогенный (гетероазеотроп) 85, 303, 306
гомогенный (гомоазеотроп) 85, 303, 306
определение состава 311 сл.
отрицательный 52, 85, 299 сл., 307, 309
положительный 52, 85, 299 сл., 307, 309
Азеотропии 300 сл.
Азеотропная смесь 299 точка 76
Анероид 439
Аргон, обогащение методом ректификации 222 сл.
Бани 398
для высокотемпературной ректификации 258
Бар 33
Баровакуумметры 440 сл.
Барометры 439
корректировка показаний 181 сл. Бензол, давление паров при различных температурах 78 Бидистиллятор воды 217 сл.
Бора фторид, концентрирование методом ректификации 222 сл.
Бюретка 328
Вакуум
измерение 267, 440 сл., 446 сл.
получение 267
регулирование 443 сл., 448, 451 сл. Вакуумметры 441 сл., 447, 451 сл. Вакуумные коммуникации, определение размеров 265 сл.
Вакуускопы 447, 449
Вентиль газовый проходной 214 Вода — фенол, зависимость состава азеотропа от давления 306
Время разгонки 127
пусковое 127
Высота единицы переноса (ВЕП) 99, 124
Высота, эквивалентная теоретической ступени (ВЭТС) 96 сл., 124, 130, 141
зависимость от диаметра колонны 137
----рабочей высоты колонны 138 сл.
колонн безнасадочиых 336
— с вращающимися контактными устройствами 363
колонн насадочных 136, 162 сл.
— с неподвижными контактными устройствами 354 сл.
— для полумикроректификации 200
насадок 120, 130 сл., 412, 414
Вытяжной шкаф 471
Вязкость, определение 462
Газовая смесь
давление, общее и парциальное 72 сл., 77 сл., 81 сл., 295 сл.
разделение ректификацией 250, 262, 292
Герметичность
кранов и шлифовых соединений 31 сл., 268, 477 сл.
установок 268 сл., 480
Гидравлическое сопротивление колонны 152, 164 сл. — насадочной 368 — тарельчатой 345, 349 — трубчатой щелевой 341 насадки 166 сл.
Головки ректификационных колони
206, 248, 378—386
азеотропные 320 дистилляционные 24 для парциальной конденсации 248
Градус Кельвина 33
Давление 33
газовой (паровой) смеси
----общее и парциальное
72 сл., 77сл., 81 сл., 295сл.
512
измерение и регулирование 433 сл., 438 сл., 443 сл.
насыщенных паров, определение 54 сл., 60 сл.
остаточное, измерение и регулирование 267, 440 сл., 443 сл.,
446 сл„ 448, 451 сл.
рабочее при перегонке 53 сл.,
151 сл., 262, 438
Дефлегматоры 26, 245 сл., 369
Дефлегмации 23, 245
Джоуль 33
Диаграмма
равновесия пар—жидкость 51 сл., см. также Кривая равновесия пар — жидкость разгонки 185 сл. — бензиновой фракции 180 — смеси жирных кислот 184, 244, 270, 275
--------- растворителей 206 Дистиллят 38
определение физико-химических свойств 179 сл., 458 сл.
отбор 206, 329 сл., 390 сл., 453 — при низкотемпературной ректификации 250, 253
Дистилляторы воды 216 сл. для молекулярной дистилляции 283 сл.
пленочный Креля 273 сл.
Дистилляционная способность 290 Дистилляционные
приборы, с колбой Кляйзена 326 сл.
-----Энглера 326 установки 25, 328 сл. — автоматизированные 419, 422 — для молекулирной дистилляции 286 сл.
— для перегонки веществ с высокой температурой плавления 259 — с пленочным испарителем 277сл.
— с трубчатой колонной 241 Дистилляция 19 сл., 39 сл., 175 сл., 327 сл.
вакуумная 263—269
с восходящим или нисходящим потоком паров 21
молекулярная 262 сл., 280—292, 438
«на коротком пути» 284 ртути 260 сл.
тепловой баланс 175 сл.
по Энглеру 40, 207, 326
Дозирование газов 463 сл. жидкостей 463 сл.
Жидкость
взаимная растворимость 50 измерение расхода 463 сл. Жирные кислоты
разделение непрерывной ректификацией 133 сл., 243 сл.
— парциальной конденсацией 248 сл.
— перегонкой с водяным паром 296 сл.
— пленочной дистилляцией 275 — расширительной перегонкой 270 сл.
Закон
Дальтона 72
Рауля 77, 81, 308
«Захват» жидкости 149
Захлебывание колонны 158 сл.
Идеальная смесь
летучесть 81 — относительная 84 сл. построение кривой равновесия 77 сл., 86 сл.
Изотенископ 58 сл.
Изотопы, разделение методом ректификации 219 сл.
Испарение 70
скорость 280 сл.
пленочное 272 сл.
Испарители для молекулирной дистилляции 286
для пленочной перегонки 273 сл. циркуляционные 211 сл., 400
513
Каналообразование 43, 119 сл.
Капельницы 380, 465
Кипение
бинарных смесей 52
способы стабилизации 330, 481
температура, определение 55 сл., 61 сл., 182 сл., 295 сл.
Кипятильники погружные 396
Клапаны 334 сл.
Колбы для перегонки 26, 387 сл.
Кляйзена 26, 326 сл.
Кольца Рашига 27, 120, 144, 151, 409
Конденсаторы 369
Конденсация 22 сл., 369
при высокотемпературной ректификации 258 сл.
краевая 245
парциальная 245 сл.
Концентрация 34 сл.
Коэффициент
активности 84 сл.
использования насадочной колонны 44
неравномерности орошения 44
обогащения 112 сл.
полезного действия колонны 148, 349
-----тарелки 136, 161
разделения 112
теплопередачи для холодильников 373 сл.
Краны 332 сл.
вакуумные 267
герметизация 478
для работы под давлением 292 регулирующие 381 сл.
Кривая
«выделении» 290 сл.
давления паров 53, 74
----- построение 63 сл.
равновесия пар—жидкость 74 сл.
------- дифференциальная 109
-------построение
---------графический способ 79 ---------- расчет 76 сл.
--------- экспериментальное определение 86 сл.
------- смеси бензол—гептан
109
Кривая
--------- бензол—толуол 98
---------жирных кислот 105
---------ж-крезол—и-крезол
109
---------толуол—н-октан 109
разгонки 93
Куб 387 сл., 481
загрузка 480
обогрев 22, 394 сл., 455 сл.
— при высокотемпературной ректификации 258
для периодической разгонки 209
Лаборатория для перегонки, планировка помещения 469 сл.
Летучесть 81
относительная 81 сл., 115
Манометры 439 сл., 447 сл., 456 сл.
Мак-Леода 442, 447 сл.
Массовые
доля 34
проценты 34 сл.
Массообмен 38
в насадочной колонне 99, 119
в тарельчатой колонне 99, 119 фактор интенсивности 127 сл.
«Мертвый» объем 205 сл.
Металлы, очистка высокотемпературной ректификацией 259
Мнкродистилляция 196 сл.
молекулярная 199 сл.
Мнкроперегонка 195 сл.
вакуумная 198
Микроректификация 196 сл.
Многокомпонентная смесь 131 сл.
расширительная перегонка 271 ректификация 131 сл., 239
— аналитическая 203
— расчет процесса на ЭВМ 193
средняя масса моля 34
Молекулярная дистилляция 262 сл.,
280 сл., 438
микродистилляции 199 сл.
514
Мольные
доля 34 сл.
проценты 34 сл.
Муфты 331
Нагревание исходной смеси н кубовой жидкости 398 сл.
при мнкроперегонке 201
Нагревательные банн 258, 398 приборы электрические 393 сл. элементы 403 сл., 406
Нагрузка ректификационных колонн 153 сл., 164
расчет для периодической ректификации 188
регулирование 456 сл.
Насадка 120, 165, 352 сл., 407—415 влияние материала на эффективность разделения 415 гидравлическое сопротивление 166 сл.
доля активной и смоченной поверхности 46
очистка 155, 480 пакетная 42, 358 пленочная 359 пленочное течение жидкости 48 сл.
для полумикроперегонкн 200 распределение жидкости 41 сл. регулярная 42, 48 смачивание 48, 121, 415 Стедмана 355 сл.
укладка 139, 155, 343 сл. «Хэли-грид» 170, 250, 354 сл. Насосы
вакуумные 267 сл. дозировочные 213 сл., 466 сл. установка в помещении 469 Номограмма
для корректировки показаний барометра 181 сл.
----термометра 182 сл.
для определения размеров вакуумных коммуникаций 265 сл.
— скорости паров для насадок 172
Номограмма
— числа теоретических ступеней разделения 116, 256 сл.
для пересчета концентраций 35 сл.
Ньютон 33
Обогрев
кожуха колонны 403 сл.
куба 22, 394 сл., 455 сл.
при высокотемпературной ректификации 258
Объем паров, расчет 71
Объемные
доля 34
проценты 34 сл.
Орошающая жидкость 148 сл.
Отбор дистиллята 329 сл., 390, 453 при аналитической ректификации 206
при низкотемпературной ректификации 250, 253
Относительная летучесть 81 сл.
для бинарной смеси 115
Параметр разделения 226 сл.
Парциальная конденсация 245 сл. Паскаль 33
Перегонка 19 сл., 32, 38
азеотропная 299 сл., 304—314, 320
вакуумная 263—269, 482
взрывоопасных веществ 482
с водяным паром 51, 294 ' сл. высокотемпературная 257—262 давление рабочее 53 сл., 151 сл., 262, 438
изотермическая 249, 262
наработочная 207 сл., 216 сл. непрерывная 40, см. также Ректификация непрерывная
в парах носителя 51, 294—299 периодическая 40, 234 сл.
пленочная 262, 272—280, 438
полунепрерывная 235 препаративная 207 сл.
простая 38 сл., 195, 234
515
расчет процесса на ЭВМ 190 сл. расширительная 262, 269—271, 438 солевая 321—323 стандартизация методов 28 сл. тепловой баланс 175 сл.
токсичных веществ 469 сл., 482 фракционная 195
экстрактивная 299, 303, 314—321 Перепад давления
в вакуумном трубопроводе 265 в колонне 152, 164, 341, 364, см.
также Гидравлическое сопроти-леиие колонны
Переходы 331
Пилотная установка 11, 208, 211—216 Плотность
жидкости, определение 462 паров, расчет 71 сл.
Показатель преломления, определение 180, 459 сл.
Полумикроперегонка 195—202 Полумикроректификация, см. Полуми-кроперегонка
установки 342 сл., 422
Полюсное расстояние 95
Правило Дюринга 61
Приемники дистиллята 387, 390 сл.
Приставки для дистилляционных аппаратов 328 сл., 376—378
Пристеночный эффект 43, 137 Пробоотборники 156
Рабочая задержка 149
Разгонка 40
.. время 127
фракционная 234
Разделяющая способность ректификационной колонны 93, 137, 338 сл.
зависимость от нагрузки и рабочего давления 153
связь с гидравлическим сопротивлением 166 сл.
Разделяющий агент 301, 304 для азеотропной перегонки 304, 309 сл.
для экстрактивной перегонки 314 сл.
Разъемные соединения 30, 32
516
Растворы, классификация 53
Расходомеры 463
Регуляторы давления 443 сл., 451 сл.
Ректификатор-автомат 420 сл.
Ректификационная колонна 25 сл., 335 сл.
безнасадочная 335—343
с вращающей лентой 200, 203, 360, 362, 364 сл.
— спиралью 361
с вращающимися и неподвижными пластинами 360 сл.
— контактными устройствами 359—369
— щетками 277
с вращающимся конденсатором
360 сл., 363 сл.
— цилиндром 360, 363 сл.
высота рабочая 138 сл., 171, 189 гидравлическое сопротивление
152, 164 сл., 341, 345, 349, 368
диаметр 164, 137 сл., 189 динамические характеристики
49 сл.
с елочным дефлегматором 336 сл.
захлебывание 158 сл.
испытание 136, 145, 155 сл.,
189 сл.
исчерпывающая часть 103
с колпачковыми тарелками 26, 42, 210 сл., 241, 345 сл.
с кольцевой щелью, см. трубчатая щелевая
многотрубчатая 338 сл.
для молекулярной дистилляции
283 сл.
нагрузка 153 сл., 164, 188, 456 сл.
с насадкой Стендмана 353 , 356 насадочная 42, 99, 118 сл., 162 сл., 344—345
— для низкотемпературной ректификации 250 сл.
с неподвижными контактными устройствами 352—359
для низкотемпературной ректификации 252 сл.
с орошаемой насадкой 42
Ректификационная колонна
с орошаемыми пластинами 353, 356 сл.
с перемешивающейся пленкой 275 сл.
перепад давления 152, 164, 168, 341, 364, см. также гидравлическое сопротивление
пленочные 42, см. также Испарители для пленочной перегонки с проволочной спиралью 353 сл. разделяющая способность 93, 137,
153, 166 сл., 338 сл.
с регулярной насадкой 42, 352 роторная 278, см. также с вращающимися контактными устройствами
роторно-спиральная 361
с ситчатыми тарелками 42, 348 сл.
«со смоченными стенками» 353
со спиралью из металлической сетки 353 сл.
со стеклянной спиралью 353 тарельчатая 26, 42, 99, 150, 161, 164, 209 сл., 240 сл., 345—352 тарельчато-насадочная 240, 335
> тепловая изоляция 177 сл., 401 сл. трубчатая змеевиковая 337 сл. — щелевая 200, 204, 338, 340 сл., 365
удерживающая способность 127, 149 сл., 161, 200, 335, 354 сл., 363
с фонтанирующими тарелками 346 Ректификационные установки
автоматизированные 417 сл., 422 сл., 342
для азеотропной ректификации 302 для аналитической ректификации 205 сл.
вакуумные
— непрерывного действия 25, 104, 214, 236
— полунепрерывного действия 235 герметизация 477—479 конструкционные материалы
324 сл.
Ректификационные установки непрерывного действия 21, 25, 104, 214, 236, 240—243
для низкотемпературной ректификации 223, 251 сл., 423
пилотные 11, 208, 211—216 для полумикроректификации 342 сл.
полупромышленные 208—211, 215 для получения тяжелой воды
229 сл.
для ректификации под давлением 292 сл.
— с перегретым водяным паром 298
сборка 330 сл., 474 сл. с трубчатой колонной 241 эксплуатация 244 сл., 480 сл.' для экстрактивной ректификации 303
Ректификация 32, 38 сл.
азеотропная 51, 299 сл., см. также
Перегонка азеотропная азеотропноэкстрактивная 299 аналитическая 202—207 вакуумная 263—269 высокотемпературная 249, 258 сл. газов 250, 262, 292 под давлением 250,292 сл., 438сл. изотопов 219 сл.
непрерывная 103 сл., 133, 235— 245, 398 сл.
низкотемпературная 249, 250— 256, 422 сл. .
— разделение йзотопов 222 Сл. «обменная» 221
периодическая — расчет процесса разделения 186 сл., 239 сл.
--- числа теоретических ступеней . разделения 100 сл., 111 сл., 186
пленочная 438 по принципу «падающей пленки» 286 сл.
пробная 207 расчет процесса на ЭВМ 190 сл* тепловой баланс 175 сл.. ,
517
«термическая» 365 циклический метод 240 экстрактивная 299, 303, см. также Перегонка экстрактивная
Реле времени 453 сл.
Реометры 463
Рефрактометр 459 сл.
Ротаметры 463 сл.
Ртуть, дистилляция 260 сл.
Сборники дистиллята 328 сл., 387, 390 сл.
Скорость паров в колонне 154 сл., 164 сл., 172, 343 константа 170 сл.
Смазки жировые 478 сл.
Среднее относительное обогащение 136
Стандартизация единиц измерения 33 методов перегонки 31 термометров 429 физических величин 33 шлифов 28 сл.
Стекло как конструкционный материал 211, 324 сл.
Стенды для ректификационной аппаратуры 472 сл.
Степень перемешивания 44 сл.
Тарелка ректификационной колонны 39 сл.
барботажная 345
колпачковая 345, 348
ситчатая 345, 348
теоретическая 95 сл.
фонтанирующая 345
Температура
измерение 428—434
затвердевания, определение 458 сл.
кипения, определение 55 сл.,
61 сл., 182 сл., 295 сл.
регулирование 434—438
Теоретическая ступень разделения 96, 123 сл.
Теплоизоляция 393 сл., 401 сл.
Теплоноситель 397 сл.
518
Теплообмен в колонне 119
Термометры
контактные 434 сл.
корректировка показаний 181 сл. проверка градуировки 431 сл. стеклянные 429 сл.
сопротивления 428 сл., 432 сл.
Термопары 432 сл.
Течеискатели 268 сл.
Толуол, давление паров при различных температурах 78
Трубопроводы для ректификационных установок соединительные элементы 406 теплоизоляция 406
Тяжелая вода, получение ректификацией 224 сл.
Углерод, разделение изотопов ректификацией 233 сл.
Удерживающая способность ректификационной колонны 127, 161 динамическая 149 сл.
колонн безнасадочных 335 сл.
— с вращающимися контактными устройствами 363
— с неподвижными контактными -устройствами 354 сл.
— тарельчатых 150, 161 колонок для полумикроректификации 200
общая 149, 151 статическая 149 сл.
Уравнение
Антуана, зависимость между давлением насыщенных паров и температурой 62 сл.
Ваи-дер-Ваальса 70
Вебера для расчета пускового времени 127
Клаузиуса—Клапейрона 64 сл.
Мерча для расчета ВЭТС 141, 144 состояния 70 сл.
рабочей линии укрепляющей части колонны 98 сл.
Фенске для определения числа теоретических ступеней разделения 115 сл., 135
Фазовая диаграмма
р—х 70 сл.
/—х—у 74 сл.
Фазовое равновесие, методы исследования 86 сл.
Фактор интенсивности массообмена 127 сл.
Фенолы, разделение ректификацией 243 сл.
Флегма 39
«дикая» 178, 403
Флегмовое число 97, 145 сл.
бесконечное 97, 145
измерение 379
для низкотемпературной ректификации 255, 257
расчет для непрерывной ректификации 103 сл.
----периодической ректификации 100 сл., 187
регулирование 379, 453 сл.
Форштос 328, 376
Хладоагент 369
Хлор, получение изотопов ректификацией 233 сл.
Холодильники 22 сл., 328, 369—375
Число единиц переноса 122
определение 123 сл.
Число теоретических ступеней разделения
определение, аналитические методы 111 сл., 160
— при конечном флегмовом числе 145
— метод Мак-Кэба и Тиле 100 сл., 103 сл., 107 сл.
-----Оболоицева и Фроста 160 ---- Роуза 113 сл.
Число теоретических ступеней разделения
— для насадки Стедмана 356
— для непрерывной ректификации 103 сл.
— для низкотемпературной ректификации 255 сл.
— для периодической ректификации 100 сл., 186 сл.
— для ректификации азеотропных смесей 313
---многокомпонентных смесей 131 сл.
— по уравнению Фенске 115 сл., 135
— по фактору обогащенйя 112 сл.
— на ЭВМ 192
— для экстрактивной перегонки 318
Число флегмовое см. Флегмовое число эквивалентных теоретических ступеней разделения 147
Шлифы 28 сл.
для вакуумных работ 267 герметизация 478 сл.
смазки 478 сл.
соединения 31, 330, 474 сл.
Штативы 476 сл.
Эбуллиометр 55 сл.
Эбуллиоскоп 55 сл.
ЭВМ для расчета ректификационных колонн 190
Электронагреватели 394
Эталонная смесь 140 сл., 156
Этанол, абсолютирование азеотропной ректификацией 313 сл.
Этанол — вода, зависимость состава азеотропа от давления 306
Эффект пристеночный 43, 137
Эрих Крель
РУКОВОДСТВО ПО ЛАБОРАТОРНОЙ ПЕРЕГОНКЕ
Редактор
Л. Н. ЛАРИЧЕВА
Художественный редактор
Н. В. НОСОВ
Технический редактор
В. М. СКИТИНА
Художник
А. Я. МИХАЙЛОВ
Корректоры:
О. И. ГОЛУБЕВА, И. М. АВЕЙДЕ
ИБ № 854
Сдано в наб. 17.08.79. Подп. в печ. 24.03.80.
Формат бумаги 60X 90*/,,.
Бумага тип. № 2. Гари, литературная.
Печать высокая. Усл. печ. л. 32,5.
Уч.-изд. л. 34,87. Тираж 7000 экз.
Заказ № 1589. Цена 3 р. 30 к.
Изд. № 1780.
Ордена «Знак Почета» издательство «Химия».
107076, Москва, Стромынка, 13
Ленинградская типография № 6 Ленинградского производственного объединения «Техническая книга» Союзполиграфпрома при Государственном комитете СССР по делам издательств, полиграфии и книжной торговли.
193144, Ленинград, С-144, ул. Моисеенко, 10.
Э. Кроль
Handbuch
der Laboratoriumsdestillation
Mit einer Einfiihrung in die Pilotdestillation
von Erich Kreil
3.,bearbeitete und erweiterte Auflage
VEB Deutscher Verlag der Wissenschaften
Berlin 1976
Э.Крель
РУКОВОДСТВО ПО ЛАБОРАТОРНОЙ ПЕРЕГОНКЕ
Перевод с немецкого канд. техн, наук В.И.Чернышева канд. техн, наук А.В. Шафрановского
Под редакцией д ра техн, наук В.М.Олевского
Москва «Химия» 1980
УДК 542.48
Крель Э.
Руководство по лабораторной перегонке — Пер. с нем./Под ред. В. М. Олевского — М.:Химия, 1980.— 520 с., ил.
Книга представляет собой руководство по технике лабораторной перегонки. В ней изложены физические основы процессов дистилляции и ректификации, описаны различные методы перегонки и соответствующая аппаратура, а также контрольно-измерительные устройства. Один из разделов книги посвящен вопросам ректификации на пилотных (опытнопромышленных) установках.
Книга содержит графики, таблицы, номограммы, облегчающие математическую обработку данных по лабораторной перегонке и расчет применяемой аппаратуры.
Книга предназначена для научных сотрудников, инженеров н техников, занимающихся проведением дистилляции и ректификации в лабораторных и промышленных условиях. Она может быть полезна для учащихся высших и средних технических учебных заведений, а также аспирантов н преподавателей.
520 с.; 70 табл.; 434 рнс.; список литературы 918 ссылок.
20507-003
К050 (01)-80
3.80.1801000000
© VEB Deutscher Verlag der Wissenschaften, Berlin 1976
© Перевод на русский язык. Издательство «Химия», 1980 г.
СОДЕРЖАНИЕ
Предисловие редактора ........................................... Ю
Предисловие автора к третьему немецкому изданию ................. И
Основные обозначения и сокращения .............................. 13
1. Введение.................................................... 15
2. Из истории лабораторной перегонки........................... 19
3. Стандартизация в технике перегонки. Способы выражения концентрации ................................. 28
3.1. Стандартизация методов перегонки и применяемых приборов ... 28
3.2. Определение основных понятий .............................. 32
3.3. Единицы измерения ......................................... 33
3.4. Способы выражения и пересчета концентрации................. 34
4. Физические основы процесса разделения .... 38
4.1. Принципы дистилляции и ректификации ....................... 38
4.2. Особенности движения жидкой фазы в колоннах с орошаемой насадкой ......................................................... 41
4.2.1. Распределение жидкости по насадке ....................... 41
4.2.2. Пленочное течение жидкости .......................... . 48
4.2.3. Динамические характеристики колонн ...................... 49
4.3. Разделение смесей взаимно растворимых компонентов.......... 50
4.4. Зависимость между давлением пара и температурой р—t........ 53
4.4.1. Экспериментальное определение давления насыщенных паров 54
4.4.2. Расчет и способы представления данных по давлению насыщенных паров .......................................................... 60
4.5. Уравнение состояния и фазовая диаграмма р—х................ 70
4.6. Фазовая диаграмма t—х—у и кривая равновесия у*—х .......... 74
4.6.1. Расчет кривой равновесия ................................ 76
4.6.2. Летучесть, относительная летучесть и коэффициент активности 81
4.6.3. Экспериментальное определение кривых равновесия.......... 86
4.7. Число теоретических ступеней разделения (число теоретических тарелок) ....................................................... 93
5
VI '
4.7.1. Расчет числа теоретических ступеней разделения по методу Мак-Кэба и Тиле для периодической ректификации .......................... 1W-
4.7.2. Расчет числа теоретических ступеней разделения по методу Мак-
Кэба и Тиле для непрерывной ректификации....................... 103
4.7.3. Определение числа теоретических ступеней разделения по методу Мак-Кэба и Тиле для смесей с кривыми равновесия, имеющими
точку перегиба и азеотропную точку .......................... 107
4.7.4. Определение числа теоретических ступеней разделения для смесей со слабо выпуклыми кривыми равновесия и при небольшом расстоянии между кривой равновесия и рабочей линией .... 108
4.7.5. Аналитические методы определения числа теоретических ступеней разделения для периодической ректификации ........................... 111
4.7.5.1. Определение числа теоретических ступеней разделения по разности температур кипения ........................................... 111
4.7.5.2. Расчет числа теоретических ступеней разделения с помощью фактора обогащения и формул Роуза .................................. 112
4.7.5.3. Расчет минимального числа теоретических ступеней разделения по уравнению Фенске для идеальных смесей при v = со . . . 115
4.7.5.4. Другие методы расчета числа теоретических ступеней разделения и их сравнительная оценка . .................................... 117
4.8. Теория процессов, протекающих в насадочной колонне.............. 118
4.8.1. Процесс разделения в насадочной колонне....................... 118
4.8.2. Определение числа единиц переноса ............................ 122
4.8.3. Интенсивность противоточного массообмена и время, необходимое для разгонки .................................................... 127
4.9. Определение числа теоретических ступеней разделения при периодической и непрерывной ректификации многокомпонентных смесей 131
4.10. Аппаратурно-технологические параметры тарельчатых и насадочных колонн. Испытания колонн ........................................ 135
4.10.1. Диаметр колонны . .......................................... 137
4.10.2. Рабочая высота колонны и способы укладки насадочных тел . .. . 138
4.10.3. Эталонная смесь и состав загруженной жидкости............... 139
4.10.4. Флегмовое число и количество орошающей жидкости............ 145
4.10.5. Общая, статическая и динамическая удерживающая способность колонны ............................................................ 149
4.10.6. Рабочее давление ........................................... 151
4.10.7. Нагрузка и скорость потока паров ........................... 153
4.10.8. Проведение испытаний ....................................... 155
4.10.9. Некоторые данные по разделяющей способности насадочных и тарельчатых колонн ................................................. 161
4.11. Гидравлическое сопротивление, предельная скорость паров и расчет основных размеров колонны . ................................. 164
4.12. Тепловые расчеты ......................................... 175
4.13. Характеристика разделяемых веществ и получение кривых разгонки ............................................................... 179
4.14. Примерная последовательность расчета процесса разделения . . . 186
4.14.1. Периодическая ректификация при атмосферном давлении . . . 186
4.14.2. Непрерывная ректификация ................................. 190
4.14.3. Вакуумная перегонка ...................................... 190
4.15. Использование ЭВМ при расчете колонн ....................... 190
6
5. Обычные методы перегонки..................... 195
5.1. Классификация методов перегонки п« количеству перерабатываемого продукта .................................................... 195
5.1.1. Микро- и полумикроперегонка ............................... 195
5.1.2. Аналитическая ректификация . ........................ 202
5.1.3. Препаративная и наработочная перегонка..................... 207
5.1.3.1. Полупромышленные установки , . .......................... 208
5.1.3.2. Пилотные установки ...................................... 211
5.1.3.3. Получение дистиллированной воды ......................... 216
5.1.4. Разделение изотопов ....................................... 219
5.1.4.1. Разделение изотопов низкотемпературной ректификацией .... 222
5.1.4.2. Получение D и 18О путем ректификации воды................ 224
5.1.4.3. Получение других изотопов .............................. 233
5.2. Классификация методов перегонки по способу проведения процесса 234
5.2.1. Периодическая и полунепрерывная перегонка.................. 234
5.2.2. Непрерывная ректификация. , . ,. , ........................ 235
5.2.2.1. Области применения непрерывной ректификации.............. 237
5.2.2.2. Теоретические расчеты ................................... 239
5.2.2.3. Аппаратура для непрерывной ректификации.................. 240
5.2.2.4. Примеры непрерывной лабораторной ректификации............ 243
5.2.2.5. Выведение ректификационных установок на стационарный режим 244
5.2.3. Разделение методом парциальной конденсации ................ 245
5.3. Классификация методов перегонки по температуре процесса .... 249
5.3.1. Низкотемпературная ректификация ........................... 250
5.3.2. Высокотемпературная и изотермическая перегонка............. 257
5.4. Классификация методов перегонки по рабочему давлению......... 262
5.4.1. Вакуумная дистилляция и ректификация ...................... 263
5.4.2. Расширительная перегонка .................................. 269
5.4.3. Пленочная перегонка (пленочное испарение).................. 272
5.4.4. Молекулярная дистилляция .................................. 280
5.4.5. Ректификация под давлением ................................ 292
6. Селективные методы перегонки ......... 294
6.1. Перегонка в парах носителя ............................... 294
6.2. Азеотропная и экстрактивная перегонка .................... 299
6.2.1. Азеотропная перегонка .................................. 304
6.2.2. Экстрактивная перегонка ................................ 314
6.3. Солевая перегонка. . ................................... 321
7
7. Материалы и аппаратура..................................... 324
7.1. Материалы для изготовления ректификационной аппаратуры . . . 324
7.2. Стандартные и сборные приборы ............................. 326
7.2.1. Краны и клапаны ......................................... 332
7.3. Ректификационные колонны .................................. 335
7.3.1. Безнасадочные колонны ................................... 335
7.3.2. Насадочные колонны ...................................... 344
7.3.3. Тарельчатые колонны ..................................... 345
7.3.4. Колонны с неподвижными контактными устройствами.......... 352
7.3.5. Колонны с вращающимися контактными устройствами ......... 359
7.4. Холодильники и дефлегматоры ............................... 369
7.5. Форштосы, приставки для дистилляционных аппаратов и головки колонн ......................................................... 376
7.5.1. Форштосы ................................................ 376
7.5.2. Приставки для дистилляционных аппаратов.................. 376
7.5.3. Головки колонн .......................................... 378
7.6. Кубы, приемники и сборники фракций дистиллята.............. 387
7.6.1. Кубы .................................................... 387
7.6.2. Приемники и сборники фракций дистиллята.................. 390
7.7. Нагревательные приборы и теплоизоляция .................... 393
7.7.1. Обогрев кубов ........................................... 394
7.7.2. Нагревание исходной смеси и кубовой жидкости при непрерывной ректификации ................................................... 398
7.7.3. Теплоизоляция............................................ 401
7.8. Насадки ................................................... 407
7.8.1. Формы выполнения элементов насадок ...................... 407
7.8.2. Влияние материала насадки на эффективность разделения . . . 415
8. Автоматические установки, измерительные и регулирующие устройства........................................417
8.1. Автоматические установки .................................. 417
8.1.1. Автоматизированные установки для стандартного дистилляционного анализа ................................................... 419
8.1.2. Полностью автоматизированные установки для фракционной перегонки ...................................................... 422
8.2. Измерение и регулирование температуры ..................... 428
8.2.1. Измерение температуры ................................... 428
8.2.2. Регулирование температуры ............................... 434
8.3. Измерение и регулирование давления ........................ 438
8.3.1. Измерение и регулирование давления в областях выше 760 и от 760 до 1 мм рт. ст.............................................. 438
8
8.3.1.1. Методы частичного вакуумирования ......................... 443
8.3.2. Измерение и регулирование давления в интервале от 1 до 10“8 мм рт. ст..................................................... 446
8.3.2.1. Компрессионный манометр Мак-Леода ........................ 448
8.3.2.2. Регулирование давления в интервале от 100 до 10"в мм рт. ст. 451
8.4. Регулирование флегмового числа и скорости выкипания........... 453
8.4.1. Реле времени для регулирования флегмового числа............. 453
8.4.2. Регулирование скорости выкипания............................ 455
8.5. Измерение физико-химических свойств дистиллята в процессе ректификации ......................................................... 458
8.5.1. Измерение температуры затвердевания ..................... 458
8.5.2. Измерение показателя преломления ..................... 459
8.5.3. Измерение диэлектрической проницаемости..................... 461
8.5.4. Измерение других физико-химических свойств ................. 462
8.6. Измерение расхода и дозирование газов и жидкостей............. 463
9. Оборудование лаборатории. Ввод в эксплуатацию ректификационных установок ................. 469
9.1. Планировка помещения ................................... 469
9.2. Стенды и коммуникации .................................. 472
9.3. Сборка установок ....................................... 474
9.4. Герметизация установок ................................. 477
9.5. Ввод ректификационных установок в эксплуатацию ......... 480
9.6. Техника безопасности ................................... 481
Литература .................................................. 483
Предметный указатель ........................................ 512
ПРЕДИСЛОВИЕ РЕДАКТОРА
Первое издание монографии Э. Креля «Руководство по лабораторной перегонке», опубликованное Издательством иностранной литературы в 1960 г. (перевод с 1-го немецкого издания 1958 г.), получило широкое одобрение советских специалистов.
За прошедшие 20 лет литература по лабораторной перегонке существенно обогатилась, расширилась область применения ректификации, в связи с чем выпуск перевода третьего немецкого издания книги Э. Креля «Руководство по лабораторной перегонке» является своевременным.
При подготовке третьего немецкого издания автор в значительной мере учел недочеты, имевшиеся в первых двух изданиях. Как особо положительный момент следует отметить включение раздела, в котором рассматривается ректификация на укрупненных (пилотных) установках, что представляет большой интерес в качестве ступени для перехода к промышленным агрегатам.
Полагаю, что настоящее издание монографии Э. Креля послужит дальнейшему прогрессу в области техники перегонки, приобретающей особое значение в борьбе за повышение качества продукции на предприятиях.
Шафрановский А. В. перевел главы 1, 2, 3, 4, 6 и начало главы 5 по раздел 5.1.4.1. Чернышев В. И. перевел главы 7, 8, 9 и конец главы 5, начиная с раздела 5.1.4.2.
В. ОЛЕВСКИЙ
ПРЕДИСЛОВИЕ АВТОРА К ТРЕТЬЕМУ НЕМЕЦКОМУ .-ИЗДАНИЮ
Первое издание этой книги, вышедшее в 1958 г., разошлось сравнительно быстро и уже в 1960 г. возникла необходимость во втором издании. Книга нашла признание и была переведена на русский, английский и венгерский языки.
Цель третьего издания (как и предыдущих) — дать возможно более полное введение в технику лабораторной перегонки; в нем представлены новейшие научные данные и достижения; приведена специальная литература по 1975 г. включительно. При составлении книги автор придерживался мнения, что излагать следует только общепризнанные факты, поскольку на многие вопросы дистилляции и ректификации до сих пор еще нет ясного и окончательного ответа. Однако с целью стимулирования дальнейших исследований в книге отражены также и тенденции, наметившиеся в развитии техники перегонки.
Разумеется, ввиду наличия обширного материала к его отбору необходимо было отнестись критически. Автор стремился просто и понятно описать уравнения и расчетные методы, облегчающие практическую работу в лаборатории. Читатели, интересующиеся вопросами математической обработки и другими специальными вопросами и желающие углубить приобретенные знания, найдут в конце книги обширные списки рекомендуемой литературы.
Расположение материала, принятое в первом издании, в основном сохранено. Дополнительно рассмотрены вопросы ректификации на пилотных * установках (разд. 5.1.3.2). Содержанием раздела 4.2 в третьем издании является гидродинамика потоков в насадочных колоннах. Гл. 8 значительно сокращена ввиду того, что стандартные детали дистилляционных и ректификационных „ приборов и соответствующие контрольно-измерительные приборы уже нашли достаточно широкое применение в лабораторной практике. Таблицы, ранее приводившиеся в приложении, в третьем издании включены в текст. Литературные ссылки распределены по главам и дополнены новыми важнейшими работами; в списки литературы включено по возможности больше обзорных статей.
Книга рассчитана, главным образом, на физиков, химиков и инженеров, занятых в химической промышленности и научно
* Под пилотными установками автор подразумевает малопроизводительные . опытно-промышленные установки. — Прим. ред.
11
исследовательских центрах, в задачу которых входит проведение дистилляции и ректификации в лабораторных и промышленных условиях. Учащиеся высших и средних технических учебных заведений, химики-техники и лаборанты также найдут в этой книге ответы на многие вопросы, связанные с практикой лабораторной перегонки и с расчетами процессов разделения.
Автор надеется, что данное руководство послужит советчиком и путеводителем при разработке более совершенных и экономичных методов разделения для всех, кому приходится заниматься перегонкой в лабораторных условиях.
Автор выражает свою особую благодарность профессору доктору наук Кл. Ветцелю за его благосклонное внимание к этой книге, за его ценные указания и постоянную поддержку. Автор приносит благодарность коллегам доктору Г. Штаге и Е. Гибе-леру за многочисленные замечания, а также благодарит фирмы, выпускающие лабораторные приборы и изделия из стекла, за любезное представление каталогов и другой технической документации. Автор выражает также благодарность издательству за образцовое оформление книги и особенно отделу химии за тщательное редактирование рукописи.
Автор надеется, что и третье издание этой книги окажет содействие дальнейшему развитию техники перегонки на лабораторных и пилотных установках, а также послужит учебным и рабочим пособием при решении задач разделения путем дистилляции и ректификации в лабораториях и в опытных производствах химической промышленности и научно-исследовательских центров, а также в высших и средних учебных заведениях.
БЕРЛИН, ЛЕТО 1975 г.
Доктор технических наук ЭРИХ КРЕЛЬ
ОСНОВНЫЕ ОБОЗНАЧЕНИЯ И СОКРАЩЕНИЯ
А — количество (расход) кубового продукта при непрерывной ректификации а — полная поверхность насадки
— смоченная поверхность насадки
ае — активная поверхность насадки
В — количество жидкости в кубе
ср — теплоемкость при постоянном давлении
си — теплоемкость при постоянном объеме
D — количество паров (суммарное количество дистиллята и флегмы)
dK — диаметр колонны
dH — диаметр насадочного тела
Е — количество (расход) дистиллята
Нк — рабочая высота колонны
НуС — общая удерживающая способность колонны по жидкости
h — высота слоя насадки
I — фактор интенсивности
L — объемная плотность орошения, средняя (по поперечному сечению колонны)
L — объемная плотность орошения в определенной точке т — масса
mq — степень перемешивания
п — число теоретических ступеней разделения
пмнн — число теоретических ступеней разделения, соответствующее минимальному флегмовому числу vMHH
Пуд — число теоретических ступеней разделения, приходящееся на Гм рабочей высоты колонны
р — общее давление паров смеси, рабочее давление процесса перегонки Р° — давление паров чистого вещества
р{ — парциальное давление /-го компонента
Q — количество тепла
Т — термодинамическая (абсолютная) температура
t — температура в градусах Цельсия
V — объем
v — флегмовое число
vMHH — минимальное флегмовое число
vpa6 — рабочее флегмовое число
wn — скорость потока паров
х — концентрация легколетучего компонента в растворе (мольная доля) хд — концентрация легколетучего компонента в кубовой жидкости хд — мольная доля легколетучего компонента в кубовой жидкости
Х£ — мольная доля легколетучего компонента в дистилляте
xz — концентрация легколетучего компонента в питающей жидкости (исходной смеси)
у — концентрация легколетучего компонента в парах
у* — равновесная концентрация легколетучего компонента в парах
Z — количество (расход) питающей жидкости при непрерывной ректификации
13
a — относительная летучесть (отношение давлений паров чистых компонентов)
у — коэффициент активности
е — доля свободного объема (насадки)
6 — характеристическая температура (приведенная температура)
X — коэффициент теплопроводности
|х — динамическая вязкость
л — приведенное давление
р — плотность
о — поверхностное натяжение
т — время
Ф — приведенный объем
Ф — краевой угол смачивания
фю, фс — доля смоченной или активной поверхности насадки
ВЭТС — высота, эквивалентная теоретической ступени разделения
ВЕП — высота единицы переноса
УС — удерживающая способность колонны по жидкости
ЧЕП — число единиц переноса
Введение
Дистилляция и ректификация как методы физического разделения нашли весьма широкое применение в химической промышленности, и в том числе в заводских и исследовательских лабораториях. Однако следует отметить, для лабораторной перегонки часто еще используют малоэффективную с современной точки зрения аппаратуру. К тому же в большинстве случаев не проводят расчетов процесса разделения, а работают чисто эмпирически, руководствуясь опытными данными.
За последние 30 лет проведена большая исследовательская работа по усовершенствованию техники лабораторной перегонки. Теперь в нашем распоряжении имеются современные приборы, изготовленные из стандартных деталей, а также полностью автоматизированные и высоковакуумные установки; разработаны методы расчетов процесса перегонки; лабораторные способы разделения включают разнообразные методы перегонки: от микроректификации с загрузкой менее 1 г до непрерывных процессов с пропускной способностью до 5 л/ч, от низкотемпературной ректификации сжиженных газов до высокотемпературной разгонки смол, от перегонки при атмосферном давлении до молекулярной дистилляции при остаточном давлении ниже 10-4 мм рт. ст. Усовершенствованы селективные методы разделения; путем изменения соотношения парциальных давлений компонентов в парах удается разделять такие смеси, которые до сих пор не поддавались разделению обычными методами.
Классические работы по лабораторной перегонке Рехенберга [1] и Янга [2] содержат превосходные обзоры по теории и практике перегонки, включая описание промышленных установок, однако оии уже во многих отношениях устарели; это особенно касается технологических расчетов и аппаратуры. В монографиях Торманна [3], а также Беджера и Мак-Кэба [4], изданных к началу тридцатых годов, описаны до сих пор еще применяемые графические методы расчета по. Мак-Кэбу и Тиле [5]. Эти монографии представляют особую ценность благодаря ясности изложения. Однако они посвящены исключительно процессам дистилляции и ректификации в промышленных условиях и не содержат рекомендаций по лабораторной перегонке. Работы Янцена и его учеников послужили своеобразным стимулом к дальнейшим исследованиям в области лабораторной перегонки. В монографии [6], выпущенной в 1932 г. Германским обществом химического машиностроения (Dechema), Янцен подробно изложил сформулированные нм еще в 1923 г. основные требования, которые следует предъявлять к ректификационной колонне. Эти требования сохраняют свое значение еще и сегодня.
Работы Киршбаума [7], Гиюла [8] и Якобса [9] посвящены проблемам промышленной ректификации, как н монография Робинсона и Джиллиленда [10], в которой подробно изложены теоретические основы перегонки и рассмотрены сложные проблемы разделения многокомпонентных смесей, а также методы азеотропной и экстрактивной ректификации. Справочник Перри для инженеров-химиков [10 а] включает главу «Перегонка» с многочисленными примерами, таблицами н номограммами для расчета промышленных установок. В справочнике почти нет сведений о лабораторной перегонке.
15
Указанные выше книги предполагают наличие у читателя теоретических знаний и практического опыта в области ректификации. Те же требования предъявляются к читателю в более обширном труде Шульце и Штаге [11] по проблемам ректификации, в брошюре по обмену опытом «Методы работы и приборы для дистилляции и ректификации» Германского общества химического машиностроения, подготовленной Торманном [ 12 ] и охватывающей лабораторные методы перегонки, а также в брошюре «Ректификация в промышленности» [13], в основу которой положены производственные данные.
О темпах развития техники перегонки в период 1920—1944 г. дает представление литературный обзор Штаге и Шульце [14], который содержит ссылки на 2300 важнейших работ по теории и методам дистилляции и ректификации, а также применяемой аппаратуре. Всю мировую литературу по технике перегонки за 1941—1945 г. и 1946—1952 г. А. и Е. Роуз [15] отразили в кратких рефератах, которые за первый период времени охватывают 1000 публикаций, а за второй 5000 публикаций. Обзор за два года приведен в сборнике «Успехи технологии» [16] и, в частности, в его разделах «Дистилляция и ректификация», «Ректификация при низких температурах», «Массопередача», «Данные по равновесию для одно- и многокомпонентных смесей», а также в разделе «Вакуумная техника и методы работы под вакуумом». Уэльш [17] ежегодно публикует критические обзоры технической литературы под рубрикой «Unit Operations Review» *. Ценные указания о константах индивидуальных веществ и методах их расчета можно найти в пособии Орличека и Пёлля [18] для инженеров-нефтяников, в частности в специальном разделе книги приводятся данные по температурам кипения смесей и фазовым равновесиям. Современный уровень перегонки как в лабораторных, так и в промышленных условиях отражен Зигвартом [19] в превосходном обзоре, составленном для Ульмановской энциклопедии технической химии. Теория перегонки и экстракции с позиций термодинамики изложена в монографиях Кортюма и Бухгольц-Майзенхаймера [20] и Бошняковича [21].
Известные каждому изучающему химию книги Гаттерманна и Виланда [22] и Виттенбергера [23] дают практические сведения для химиков-органиков, однако по практике перегонки в них содержатся, к сожалению, лишь очень краткие обзоры. В книгах Вейганда [24] и Бернхауэра [25] по методам лабораторной техники кратко описаны также и современные приборы и методы; однако до сих пор не было исчерпывающего изложения проблем лабораторной перегонки, в том числе и методов расчета. Лишь в работах Карни [26] и Розенгарта [27], а также Роуза с сотр. [28] систематически изложены специфические вопросы лабораторной перегонки. Работа Вебера [29] посвящена получению эфирных масел с помощью перегонки, в частности перегонке с водяным паром. Для четвертого издания книги Губен-Вейля Зигварт [30] написал главу «Дистилляция и ректификация»- В этой главе дан превосходный обзор с многочисленными примерами из лабораторной практики и приведены 13 задач разделения с намеренно усложненными условиями процесса. Вышли в свет небольшие книги Цуидервега [31] и Коулсона и Херингтона [32], в которых кратко изложены основные принципы перегонки. В монографии Букала, Маевского и Родзинского [33] подробно рассматривается применение в лаборатории фракционной дистилляции и ректификации.
Начиная с 1960 г. резко возросло число публикаций по теоретическим основам процессов дистилляции и ректификации. Поскольку объем реферативных сборников постоянно растет, информацию теперь обрабатывают, широко применяя вычислительную технику. Тем не менее в ряде журналов еще появляются периодические литературные обзоры по вопросам перегонки [34]. Экспресс-информацию содержат реферативные карточки и доклады, обобщающие научные достижения [35, 16]. Назрела необходимость в издании для студентов обстоятельных руководств по технике лабораторных работ, включающих также разделы по методам дистилляции и ректификации [36], поскольку в большинстве имеющихся пособий методы разделения освещены недостаточно [37].
В ряде монографий обсуждаются специальные вопросы перегонки. Так, книга Рёка [38] является превосходным введением в экстрактивную и азеотроп
* «Обзор элементарных процессов». —Прим. ред.
16
ную ректификацию, а в книгах Штаге с сотр. [39] в доступной форме изложена методика экспериментального исследования фазового равновесия в системе пар— жидкость на примере жирных кислот, а также приведены сведения о соотношениях нагрузок в насадочных колоннах- Опубликованный в 1960 г, классический труд Хала с сотр. [40] по вопросам теории и экспериментального исследования равновесия в системе жидкость—пар был дополнен обширным сборником равновесных данных тех же авторов [41]. Переизданы таблицы Когана и Фридмана, обобщающие данные по равновесию для более чем 2000 бинарных систем. Кроме того, те же авторы подготовили справочник по азеотропным смесям (21069 систем, содержащих от 2 до 5 компонентов) [42]. В двух монографиях Биллет подробно и поэтапно излагает основы разделения жидких смесей путем перегонки [43] и принципы оптимизации в технике ректификации (особенно вакуумной ректификации) [44]. Материал монографий-относится к промышленной ректификации, вместе с тем приведенные многочисленные расчетные методы пригодны также для лабораторной и полупромышленной ректификации. В двух солидных трудах — Оливера [45] «Диффузионные процессы разделения» и Пратта [46] «Противоточные процессы разделения» — отчетливо проявляется тенденция рассматривать все методы разделения под одним и тем же углом зрения. С теоретической точки зрения это дает большие преимущества. Поэтому Крель с сотр. [47] в учебном пособии для студентов-химиков «Введение в методы разделения» попытался дать общее представление о методах разделения и их возможных комбинациях.
В последние годы появились новые монографии, трактующие специальные проблемы ректификации, например «Основы моделирования тарельчатых колонн» Хоппе и Миттельштрасса [48], «Низкие температуры в технологии» Юнгникеля и Отто [49] и «Ректификация как метод очистки» Франка и Куче [50]. В то время как последняя работа в основном охватывает лабораторные методы, монография Олевского и Ручинского [51] ориентирована на дистилляцию и ректификацию термически нестойких продуктов в промышленных условиях*. В монографии Холло с сотр. [52] рассмотрены вопросы применения молекулярной дистилляции в лаборатории, а также в пилотных и в промышленных установках; книга содержит многочисленные литературные ссылки и обширный справочный и иллюстративный материал.
В первой части своего двухтомного труда [53] «Термодинамические основы ректификации и экстракции» Шуберт обсуждает принципы термодинамики много-«азных систем, а также вопросы классификации и разделения бинарных смесей, торая часть посвящена тройным системам, экстракционному разделению двух компонентов с помощью одного или нескольких растворителей, специальным методам селективной ректификации, а также проблемам ректификации и экстракции многокомпонентных смесей. Приведенные в этой книге частные теоретические положения, справедливые для процессов ректификации, логически следуют из строгих термодинамических принципов.
Брауэром [54] в общей форме изложены теоретические основы процессов массообмена и разделения одно-и многофазных систем. При этом рассмотрен массо-перенос в неподвижных и движущихся средах. Для изучающих ректификацию особенный интерес представляют разделы «Массопередача в неподвижных и подвижных слоях насадки», «Массоперенос через границу раздела в простых двухфазных системах» и «Массоперенос в двухфазных потоках промышленных аппаратов». Холланд [55] подробно обсуждает вопросы многокомпонентной ректификации. В своей монографии [43а] Биллет освещает вопросы применения ректификации в промышленности.
На международном симпозиуме по перегонке, состоявшемся в Брайтоне (Англия) в 1969 г., особенно четко выявилась необходимость в дальнейшей разработке теоретических основ ректификации с ориентацией на применение электронной вычислительной техники при расчетах процесса разделения [56]. На
* В позднее опубликованной монографии В. М. Олевского и В. Р. Ручинского «Роторно-пленочные тепло-и массообменные аппараты» (М., Химия, 1977 г.) описаны также лабораторные и пилотные установки для дистилляции и ректификации термически нестойких продуктов под вакуумом. —Прим. ред.
17
симпозиуме были обсуждены всевозможные исследования и разработки в области дистилляции и ректификации. Вместе с тем отчетливо было показано, что в многоплановой области перегонки по-прежнему остается еще множество нерешенных проблем. К такому выводу приходит и Биллет [57], который освещает современный уровень и перспективы развития техники дистилляции и ректификации, проводя сравнение с другими методами разделения, основанными на тепловом воздействии на смесь веществ. Он подчеркивает, что ввиду ограниченности наших знаний о процессах массообмена и пропускной способности колонн затрудняется расчет оптимальных параметров и поэтому необходимы дальнейшие обширные и детальные исследования экспериментального и теоретического характера.
В результате бурного развития техники лабораторной перегонки за последние 30 лет и углубления специализации назрела необходимость в создании руководства по перегонке для лабораторий и опытных производств, рассчитанного на читателя, не имеющего специальных знаний в этой области. Такое руководство должно включать, методику определения давления паров и кривых равновесия, а также подробное описание непрерывных и селективных методов перегонки и рекомендации по применению контрольно-измерительной и регулирующей аппаратуры. Подобный обобщающий труд должен способствовать устранению многих неверных представлений о данном процессе разделения, подверженном влиянию многочисленных факторов, и послужить обстоятельным руководством при практическом проведении различных, в том числе и специальных, видов перегонки
Вторая глава настоящей книги «Из истории лабораторной перегонки» одновременно знакомит читателей с общими принципами перегонки. В третьей главе уточняются основные понятия, вводятся единицы измерения и условные обозначения, при этом особое внимание уделяется стандартизации, которая дает возможность за счет унификации определенных приборов и методик получать сопоставимые результаты, служащие фундаментом для дальнейших научных исследований. В главах 4—6 сначала изложены физические основы процесса перегонки и приведена классификация разделяемых смесей, после чего разносторонне рассмотрены обычные и селективные методы перегонки, с помощью которых можно решать самые разнообразные задачи разделения. В главах 7 и 8 описываются необходимые для проведения перегонки приборы и установки, включая вспомогательное оборудование, а также контрольно-измерительную и регулирующую аппаратуру. Наконец, девятая глава касается вопросов, которые следует принимать во внимание при оборудовании лабораторий дистилляции и ректификации и при вводе установок в эксплуатацию.
При изложении материала особое значение придавалось тому, чтобы процессы лабораторной и полупромышленной дистилляции и ректификации рассматривались во взаимосвязи с задачами промышленной перегонки, по отношению к которой эти процессы часто являются предварительным этапом. Ранее в лабораториях нередко разрабатывали методы дистилляции и ректификации, не имеющие никакой взаимосвязи с промышленностью. В итоге при переносе данных лабораторных исследований в промышленные и даже полупромышленные условия часто возникали значительные трудности. Если же при разработке новых способов, представляющих интерес для промышленности, с самого начала рассчитывать на их промышленное внедрение, то можно сэкономить значительные средства и много времени; при этом лабораторные данные почти без коорректи-ровки найдут применение при расчете промышленных процессов. Такой подход не должен, конечно, исключать тех случаев, когда в лаборатории намеренно подбирают условия, совершенно не экономичные для промышленной ректификации (например, при аналитических разгонках). Только глубокое знание закономерностей процесса ректификации обеспечит подбор оптимальных условий при решении любой задачи разделения, и данная книга должна способствовать приобретению этих знаний. '
Условные обозначения физико-химических величин и единицы измерения, использованные в прежнем издании, остались в основном без изменения. Однако сделана попытка привести условные обозначения в соответствие с международной практикой. Библиографические данные упорядочены по главам и приведены в списке литературы. Рисунки, формулы и таблицы пронумерованы по порядку с начала до конца книги,
18
2
Из истории лабораторной перегонки
Проследить, как на протяжении веков развивалась какая-либо химическая отрасль, интересно не только с исторической точки зрения; сравнение с современным уровнем развития часто приводило к ценным начинаниям в научно-исследовательской деятельности сегодняшнего дня. Исторический обзор особенно полезен при ознакомлении с такой специальной отраслью химической технологии, какой является перегонка. При этом мы становимся свидетелями того, как человеческая мысль на основе уже достигнутого постоянно находит все новые пути к повышению экономичности технологических процессов и производительности применяемой аппаратуры.
Шеленц [1], Гильдемайстер [2] и Рехенберг [3] проанализировали многочисленные исторические сведения, позволяющие проследить развитие методов перегонки на примере получения эфирных масел. Андервуд [4] дает краткий обзор развития техники перегонки вплоть до 19 века. В своей превосходной книге Фобе [5] подробно излагает историю этого своего рода искусства с самого его зарождения и вплоть до смерти Селлье-Блюменталя. Этот французский инженер умер в Брюсселе в 1840 г. Он был одним из самых талантливых конструкторов ректификационных колонн начала 19 века. Другие исторические сведения можно найти в разрозненном виде в многочисленных работах по истории алхимии и химии. Из книг, вышедших за последние десятилетия, следует отметить двухтомный труд Фобса и Дийкстеруиза [6] по истории естественных и технических наук, в котором общедоступно излагаются этапы развития техники, астрономии, математики, химии и физики, причем особое внимание уделено физике.
В. Шнайдер [7] составил словарь алхимических и фармацевтических терминов, который облегчает изучение старинных трудов.
В настоящем обзоре автор не стремился дать возможно более полную хронологию событий и лингвистически объяснить происхождение специальных терминов, он скорее пытался точно проследить этапы развития лабораторных методов и аппаратов и сопоставить их с современным уровнем техники. При этом внимание уделялось также и полупромышленным установкам.
Перегонка — это своего рода искусство, к которому еще до новой эры приобщились древние египтяне. Жрецы совершенствовали и оберегали это искусство в храмах как тайную науку. Индусы, персы и китайцы также, по-видимому, уже с древнейших времен использовали дистилляционные приборы. По мнению Шеленца [1], изобретение дистилляции следует приписать персам, которые возможно сначала использовали ее при получении розовой воды. Кроме того, он полагает, что в принципе дистилляция аналогична сухой перегонке дерева, с чем согласен также и Липпман [8—10], который сообщает, что сведения о перегонке с нисходящим'потоком газовой фазы можно найти еще в папирусе Эберса («Papyrus Ebers»), относящемся примерно к 1500 г. до нашей эры. Поэтому можно считать, что история перегонки насчитывает почти 3500 лет. Следует учесть, что в те времена слово «дистилляция» применяли в более широком смысле для обозначения всех известных тогда методов разделения. Это слово можно перевести как «разделение по каплям»*. На языке алхимиков это означало выде-
* Это слово (от лат. distillatio) можно также перевести как «стекание по ('каплям», что точнее соответствует специфике мелкомасштабных процессов разделения, когда целевой продукт собирают по каплям в приемные сосуды. — Прим. ред.
19
Рис. 1.
«Шлем» (alembic) на печи в окружении магических знаков.
Из трактата египетского алхимика Клеопатры «Приготовление золота» (II век н. э.).
ление более или менее ценных веществ. Поэтому термином «дистилляция» обозначали также фильтрацию, кристаллизацию, экстракцию, а также отжимание масла.
Мы будем рассматривать историю дистилляции (перегонки) только в современном значении этого слова, подразумевая под дистилляцией метод разделения веществ, основанный на испарении жидкости и конденсации образующихся паров. Разумеется, по данному определению дистилляция с нисходящим потоком паров представляет собой разновидность обычной дистилляции в современном ее понимании.
Примерами самого древнего применения перегонки являются получение розового и других эфирных масел, дистиллированной воды для мореплавателей (Аристотель сообщает о получении пресной воды из морской), а также приготовление алхимиками большого числа микстур и напитков. На рис. 1 показан «шлем» (alembic), установленный на печи; «шлем» окружен магическими знаками, которым при проведении перегонки в древности приписывалась особая роль. Типичный дистилляционный прибор того времени показан на рис. 2а. Он представляет собой стеклянный прибор с песочной или водяной баней. Приборы подобного устройства применяют еще и сегодня, что иллюстрирует рис. 26, на котором представлен прибор для перегонки ртути. Четыре основных элемента прибора: баня, куб (curcurbita, колба), головка (alembicus, шлем) и приемник дистиллята (receptacula) до настоящего времени сохранились практически в первоздан-
Рис. 2.
Простые перегонные приборы типовых конструкций:
а — стеклянный дистилляционный прибор с песочной или водяной баней (II век н. э.); б — прибор для вакуумной дистилляции ртути с расширением в виде кармана для сбора и отвода дистиллята (XX век).
20
Рис. 3.
б
1952 г.) снова применен этот
Приборы для перегонки по методу «per descensum» (с нисходящим потоком паров):
а — прибор для сухой перегонки коры и кореньев пряных растений (1300 г.); б — аппарат Блоума для дистилляции воды (XX век).
ном виде. Интересно отметить, что и расширение в виде кольцевого кармана для сбора и отвода дистиллята до сих пор еще используют в различных Дистилляционных приборах. В качестве конструкционных материалов для приборов в древние времена и в средние века кроме меди применяли в основном стекло и керамику-
Известные до 1300 г. методы перегонки в основном можно подразделить на дистилляцию с восходящим потоком паров (per ascensum) и на дистилляцию с нисходящим потоком паров (per descensum). Второй способ (per descensum) после 1880 г. был временно забыт, хотя он и представляет собой оптимальное решение для ряда случаев разделения (рис. За). В одном из аппаратов для дистилляции воды (выпуск принцип, как обеспечивающий экономное ис
пользование тепла (рис. 36)
Вскоре после изобретения книгопечатания появились книги по дистилляции,
в которых весьма наглядно отражен уровень развития техники перегонки в средние века. Имеются в виду главным образом следующие труды: Шрик «Указатель перегнанных вод» (1483 г.); Брунсвиг «Книга об истинном искусстве дистилляции
Рис. 4.
Установки с обогревательной системой, расположенной вне куба:
а — деревянный перегонный аппарат со змеевиковым холодильником и вынесенной топкой (XVII век); б — промышленная ректификационная установка непрерывного действия с вынесенным кипятильником (XX век).
21
Рис. 5.
Террасообразная печь для перегонки.
Внутри конического корпуса установлена топка, а по бокам расположены газоходы, в которых поддерживается рабочая температура. Керамические полки служат для размещения перегонных кубов (XVI век).
отдельных веществ» (1500 г.); Брунсвиг «Книга о подлинном искусстве дистилляции» (1507 г.); Ульштад«Сое1ит philosophorum» (1528 г.); Рифф «Новая большая книга по дистилляции», излагающая хорошо обоснованные методы искусной дистилляции (1536) г.
Начиная с XVI века наблюдается большее
разнообразие применяемых систем обогрева перегонных кубов. Кубы обогревали] с помощью воздушной, песочной или зольной бани, для обогрева применяли также и восковые свечи. Чтобы обеспгчить непрерывный режим работы, печи оснащались шахтными топками. Иногда применяли и весьма причудливые средства, например кислое тесто или баню с отжатой фруктовой мякотью, выделяющими тепло при брожении. В жарких странах для нагревания иногда применяли фокусирующие зеркала. В связи с этим интересно отметить, что в 1943 г. был выдан патент на устройство для обогрева перегонных аппаратов с помощью фокусирующего зеркала в форме параллелепипеда.
В промышленных установках часто требуется отделять нагреватель от собственно куба (рис. 46). Такое исполнение было предложено еще Глаубером (1604—1668 г.) (рис. 4а). При переработке значительных количеств дистиллята еще в XVI веке было принято проводить перегонку одновременно на нескольких Дистилляционных приборах, устанавливаемых в одной террасообразной печи (рис. 5).
Первые попытки применить теплоизоляцию, а именно шерсть животных, замешанную на глине, принадлежат еще Люлли (1415 г.). В XVI веке были установлены преимущества ступенчатого обогрева и уже тогда стали применять для обогрева тепло конденсирующегося водяного пара. Применение водяного пара в качестве теплоносителя в широких масштабах началось только после 1800 г.
Примерно до 1300 г. из способов конденсации паров был известен только прием, основанный на воздушном охлаждении. Для лучшего охлаждения паровую
Рис. 6.
Охлаждающая труба в форме змеевика: в полупромышленном н лабораторном дистилляционных аппаратах (а), в качестве конденсаторов с водяным и с воздушным охлаждением (XVI век) н в холодильнике Димрота (б) со стандартными шлифами (XX век).
22
Рис. 7.
Перегонные аппараты с удлиненной паровой трубой:
а — дистилляционные приборы, применявшиеся в химико-фармацевтической отрасли [большой аппарат с головкой в форме кулича и малый аппарат со «шлемом» установлены наГпечах и снабжены зигзагообразными паровыми трубками с воздушным охлаждением; приемник дистиллята закреплен на передвижном штативе (1709 г.)]; б — ректификационная колонна Янцеиа с корпусом в виде спиральной трубки (XX век).
трубу удлиняли, а позднее ее стали пропускать через бочку или чан с водой. Позже обнаружили, что наиболее выгодной формой охлаждаемой трубы является змеевик (рис. 6а), который и сейчас используется в конденсаторе Димрота (рис. 66). В XVI веке широко использовался непрерывный способ водяного охлаждения. Бы-
ло установлено обогащающее действие удлиненной паровой трубы, не имеющей тепловой изоляции (1709 г.), и частичной конденсации* (1648 г) (рис. 7, а). Аналогичный современный прибор показан на рис. 76. В 1770 г. Писсонье ввел принцип противотока при охлаждении, который и теперь еще используется, например в известном холодильнике Либиха (рис. 8, 6), применявшемся еще Дарио (1533—1594 г.) (рис. 8а).
Развитие техники перегонки в период с XVI по XIX век шло по пути совершенствования аппаратурного оформления. Еще в середине XVI века было установлено, что металлические кубы легко корродируют, и поэтому предпочтение стали оказывать приборам из стекла и керамики. Благодаря Кункелю (1638— 1703 г.) стекло стало преобладающим материалом в лаборатории и осталось таковым до настоящего времени. На рис. 9 представлены разнообразные дистилляционные головки («шлемы»), применявшиеся в то время. Уже тогда в употреблении были и приборы в миниатюрном исполнении (см. рис. 9 п). О том, как выглядела лаборатория алхимика примерно в 1700 г., дает представление
* Т. е. дефлегмации, когда часть паров конденсируется еще до ввода в конденсатор, а образовавшийся при этом конденсат возвращается в перегонную колбу.—Прим. ред.
Рис. 8.
Противоточные холодильники:
а — холодильник Дарио с непрерывным охлаждением (XVI век); б — холодильник Либиха (XIX век).
23
Рис. 9.
Головин («шлемы») дистилляционных приборов по данным Андреаса Ли-бау (около 1600 г.):
а — с длинным носиком; б — с коротким носиком; в, г, з — глухне; д — из олова; е, ж — сублимационные; а, у — глухне, с тубусом; к — колоколообразная; л — в виде тиары; м. — оловянная с оттянутым иосиком; н — с патрубком; о — с охлаждаемым устройством; п — в миниатюрном исполнении; р — глухая с носиком; с — тройная глухая; т — тройная с ио-снкамн.
Рис. 10.
Лаборатория аптеки при монастыре капуцинов в Париже (около 1700 г,).
рис. 10, изображающий лабораторию аптеки при монастыре капуцинов в Париже. По обилию дистилляционных приборов можно догадаться, что дистилляция занимала большое место в лабораторной практике. До XVIII века типичные лабораторные приборы практически ничем не отличались от самых древних образцов, но уже в середине XVIII века химики, как например Бом и Вульф, начали создавать новые модификации приборов. Систематические исследования процесса перегонки впервые были начаты английским физиком Бойлем (1627—1691 г.), который уже ставил опыты с дистилляцией под вакуумом и при давлении выше атмосферного (рис. 11). В середине XVIII века был создан ряд типовых установок, которые нашли применение во многих странах (рис. 12). Перегонные кубы имели диаметр 45—75 см и высоту 90—120 см.
Спиртовая промышленность Франции стимулировала бурное развитие техники перегонки, начавшееся в XIX веке при активном участии французских инженеров. В результате ряда усовершенствований (Адам, Берар, Перье) Селлье-Блюменталю в 1813 г. был выдан патент на ректификационную установку непрерывного действия, которая в принципе вполне соответствует современным аппаратам (рис. 13). Аппараты для вакуумной перегонки сконструировал в 1828 г. Триттон. Следующим достижением была колонна с ситчатыми тарелками, предложенная Коффи в 1830 г. Принцип барботажных {тарелок использован Шам-поннуа в промышленных аппаратах (рис. 14а). Этот принцип ' -----х лежит в основе современной промышленной ректификации
наряду с принципом орошаемой насадки, предложенной Игле-сом. В 1873 г. ои впервые применил шары в качестве насадки.
Рис. И.
Прибор Бойля для простой вакуумной перегонки (1627-—1691 г.).
24
Рис. 12.
Полупромышленные Дистилляционные установки XIX века, применявшиеся: в Германии (а); в Китае (б) и в Болгарии (в).
На основании изложенного выше можно прийти к заключению, что в древности и в средние века, несмотря на простоту применявшихся аппаратов, уже были известны основные принципы перегонки. До середины XIX века лабораторные и полупромышленные аппараты различались только размерами. Лишь в период бурного развития органической химии в середине XIX века были созданы дистилляционные и ректификационные приборы, специально предназначенные для экспериментальных исследований в химических лабораториях. Такие выдающиеся химики этого периода (по 1900 г.), как Кляйзен, Димрот, Глинский, Хемпель, Либих, Мичерлих, Мор и Вурц, создали ряд приборов для лабораторной перегонки. В качестве куба и приемника стали применять реторту, а от дистилляционной колбы с боковой трубкой для воздушного охлаждения через колбу Кляйзена и приставку с шариковым дефлегматором Вурца пришли к барботажной колонне. Приставки Линнеманна, Глинского, а также Ле-Беля и Хеннингера являются прототипами колпачковой колонны [11] (рис. 146 и 15). Колонна с насадкой из стеклянных шаров была введена в лабораторную практику Хем-пелем в 1881 г.
В конце XIX века назрела необходимость сравнить по разделяющей способности уже имевшиеся в большом числе дефлегматоры, выполняемые в виде приставок к дистилляционным кубам. Крайс, Янг и Фридрихе [12] дополнительно исследовали также эффективность холо-
Рис. 13.
Непрерывно действующая ректификационная __ . - .- ................. установка Селлье—Блюменталя (1813 г.).
25
Рис. 14.
Колпачковые колонны:
а — колонна Шампониуа (1854 г.); б — стеклянная колонна Брууна с 20 та* релками и вакуумированной рубашкой (1931 г.).
Рис. 15.
Этапы развития колодны с колпачковыми тарелками:
а ~ перегонная колба с боковой трубкой для отвода паров; б — колба Кляй* зека; в — дефлегматоры с шарообразными расширениями и без расширения; г — шариковый дефлегматор Вюрца (1854 г.); д — дефлегматор Линнеманна с ситчатыми тарелками (1871 г.); е — дефлегматор Глинского с ситчатыми тарелками (1873 г.); тс — барботажная колонна Ле-Беля— Хеннингера (1875 г.); з — ректификационная колонна Янга—Томаса (1889 г.); и — тарельчатая колонна Баума (1910 г.); к — колпачковая колонна Брууна (1931 г.).
Рис. 16.
Перегонные аппараты образца 1920 г.:
а — вакуумная ректификационная установка Рехенберга с колонной Хемпеля и приемником дистиллята Бертрана; б — ректификационная колонна Эльснерса для работы при атмосферном давлении и под вакуумом.
26
дильников для конденсации паров. Наряду с холодильником Либиха приобретает известность холодильник Димрота с охлаждаемым змеевиком (см. рис. 66), и уже применяется эффективный холодильник Мичерлиха для низкоки-пящих веществ, послуживший прототипом для ряда аналогичных устройств.
На период с 1900 по 1920 г. приходятся многочисленные изобретения, применяемые и сегодня, например колонна Янцена (см. рис. 76), насадочные кольца Рашига (1916 г.) и насадочные кольца «Прим» (1919 г.). Был усовершенствован обогрев аппаратов и создан регулятор давления для вакуумной перегонки. На рис. 16а показана установка для вакуумной ректификации, которой пользовался известный специалист по перегонке Рехенберг [3] в период с 1900 по 1920 г. Эта установка, как и ректификационная колонна Эльснерса (рис. 166), демонстрирует уровень развития техники перегонки в 1920 г.
Теоретические труды Янцена [13] и его школы, проводивших систематическую научно-исследовательскую работу по изучению физических основ процесса ректификации, послужили фундаментом Для бурного развития этой отрасли техники после 1920 г. Период с 1920 г. и по настоящее время еще слишком мал, чтобы ему можно было дать историческую оценку. Однако мы видим, что корни современной техники перегонки с ее сложными аппаратами и методами уходят глубоко в прошлое, когда уже были известны ее основные принципы [14].
3
Стандартизация
в технике перегонки.
Способы выражения концентрации
3.1. Стандартизация методов перегонки и применяемых приборов
Научное и технико-экономическое значение стандартизации настолько общеизвестно, что, по-видимому, нет нужды еще раз это подробно обосновывать. Однако следует постоянно пропагандировать применение стандартных приборов. Во всех странах прослеживается тенденция к созданию стандартных приборов для различных процессов разделения. Благодаря серийному производству такие приборы стоят сравнительно дешево, они всегда имеются в наличии и позволяют получать сопоставимые результаты. Начало стандартизации было положено известными приборами Эиглера и Американского общества по испытанию материалов [ASTMJ. Аналогичный прибор можно собрать из стандартных деталей, например, для работы под вакуумом (рис. 17). В работах [1, 2] описаны стандартизованные приборы, их детали, а также специальные методы перегонки.
Основное преимущество метода блочной сборки состоит в том. что он позволяет надежно и быстро собирать нужные установки и обходиться при возможности многих комбинаций без применения специальной аппаратуры. При этом дополнительно появляется возможность в мельчайших деталях копировать конструкцию промышленных установок. При ремонте прибора в этом случае требуется лишь заменить вышедшую из строя деталь.
В наше время в любой лаборатории органической химии и в том числе в любой лаборатории, занимающейся процессами перегонки, применяют почти исключительно аппаратуру со стеклянными шлифами (табл. 1). Телле [3] подготовил указатель аппаратуры для химических лабораторий в соответствии с требованиями стандарта TGL, действующего в ГДР.
С начала XX века в качестве взаимозаменяемых соединений нашли применение главным образом конические шлифы (рис. 18); сферические шлифы (рис. 19) используют пока в тех приборах, в которых использование конических шлифов давало бы слишком жесткое соединение. В ГДР сферические шлифы изготавливают в соответствии со стандартами TGL 20678 и DIN 12244. Прецизионные сферические шлифы с номинальными диаметрами от 25 до 150 мм фирмы Бюхи (г. Флавиль, Швейцария) обеспечивают герметичное вакуумное уплотнение без применения специальной смазки.
В стандарте DIN 12242 дается подробное описание различных типов шлифов и возможных областей их применения для лабораторных установок. Размеры взаимозаменяемых стандартных шлифов с конусностью 1 : 10 регламентированы стандартами
28
TGL 14972 (лист 2) и DlN 12242. Для лабораторной перегонки интерес представляют шлифы рядов 1 и 2, из которых преимущественно используют шлифы NS 14,5/23, 29/32 и 45/40. Для микро- и полумикроприборов находят применение также шлифы NS 7,5/16, 10/19 и 19/26. Целесообразно было бы ограничиться применением в лабораторных приборах общего назначения шлифов NS 14,5/23, приспособленных также для термометров (TGL 40—339, DlN 12784), и шлифов NS 29/32. Тем самым значительно улучшилась бы взаимозаменяемость стандартных деталей и расширилась бы возможность их комбинирования. В приборах «Дестинорм», разработанных автором (см. гл. 7), этот принцип выдержан и отлично себя оправдал. Специально для тех случаев, когда требуется облегченная разборка шлифового
Таблица 1.
Размеры применяемых в ГДР стандартных шлифов для взаимозаменяемых соединений.
А — конические шлифы NS с конусностью 1:10 (тип Кб) согласно стандартам TGL 14972 (ноябрь 1971 г.) н DIN 12242 (по рекомендациям Международной комиссии по стандартам, ISO)
Кр аткое обозначение шлифа Больший диаметр конуса, мм Длина конуса, мм Краткое обозначение шлифа Больший диаметр конуса, мм Длина конуса, мм
NS 5/13 5 13 29/32 * 29,2 32
7/16 7,5 16 34/35 34,5 35
10/19 10 19 45/40 * 45 40
12/21 12,5 21 60/46 60 46
14/23 * 14.5 23 71/51 71 51
19/26* 18,8 26 85/55 85 55
24/29 24 29 100/60 100 60
Предпочтительный размер.
Б — сферические шлифы S согласно стандартам TGL 20678 (ноябрь 1971 г.) и DIN 12244 (июль 1963 г., шаровые шлифы)
Краткое обозначение шлифа Диаметр сферического шлифа, мм Краткое обозначение шлифа Диаметр сферического шлифа, мм
TGL DIN • TGL DIN*
S 7 7,144 38 ** 40/25 38,100
13,2 13/2 12,700 41 41/25 41,275
13 13/5 12 51 51/30 50,800
19 * 19/9 19,050 64 ** 64/40 63,500
29 29/15 28,575 76 76,200
35 35/20 34,925 102 101,600
* Второе число равно внутреннему диаметру примыкающей стеклянной трубки (в мм).
** Предпочтительный размер.
29
Рис. 17.
Прибор для вакуумной дистилляции, собранный из стандартных деталей:
1 — Трехгорлая колба NS2‘9 с двумя боковыми тубусами NS14.5; 2 — термометры со стандартным шлифом NS14.5; 3 — капилляр для подсоса воздуха; 4 — угловой переходник со стандартными шлифами NS29 и 14,5;
5 — холодильник Димрота; 6 — вакуумный форштос; 7 — короткогорлая круглодонная колба.
соединения, например при перегонке высококипящих смесей и особенно при работе с высоким вакуумом, стандартами TGL 14972/3 и DIN 12243 регламентированы стандартные шлифы 1 : 5. Кроме того, главным образом в крупногаба-
с конусностью
ритных приборах применяют линзовые и плоские шлифы. Разработаны проекты стандартов на детали аппаратов и на трубопроводы промышленного назначения, выполненные из стекла [7].
Фридрихе [4] и Флиднер [5] дают обзоры способов испыта-
ния и ухода за стандартными шлифами.
Известно много традиционных методов анализа определенных смесей путем перегонки, например метод, заключающийся в исследовании процесса выкипания сырой смеси фенолсодержащих кислот. Куэнханссом с coTp.Jfl6] разработан метод экспресс-анализа бензина с температурой кипения до 180 °C, который позволяет уловить имеющиеся примеси парафинов, циклопарафинов (с 5—6-звенной углеродной цепью) и ароматических веществ. Используемый прибор комплектуется стандартной головкой (см. разд. 7.5, рис. 312). В настоящее время стандартизованы методы определения температурных пределов кипения (табл. 2); регламентированы также и основные размеры приборов.
Так как стандартные шлифы нуждаются в смазке, то предпри-
нимались неоднократные попытки создать принципиально новые соединительные элементы для стеклянных приборов. Фирма «Научно-технические стеклянные приборы» (Вертхайм) разработала бесшлифовое конусное соединение, в котором для надежной герметизации стыка вместо шлифов применено вакуумное лабиринтное уплотнение. Благодаря двум уплотнительным кольцам из тефлона полностью отпадает необходимость в смазке соединения. Сферические шлифы фирмы Бюхи также не нуждаются в каких-либо специальных уплотнениях или смазках.
Высокая точность изготовления шлифов гарантирует герметичность соединения как при высоковакууме, так и при избыточном давлении. Подобные сферические шлифы выпускаются с номинальными диаметрами 25, 50, 80 100 и 150 мм. Французский завод лабораторного стекла «Совирель» освоил выпуск разъемного соединения, в котором применены штуцеры нового типа. Гермети-
30
|S'; Рис. 18.
Стандартный конический шлиф (конусность ! : 10): / — конус шлифа; 2 — муфта шлифа.
К р“с-
'Стандартный сферический шлиф с зажимом.
Ж'Рис- 20-
«В Разъемное соединение «Совирель»:
ЯК' 1 — штуцеры С резьбой; 2 — фланцы с виуТрейней резьбой; 3 — Защитная прокладка из тефлона; 4 — промежуточное кольцо для фиксации фланцев; зЯ|г 5 — кольцо из силиконовой резины.
Изготовитель: фирма «Совирель», Левалуа, Франция.
д» Таблица 2.
нет Стандартизованные методы перегонки
Стандарт Назначение метода перегонки
TGL21120 Испытание минеральных масел, жидких топлив и аналогичных
21125 продуктов; определение температурных пределов кипения Испытание жидких топлив; определение давления паров по Рейду
0-51761 Испытание технических углеводородов бензольного ряда; определение температурных пределов кипения по Кремеру—Шпиль-
0-51786 керу Испытание жидких топлив; определение содержания неотстаи-
fDlN 51751 вающейся воды в мазутах перегонкой по ксилольному методу Определение температурных пределов кипения моторных топлив для двигателей внутреннего сгорания с посторонним зажиганием, а также бензинов
? 51752 Испытание дизельных топлив и аналогичных материалов по тем-
51761 пературным пределам кипения Проверка температурных пределов кипения по методу Кремера—
51767 Испытание нефтей, высококипящих нефтяных фракций и кубовых остатков при перегонке нефти; дробная перегонка по методу
| 51611 Г россе—Етрингхауса Низкотемпературная ректификация газов
31
чность соединения обеспечивается химически стойкой эластичной прокладкой, зажимаемой между двумя обезжиренными стеклянными поверхностями (рис. 20). Подобные соединения сохраняют герметичность как под вакуумом, так и при избыточном давлении; их термическая стойкость определяется свойствами прокладочного материала.
Крамер [16] изложил историю разработки современных уплотнительных систем. По его данным в настоящее время разъемные соединения для лабораторных приборов, выполненных из стекла, можно подразделить на шесть классов.
1. Шлифовые соединения: конические, сферические, цилиндрические, плоские и комбинированные шлифы, а также сферические фланцы.
2. Соединения с полированными контактными поверхностями: прецизионные соединения с прозрачными контактными поверхностями и калиброванные прецизионные трубы.
3. Соединения с прокладками (за исключением винтовых соединений): соединения с упругими сферическими элементами, конусные соединения с уплотнительным кольцом, плоские фланцы с уплотнительным кольцом, сферические фланцы с уплотнительным вкладышем.
4, Винтовые разъемные соединения: сальниковые уплотнения, винтовые соединения с накидной гайкой, муфтовые винтовые соединения.
5. Фланцевые болтовые соединения: пластмассовые фланцы на резьбе, фланцы с сальниковыми соединениями, накидные фланцы.
6. Другие типы соединений и вспомогательные материалы: заглушки, шланги, манжеты, сильфоны, фиксаторы шлифовых соединений, смазки для шлифов и и т. п.
По свидетельству Крамера [16] в настоящее время винтовые разъемные соединения с накидными гайками выпускаются в трех различных исполнениях фирмами Квикфит, Витег и Совирель.
3,2. ОПРЕДЕЛЕНИЕ ОСНОВНЫХ ПОНЯТИЙ
Часто приходится сталкиваться с тем, что в опубликованных работах по технике перегонки одним и тем же основным понятиям даются неодинаковые определения, что нередко приводит к недоразумениям. Даже само понятие «перегонка» применяют для обозначения ее разновидностей, весьма различающихся в техническом отношении. В связи с этим целесообразно ввести принципиальное разграничение между понятиями «дистилляция» (или «простая перегонка») и «ректификация *. Таким образом, термин «перегонка» приобретает значение собирательного понятия для обозначения методов разделения смесей жидкостей путем испарения смеси й конденсации отходящих паров.
В своей работе [7а]Хампель уделил внимание сложной проблеме «чистоты» растворителей и обсудил понятие «ультрачистый». Под этим понятием оно подразумевает предельную чистоту, которая вообще достижима на сегодняшний день. Нередко содержание тех или иных примесей должно быть уменьшено вплоть до нескольких триллионных долей. Для глубокой очистки наряду с экстракцией используют и ректификацию.
Применительно к перегонке в промышленных условиях еще в 1943 г. был разработан стандарт DIN 7052 «Разделение смесей жидкостей посредством дистилляции и ректификации». Однако он уже не отвечает современному уровню техники перегонки, и к тому же в нем не учитывается специфика лабораторной перегонки. Ввиду этого и в соответствии с предложениями автора [8], бывшего Комитета по стандартизации лабораторных приборов (рабочей комиссии «Приборы для дистилляции и ректификации») и Комитета по стандартизации в химическом машиностроении были выработаны новые определения (которые используются по мере необходимости в данной книге).
* Вместо принятых в СССР терминов «дистилляция» и «ректификация» | автор здесь и далее применяет термины соответственно прямоточная перегонка ? и противоточная перегонка. —Прим. ред.
32
3.3. ЕДИНИЦЫ ИЗМЕРЕНИЯ
Новые правила выбора единиц измерения базируются на авторитетных решениях X Генеральной конференции по мерам и весам, состоявшейся в 1954 г. В ГДР переход к новым единицам измерения начался с первого постановления от 14.08.58 г. по физико-техническим единицам измерений, которое, однако, было аннулировано новым постановлением от 31.05.67 г. В ФРГ постановление о переходе к новым единицам измерений появилось в Вестнике законов 28 июня 1970 г. В книгах Падельта и Лапорта [9], Ферстера [10], а также Хедера и Гертнера [11] приводится информация о практическом применении и сферах приложения новой международной системы единиц измерений (СИ) со сравнительным анализом старых систем. Между тем в ГДР дополнительно были утверждены новые стандарты (табл. 3).
Единицей измерения силы в СИ является ньютон (Н, м-кг/с2), который заменяет старую килограмм-силу (кгс) при следующем соотношении:
1 кгс = 1 кг-9,80665 м/с2 = 9,80665 Н
Для измерения давления жидкости и газа следует применять единицу СИ — паскаль (Па, Н/м2). Дополнительно вводится более крупная единица — бар (1 бар = Ю5 Па). Пересчет проводится по соотношениям:
1 ат = 1 кгс/см2 = 0,980665 бар =735,56 мм рт. ст.
1 мм рт. ст. = 1,333-Ю~3 бар
Применявшаяся до сих пор единица тепла — калория (кал) заменяется джоулем (Дж):
1 кал = 4,1868 Дж
Термодинамическую температуру выражают в градусах Кельвина (К). Градус Цельсия (°C) является специальной единицей для выражения температуры по шкале Цельсия. Температура по этой шкале равна Т — То, где То — температура таяния льда (плавления) при нормальном давлении равная 273,15 °C. Для разности температур обозначение «град» больше не применяется.
При составлении списка обозначений основных величин, используемых в книге, приходилось следить за тем, чтобы для обозначения различных величин не применялся один и тот же символ. Поскольку оказалось невозможным обеспечить необходимое разнообразие символов с помощью одного только немецкого алфавита, то пришлось прибегнуть к буквам греческого алфавита и индексам, расширившим возможности варьирования [12].
Таблица 3.
Стандарты иа условные обозначения, физические величины и единицы измерения
TGL 0-1304 Общепринятые условные обозначения, условные обозначения
физических величин
TGL 18-762 Лист 1 — Физические величины и единицы измерения; найме-
нования, сокращения
Лист 2 — Пояснения
DIN 1301 Единицы измерения, сокращения
DIN 1313 Способ записи применяемых в науке и технике уравнений,
связывающих физические величины
2 Э. Крсль
33
3.4. СПОСОБЫ ВЫРАЖЕНИЯ
И ПЕРЕСЧЕТА КОНЦЕНТРАЦИИ
При расчетах в технике перегонки концентрацию принято выражать в мольных долях или процентах, так как при определении объема паров, скоростей потока паров и других величин это очень упрощает вычисления. Поскольку большей частью рассматривают разделение двух компонентов, то на практике расчеты многокомпонентных смесей часто сводят к расчету бинарных смесей, принимай за второй компонент совокупность остальных компонентов смеси. В этом случае среднюю массу 1 моля смеси можно определить по уравнению:
м________ОТ1 Ч~ от2 Ч~ тз__ -1- -1- отз /1.
СР т^/М^т2/М2т3/М3 N±-{- N3 N3
где tn — масса; М.—молекулярная масса; N = mlМ — число молей соответствующего компонента.
Пример расчета средней массы 1 моля смеси
= 300 г бензола
т2 = 400 г толуола
ffZg = 500 г ксилола __________
тг + т2 -|- т3 = 1200 г смеси Mj = 78,11; М2 = 92,13;
М3 = 106,16;
m-jM-^ 300/78,11 = 3,84 т2/М2 = 400/92,13 = 4,35 щ3/Л43 = 500/106,16 = 4,71
Ml + = 12,90
Л4ср = 1200/12,9 = 93,0 г
Состав смеси всегда выражают концентрацией легколетучего компонента, если это особо не оговорено. Концентрацию указывают в объемных, массовых или мольных долях или же в объемных, массовых или мольных процентах.
Под концентрацией понимают отношение количества одного компонента к суммарному количеству всех компонентов.
Объемная доля : компонент 1 = К1/(К1 + К2) (2)
компонент 2 С2 = V2/(V1 + V2) (3)
Массовая доля : компонент 1 «1 = «1/("11 + «г) (4)
компонент 2 п2 = m3/(mi + т2) (5)
Мольная доля : компонент 1 Х1 = Mi/(Mi + M2) (6)
компонент 2 Хз = N2I(N1 + N2) (7)
При умножении этих долей на 100 получают значения концентрации в процентах.
Пример определения мольной доли и мольного процента для смеси
Компонент 1 300 г бензола mj/Mj = 3,84
Компонент 2 400 г толуола т2/Л42 = 4,35
700 г смеси М + М2 + М = 8,19
Мольная доля = 3,84/8,19 = 0,47
Мольная доля х2 = 4,35/8,19= 0,53
Мольный % компонента / 0,47-100 = 47,0% (мол.)
Мольный % компонента 2 0,53-100 = 53,0% (мол.)
34
При расчете концентраций многокомпонентных смесей знаменатель в соответствующих уравнениях увеличивается на дополнительное число слагаемых (компонентов), например для трехкомпонентной смеси:
Мольная доля xt = + Na) (8)
Мольная доля х2 = + ^з) (9)
Мольная доля ха = Na/(Ni + М2 + ^з) (10)
Лессельс [13] приводит удобную номограмму для пересчета % (масс.) в в % (мол.) и наоборот для трехкомпонентных смесей.
Для бинарных смесей пересчет концентраций проводят по следующим уравнениям, отнесенным к легколетучему компоненту 1.
Пересчет % (масс.) в % (мол.):
% (мол.) = —’ 100% ' -]- т2/Л42 (И)
Пересчет % (об.) в % (масс.):
Пересчет % % (масс.) = • 100% 1 1Р1-Ь г 2Р2 (мол.) в % (масс.) (12)
Пересчет % %(МаСС-)-Л1Л+Х2'100% (масс.) в % (об.) ' % (об.) = - 100% ^h/Pi + тз/Рз (13) (14)
Пересчет % (мол.) в % (об.):
«.«./Z.+X./p, 1Ю% (15)
Ввиду того, что такие пересчеты громоздки, были разработаны различные номограммы, обеспечивающие быстрый и простой пересчет. При переходе от мольных или массовых процентов к объемным следует учитывать, что пересчет дает правильный результат лишь в том случае, если при смешении компонентов не происходит изменения объема (отсутствует контрактация). Орличек, Пёлль и Валенда [14] разработали универсальную номограмму (рис. 21), применение которой пояснено следующей схемой:
При пересчете Вместо Q подставить Для компонента с индексом 1*** следует понимать
под Z под X
% (мол.) в % (масс.) % (об.) в % (мол.) % (об.) в % (масс.) Л41/Л42* Умол, 1/УмОЛ, 2 ** Р2/Р1 % (масс.)/100 % (об.)/100 % (об.)/100 % (мол.)/100, т. е. мольную долю % (мол.)/100, т. е. мольную долю % (масс.)/100, т. е. массовую долю
* М — молекулярная масса; ** ^мол — мольный объем;*** индексом 1 отмечают величины, относящиеся к компоненту с большей молекулярной массой.
Пользование номограммой поясняется построениями, приведенными на рис. 21, для следующего примера: скольким % (масс.) соответствуют 88% (мол.), если- компоненты имеют молекулярные массы 150 и 60?
= 150 __ 2 5 X =88% (мол.) или 0,88 мольных долей
Л42 = 60 Z = 94,8% (мол.) или 0,948 мольных долей
2*
35
Рис. 21.
Номограмма Орличека, Пелля и Валенды для пересчета концентраций.
Рис. 22.
Номограмма Бэра для пересчета мольных процентов в массовые и наоборот.
Разумеется, с помощью этой номограммы можно с таким же успехом провести обратный пересчет из % (масс.) в % (мол.), из % (мол.) в % (об.) и из % (масс.) в % (об.).
Для газообразных смесей значения концентрации, выраженные в мольных и объемных процентах, совпадают; объемы относят к атмосферному давлению, принимая, как обычно, что объем 1 моль равен 22,4 л при 0°С и 760 мм рт. ст. или 24,4 л при 15° С и 735,5 мм рт. ст.
Для пересчета из % (мол.) в % (масс.) и наоборот особенно удобна ая обеспечивает быстрое и надежное
50 круглая номограмма Бэра [15], получение результата, хотя достигаемая при этом точность не так велика, как при расчете с применением формул.
Пользование номограммой иллюстрируется примером, приведенным на рис. 22. Требуется определить, скольким мольным процентам соответствуют 30% (масс.) бензола в смеси бензол—толуол. Известно, что для бензола = 78,11, а для толуола М2 = 02,13. Тогда = 78,11/92,13 — 0,848 ~ 0,85.
Через точку 30% (масс.) на нижней полуокружности (см. рис. 22) и точку 0,85 на горизонтальной оси M-JМг проводим прямую, которая пересекает верхнюю полуокружность в точке 33,6% (мол.).
Концентрации изотопов в смеси принято выражать в атомных процентах. Например, природная вода имеет следующий состав:
99,9844% (ат.) Н
0,0156% (ат.) D
100,0000% (ат.) водорода
99,757% (ат.) 16О 0,039% (ат.) 17О 0,204% (ат.) 18О
100,000% (ат.) кислорода
4
Физические основы процесса разделения
4.1. ПРИНЦИПЫ дистилляции и ректификации
Как указывалось выше, термин «перегонка» можно заменить на «разделение по каплям» (см. гл. 2). Этот термин имеет, таким образом, собирательное значение и относится к процессам разделения смесей взаиморастворимых жидкостей путем испарения жидкости и конденсации паров, обогащенных легколетучим компонентом. Поэтому термин «перегонка» не содержит никаких пояснений относительно характера процесса. Понятия «дистилляция» (или простая перегонка) и «ректификация» четко определяют также и способ работы (рис. 23).
Со строго физической точки зрения перегонка не является обязательно процессом разделения. Например, перегонкой можно назвать также процесс, в ходе которого чистое жидкое вещество испаряют, пары этого вещества конденсируют и конденсат собирают. Ректификация с бесконечным флегмовым числом представляет собой предельный режим перегонки, при котором конденсат (дистиллят) не отбирают.
Массообмен можно описать следующим отношением:
т — К.а, Лх I ср
где т — масса переносимого вещества; К — коэффициент массопередачи, а/ — активная поверхность контакта фаз; Ал'ср — движущая сила процесса массооб-мена.
Обмен веществом происходит путем его диффузии через межфазную поверхность. На скорость массоотдачи в пределах одной фазы влияют коэффициент диффузии D, расстояние Ь, на которое передается вещество, и концентрация х. От площади межфазной поверхности at, приходящейся на единицу высоты ректификационной колонны, зависит количество вещества, переходящего из одной фазы в другую.
Движущая сила массообмена между двумя фазами определяется разностью концентраций фаз (у—х) *. При термодинамическом равновесии наступает состояние насыщения, движущая сила становится равной нулю, и процесс массообмена, протекавший до установления равновесия, прекращается.
* Т. е. разностью у* —у между концентрацией пара у*, находящегося в термодинамическом равновесии с жидкостью состава х, и средней действительной концентрацией пара у, соприкасающегося с этой жидкостью. — Прим, ред-38
Рис. 23.
Схема, поясняющая принцип перегонки: а — дистилляция (простая перегонка); б — ректификация.
’ -жшкость
При дистилляции (или простой перегонке) молекулы, отрывающиеся с поверхности испарения, движутся в одном и том же направлении до момента достижения поверхности конденсации. Отличительная же особенность ректификации состоит в том, что поток жидко-. сти (как правило сконденсированных паров) направляется навстречу поднимающемуся потоку паров. Если дистилляция состоит всего лишь из процессов испарения и конденсации, то при ректификации благодаря тесному контакту двух фаз в колонне имеет место массо- и теплообмен. Рассмотрим в общих чертах процесс, протекающий на тарелке колонны (рис. 24). При установившемся режиме составы пара и жидкости на одной и той же тарелке изменяются в направлении достижения термодинамического равновесия между ними под влиянием градиентов температур и концентраций. Вследствие переноса вещества в вертикальном направлении (парами вверх, а жидкостью вниз) это равновесие нарушается, что благоприятствует дальнейшему обогащению паров легколетучими компонентами [1]. Другими словами, поток жидкости (флегма) на своем пути из зоны более низких температур (вверх колонны) в зону более высоких температур (кипятильник) поглощает из потока паров высококипящие компоненты и выделяет легколетучие компоненты. Температурному градиенту в колонне соответствует перепад концентраций в парах Г и в жидкости. При этом в кипятильнике пар менее насыщен легколетучим компонентом, чем в головной части колонны, а ^жидкость (флегма) в верху колонны содержит больше легколетучего компонента, чем на входе в кипятильник.
Физические процессы массо- и теплообмена протекают на границе раздела фаз, поэтому очень важно создать максимально •возможную поверхность контакта. Такая поверхность контакта имеет место в полой орошаемой колонне, она может быть образована насыпной насадкой, тарелками, сетками и различными (вращающимися устройствами (см. разд. 4.3 и 7.3.).
На процесс разделения влияют многие факторы и в первую Зчередь физико-химические свойства смеси, геометрические пара-етры и колонны и ее насадки, а также условия работы колонны.
Как правило, компоненты жидкой смеси, температуры кипения которых различаются более, чем на 50 °C, мсжно до опре-
( 39
деленной степени разделить, применяя простое упаривание или дистилляцию. Поэтому дистилляцию применяют преимущественно в тех случаях, когда жидкая смесь содержит в незначительных количествах компоненты с очень высокой температурой кипения или нелетучие примеси. В качестве примера можно указать на отделение растворенных нелетучих примесей при простой перегонке воды или на очистку растворителей от высококипящих примесей. Исключением является так называемая перегонка по Энглеру [2], которую применяют для определения температурных пределов кипения смесей весьма близкокипящих компонентов с точками кипения примерно до 200 °C. В данном случае при дистилляции создаются легко воспроизводимые условия, а именно адиабатический режим без орошения. Смеси близкокипящих компонентов нельзя разделить простой перегонкой, поскольку хотя бы даже вследствие брызгоуноса высококипящий компонент может механически увлекаться парами в дистиллят.
По способу работы различают периодическую и непрерывную перегонку. Периодическая перегонка (разгонка) — это простая дистилляция или ректификация, в процессе которой содержимое куба частично или полностью отгоняется. Непрерывная перегонка —это простая дистилляция или ректификация, в процессе которой исходную смесь непрерывно вводят в перегонный аппарат, а различные выделенные из смеси продукты непрерывно выводят из аппарата.
Ректификация обеспечивает разделение компонентов и в том случае, если их температуры кипения различаются всего лишь на 0,5 °C. Применяя ректификационные колонны с числом теоретических тарелок порядка 400—500, как это требуется при разделении изотопов, удается разделять смеси компонентов с разностью температур кипения всего лишь 0,05 °C. С помощью
Гэлодка колонны
Темп, низкая Конц.бысокая
Жидкостьх^
И £ И МММ
& * А А тш
Тарелка
— пар, обогащенный легколетучим компонентом
— пар при входе на тарелку
— жидкость, обогащенная труднолетучим компонентом
- жидкость при входе на тарелку '
Куб
Темп, высокая Конц, низкая
Рис. 24.
Схема процесса разделения на тарелке ректификационной колонны.
40
Таблица 4.
Рабочая схема решения поставленной задачи разделения
Факторы, подлежащие рассмотрению Определяемые условия или рассчитываемые параметры Ссылка на соответствующий раздел
Разность температур ки- Способ перегонки: дистилляция или 4.1
пения ректификация 4.7.5.1
Кривые давления паров Оптимальное давление процесса 4.4 4.6.2 5.3 5.4
Кривая равновесия Число теоретических ступеней разделения и условия ректификации 4.6—4.7.4
Требуемая степень раз- Способ перегонки и режим процесса 4.7.5
деления 4.8
* 4.9 5 6
Требуемая пропускная Тип применяемой аппаратуры, разме- 4.11
способность ры прибора и тепловые характеристики 4.12
процесса 5.1
5.2 7
Эффективность колонны Метод испытания эффективности колонны 4.10
Автоматизация Тип контрольно-измерительной и регулирующей аппаратуры 8
[ селективных методов, а в трудных случаях дополняя ректифи-[ кацию другими методами разделения, например экстракцией, | ионообменным методом и особенно газовой хроматографией, В ,, удается разделять смеси, прежде считавшиеся неразделимыми. I/ В последующих главах более подробно рассматриваются сложные Вл" процессы ректификации, которые были теоретически детально исследованы Куном, а также Куном и Риффелем [3].
Е До проведения перегонки, дистилляции или ректификации, г прежде чем начать расчеты, необходимо решить ряд вопросов. I В табл. 4 приведена рабочая схема, которая должна помочь £ при решении поставленной задачи разделения. Более подробные i , инструкции по проведению процесса перегонки даны в разд. 4.14. £ у Сведения, касающиеся сборки установок, приведены в разд. 9.3, | а рекомендации по пуску аппаратов — в разд. 9.5.
II 4.2. ОСОБЕННОСТИ ДВИЖЕНИЯ ЖИДКОЙ
М. ФАЗЫ В КОЛОННАХ С ОРОШАЕМОЙ НАСАДКОЙ
4.2.1. РАСПРЕДЕЛЕНИЕ ЖИДКОСТИ ПО НАСАДКЕ
Для разделения веществ путем ректификации применяют в основ-йЯ'.ном следующие колонны: тарельчатые (см. разд. 7.3.3), пленоч-S ные (см. разд. 5.4.3), с регулярной насадкой (см. разд. 7.3.4), ЙЯ5 с насыпной насадкой (см. разд. 7.3.2.).
41
Рис. 25.
Схематическое изображение характера работы колонн различных типов (по данным Штаге):
а — с колпачковыми тарелками; б — с ситчатыми тарелками; в — с провальными тарелками; г — с насадкой «Спрейпак»; д — насадочная; е — пленочная.
Работа колонн этих четырех типов основана на общем принципе противоточного движения контактирующих фаз. Существенные различия в характере движения контактирующих материальных потоков определяются геометрическими особенностями применяемых типов колонн.
Колонны последних трех типов можно объединить общим понятием колонны с орошаемой насадкой. Их отличительным признаком является образование пленки жидкости в процессе встречного движения сплошных потоков . контактирующих фаз при полном отсутствии их взаимного проникновения. Напротив, тарельчатая колонна характеризуется тем, что при ее работе жидкая фаза пронизывается, более или менее раздробленными пузырьками газа, которые вновь объединяются на вышележащей тарелке (рис. 25). В качестве эталонных жидкостей для исследования массообмена в противоточных колоннах пригодны фреоны, а также растворы неорганических хлористых соединений иода [За]. При исследовании характера движения жидкой фазы применяли также радиоактивные изотопы, например при изучении процесса ректификации бутадиена был использован изотоп 82Вг [36].
Важной особенностью колонны с регулярной насадкой является то, что ее насадка имеет регулярную геометрическую форму. «Пакетная насадка» Зульцера представляет собой орошаемую насадку из проволочной сетки, снабженной гофрами, вызывающими движение контактирующих потоков по удлиненной зигзагообразной траектории. В распылительной же колонне 42
''а'"
L?
«Спрейпак» жидкая фаза под воздействием паров, обладающих р значительной кинетической энергией, приводится в распыленное Ь состояние, а газовая фаза проходит через насадку в виде сплош-Е кого потока. Уже из сопоставления этих устройств можно полу-чить представление о сложности гидродинамического режима Is в насадочной колонне. Если в колоннах с регулярной насадкой материальные потоки движутся по определенным траекториям, I то при хаотическом расположении насыпной насадки происходит | неравномерное и неупорядоченное движение обеих контакти-Е рующих фаз.
fc Для колонн с насыпной насадкой характерна йеравномер-Г ность распределения как газовой фазы, так и жидкости. Нерав-g номерное распределение жидкости вызывается следующими при-К чинами [4].
В- 1. Жидкость перетекает по насадочным телам предпочти-к‘ тельно в направлении к стенкам колонны, а не наоборот. В ре-Ц" зультате частичной конденсации паров, вызванной тепловыми № потерями, процесс накопления жидкости у стенок колонны еще к более усиливается. Этот вид неравномерности орошения назы-К’ вают пристеночным эффектом (см. разд. 4.8.1).
К • 2. Вследствие неудовлетворительной смачиваемости самих на-
IV садочных тел случайно образовавшаяся струйка жидкости не В разрушается (каналообразование, см. разд. 4.8.1).
₽> 3. Насадка может состоять из отдельных слоев с предпочти-
|Г тельными направлениями для стока жидкости. И в этом случае № имеет место каналообразование. Расслоение насадки может быть К вызвано особенностями выбранного способа загрузки насадочных тел в колонну или причинами, связанными с геометрической №• Формой насадочных тел.
Ж 4. Колонна может быть установлена не точно вертикально. Н 5. Не всегда обеспечивается равномерная подача флегмы Иг на насадку.
. Результат действия всех этих факторов можно условно предик ставить следующим образом:
Неудовлетворительное распределение = Каналообразование ф-
№Ё..' + Пристеночный эффект
Иг На пристеночный эффект оказывают влияние высота колон-Икны Нк, ее диаметр dK, а также диаметр насадочных тел da. При Игртношении djd,, 20 поток жидкости, стекающей по стенкам ИЕколонны, составляет от 10 до 20% общего количества жидкости МО зависимости от отношения высоты колонны к ее диаметру к^к/^к).
|Ик Влияние неудовлетворительного распределения жидкости тем ИВшачительнее, чем большим числом теоретических ступеней раз-Миеления обладает колонна. При коэффициенте неравномерности ИяЬрошения 10% число теоретических ступеней разделения, полу-ИЕяаемых в колонне, эквивалентной, например, 100 ступеням,
43
снижается до 30, а в колонне, эквивалентной 10 теоретическим ступеням разделения, — всего лишь до 9 ступеней. Ввиду того что в промышленности также наблюдается устойчивая тенденция к повышению эффективности колонн, вопрос распределения жидкой фазы приобретает особо важное значение.
Трудности, связанные с установкой колонны в строго вертикальном положении или с первоначальным распределением жидкости, могут быть так или иначе преодолены. Поэтому основное внимание при исследовании насадочных колонн в настоящее время уделяется тому, чтобы получить ясное представление о характере распределения жидкой фазы в ходе процесса разделения. Эффективность этого процесса снижается с увеличением как диаметра, так и высоты колонны.
Неравномерность в распределении жидкости по колонне всегда вызывает снижение числа теоретических ступеней разделения вследствие того, что соотношение расходов пара и жидкости в различных точках насадки отклоняется от нормы. Глубокий теоретический анализ влияния неравномерности распределения жидкости на эффективность колонны был выполнен Хьюбером и Хильтенбруннером [5]. Они приняли за основу модель, представляющую собой колонну с четырехугольным поперечным сечением, разделенную воображаемой продольной перегородкой на два отдельных отсека, работающих при неодинаковых нагрузках. В качестве критерия (коэффициента) неравномерности орошения I было принято относительное отклонение локальной плотности орошения от ее среднего значения, рассчитанного для поперечного сечения колонны
/ = (£ — £)/£
где L — локальная плотность орошения (в определенной точке); L — средняя (по поперечному сечению колонны) плотность орошения.
Воображаемая перегородка многократно пересекается движущимися вихрями, что связано с переносом вещества в радиальном направлении, и параллельные потоки частично смешиваются между собой в количестве, характеризуемом степенью перемешивания tnq (рис. 26).
Коэффициент использования насадочной колонны п/п0 является функцией коэффициента неравномерности орошения I и степени перемешивания tnq, отнесенной к одной теоретической ступени разделения:
п/п0 « 1/[1 + (/8/3,75/п^)]
где mq — степень перемешивания, отиесеииая к одной теоретической ступени и показывающая, какую долю от всего поперечного сечения колонны составляет площадка, на которой происходит смешение струй при движении потока в пределах ступени разделения, п0, п — число теоретических ступеней, получаемых в колонне соответственно при равномерном и при неравномерном орошении.
44
Степень перемешивания mq, отнесенную к одной ступени разделения, можно оценить на основе теоретических выкладок Хьюбера по формуле
mq = 27 (dH/dK)2
Например, для dK/da = 10 получаем mq — 0,27, а для dJdH = = 30 имеем mq = 0,025. В первом случае степень перемешивания tnq настолько велика, что даже значительное повышение коэффициента неравномерности орошения мало снижает коэффициент использования насадочной колонны. Во втором случае такая зависимость не имеет места. Этот пример иллюстрирует эмпирическое правило, которое требует, чтобы отношение dKlda находилось в интервале от 10 до 30. Поэтому представительное значение числа теоретических ступеней разделения для колонны можно получить лишь при отношении djda = 10 и при I 0.
Рис. 26.
Схема модели колонны без перемешивания в поперечном направлении по Хьюберу —Хильтеибруииеру [5].
Колонна разделена продольной перегородкой на две секции 1 н 2. Внутри каждой секции расходы газа G и жидкости L постоянны, хотя они могут быть и не одинаковыми в разных секциях. На данной схеме расходы газа в секциях 1 и 2 одинаковы, а расходы жидкости — неодинаковы.
уИ — концентрация легколетучего компонента (ЛК) в газовой фазе на входе в колонну; х0 — концентрация ЛК в жидкой фазе на входе в колонну; Х/ю, Уго — концентрация ЛК в газовой фазе на выходе из секций 1 и 2 соответственно; х1и, х2и — концентрация ЛК в жидкой фазе иа выходе из секций 1 и 2; I — критерий неравномерности распределения потоков в колонне.
Любое неравномерное распределение можно приближенно моделировать, представив колонну в виде достаточно большого числа продольных секций. Изменение концентраций показано для бесконечного числа теоретических ступеней.
Рис. 27.
Схема, иллюстрирующая явление смачивания твердого тела жидкостью (<р — краевой угол смачивания):
а - полное смачивание; б - - частичное смачивание; в — полное отсутствие смачивания.
45
Кафаров, Дорохов и Шестопалов [6] подробно исследовали взаимосвязь между нагрузками колонны по обеим фазам и различными гидродинамическими параметрами, например динамической или статической удерживающей способностью колонны (см. разд._4.10.5), продольным перемешиванием перепадом давления (разд. 4.11). Они установили л количественную связь между динамической удерживающей способностьюхи перепадом давления, а также зависимость статической удерживающей способности от нагрузки, изменявшейся в широком интервале. С использованием понятий эффективного и «мертвого» объема была выведена теоретическая модель нестационарного движения жидкости в насыпной насадке; модель была использована для предварительного расчета параметров движения жидкости. Исследована также зависимость коэффициента продольного перемешивания от нагрузок по газу и жидкости, а также от физико-химических свойств жидкости. Ионас [7] проанализировал основные факторы, приводящие к продольному перемешиванию в насадочных колоннах. В своих экспериментах Тимофеев и Аэров ([65] к гл. 7) основное внимание уделили вопросам влияния диаметра колонны на эффективность разделения.
Неодинаковое поверхностное натяжение компонентов смеси может даже заметно отразиться на ходе процесса разделения в насадочной колонне [8]. Если в процессе ректификации бинарной смеси по мере отбора дистиллята поверхностное натяжение о флегмы увеличивается (такие смеси называют положительными), то массообмен может быть значительно более интенсивным, чем при разделении смесей, при ректификации которых поверхностное натяжение флегмы постепенно уменьшается (такие смеси называют отрицательными). Если смесь отрицательна, то поток жидкости, омывающий насадку, дробится на мелкие струйки и капли. Разумеется, этот эффект проявляется лишь в том случае, когда соблюдается условие До > 3 дин/см. Смеси компонентов, для которых коэффициенты поверхностного натяжения различаются менее чем на 3 дин/см, называются нейтральными.
В технических руководствах отношение смоченной поверхности насадки aw к ее полной поверхности а называют долей смоченной поверхности
фзд, = dwl а
Для расчета доли смоченной поверхности предложено много формул, которые, однако, приводят к весьма неодинаковым результатам [9]*.
* В специальной литературе (см., например, Рамм В. М., Абсорбция газов. М., Химия, 1976) проводят различие между смоченной поверхностью aw и активной поверхностью de. Это обусловлено тем, что участки поверхности насадки, покрытые неподвижной пленкой жидкости, не участвуют в массообмене, т. е. не являются активными. Соответственно вводится понятие доли активной поверхности -- ае!а. — Прим. ред.
46
Весьма показательно, что в подобных зависимостях совершенно игнорируется влияние таких факторов, как поверхностная энергия жидких смесей, стремление жидкостей уменьшить свою поверхность (особенно при пленочном течении отрицательных жидких смесей), а также влияние краевого угла смачивания, образующегося при контакте жидкости с поверхностью твердого тела (рис. 27). Возможно, что пренебрежение указанными факторами и является причиной значительных расхождений между вычисленными и экспериментальными величинами. Комбинируя уравнения Дюпре и Янга,, приходим к следующему простому соотношению для определения работы, затрачиваемой на образование единицы площади поверхности раздела фаз (или поверхностной энергии):
^s-i = <T(l+cosq>)
Из этого уравнения следует вывод, что возрастание поверхностного натяжения и уменьшение краевого угла смачивания приводят к увеличению поверхностной энергии IK.._j.
Так как поверхностное натяжение целиком определяется природой разделяемой смеси, то при подборе материала насадки следует стремиться к тому, чтобы зависящий от природы материала краевой угол смачивания был как можно меньше. В табл. 5
Таблица 5.
Поверхностная энергия IV's-i для систем твердое тело —
жидкость (вода прн 20 °C)
Класс веществ Материал Средняя высота выступов шероховатости, мкм Кр аевой угол смачивания (р, ° ^s-r эрг/см2
Металлы Вольфрам 62 109,0
Платина 71 96,5
Медь 0,1—0,5 84 80,4
Алюминий 85 79,2
Никель 86 77,9
Сплавы Сталь St.37 79 86,7
Сталь X-10-CnNiTi-18,9 0,1—0,5 83 81,7
Фосфористая бронза 84 80,4
Полимеры Полиамид 72 96,3
Полимер сложного эфира 72 96,3
Пол иметилм ета кр и л ат 72 96,3
Поливинилхлорид 73 94,1
Поликарбонат Менее 0,1 77 89,2
Полистирол 80 85,2
Полипропилен 90 72,8
Полиэтилен 90 72,8
Тефлон 106 52,5
Керамика Фарфор 5-15 50 119,0
Стекло 0,1 20 140,8
47
приведены рассчитанные значения поверхностной энергии для различных систем твердое тело—жидкость [11].
Очевидно, чем больше поверхностная энергия, тем более высокую стабильность пленки жидкости следует ожидать при смачивании твердого тела, но тем труднее, однако, добиться полного смачивания жидкой фазой элемента насадки [11]. Предварительным затоплением насадки (см. разд. 4.10.8) и выбором оптимальной конфигурации рабочей поверхности насадки можно значительно улучшить ее смачиваемость [9]. Титов и Зельвен-ский [10] предложили три метода расчета активной поверхности ае в колоннах с насыпной насадкой. Получены графические зависимости доли активной поверхности, высоты единицы переноса и коэффициентов массопередачи от плотности орошения.
Квасняк [11а ] исследовал эффекты конденсации и испарения при ректификации в насадочных колоннах, предположив, что в любом поперечном сечении колонны между паром и жидкостью всегда имеется разность температур. Поэтому несмоченные участки поверхности насадки можно рассматривать как поверхность теплообмена. Элементы сравниваемых насадок имели идентичную конфигурацию, но одни элементы представляли собой сплошные медные пластинки, а другие — пластмассовые пластинки, облицованные медью, благодаря чему обеспечивались различные коэффициенты теплопроводности. Пластинки были размещены в насадке так, что их нижняя сторона в процессе ректификации не орошалась. Насадки очень сильно различались по разделяющей способности, что можно объяснить эффектами конденсации и испарения, возникающими на сплошных медных пластинках. Влияние подобных эффектов следует всегда учитывать. Основываясь на этих результатах, Квасняк разработал новую регулярную насадку, состоящую из зигзагообразно изогнутых и различно ориентированных металлических листов. Такая конструкция обеспечивает дополнительную турбулизацию жидкой и газовой фаз и лучшую смачиваемость рабочей поверхности.
4.2.2. ПЛЕНОЧНОЕ ТЕЧЕНИЕ ЖИДКОСТИ
Йилмаз и Брауэр [116] при теоретическом анализе гидродинамических закономерностей движения жидкости в слоях насыпной насадки использовали математическую модель, соответствующую неустановившемуся пленочному безволновому течению вдоль гладкой пластины. Теоретические выкладки были подтверждены экспериментом. Исследование условий волнообразования на поверхности ускоренно движущейся пленки дали материал для построения универсальной диаграммы, представляющей различные режимы движения жидкости в насадках. Из диаграммы следует, в частности, что пленочное течение в слоях насадки является практически всегда ламинарным и безволновым. Эти данные послужили фундаментом при выводе теоретически обос
48
нованных зависимостей для расчета динамической удерживающей способности жидкости в слоях насадки из колец Рашига (см. разд. 4.10.5).
Кюнне [Не] подробно исследовал гидравлику противоточных колонн с насадкой из орошаемых пластин. Опыты были проведены при комнатной температуре и атмосферном давлении с применением системы воздух—вода. Кюнне приводит методику измерений и предлагает метод расчета гидравлического сопротивления и верхней предельной нагрузки по газовой фазе. Один из методов экспериментального определения площади межфазной поверхности в системе газ—жидкость для пленочных колонн предложен Антоновым с сотр. [11 з].
На основе теоретической модели, которая описывает движение пленок жидкости, механически перемешиваемых в колоннах с вращающейся насадкой, Диц с сотр. [11г] вывел уравнения для расчета толщины пленки. Уравнения базируются на ряде упрощающих посылок и справедливы в том случае, когда толщина пленки жидкости не превышает величины зазора между ротором и корпусом аппарата:
6 = /p + J/flK|-i/8p Р = Х2НГ1/(4Р«''г)
& = Г/(ХР£)
В = ^/(gn)
где 8 — средняя толщина пленки; % = 0,304; rt — внутренний радиус колонны; р — плотность жидкости; п — число оборотов ротора в 1 мин; Г — массовая линейная плотность орошения; jt — динамическая вязкость; г2 — расстояние от наружной кромки скребка до оси аппарата.
Годау [11д ] исследовал некоторые закономерности движения жидкостей в пленочных испарителях. Биллет [lie] анализирует непрерывный процесс пленочной дистилляции на примере пленочного испарителя «Липотерм», оперируя критериями подобия и приводя математические зависимости. Вычисления с помощью полученных уравнений показывают, что для простой перегонки с дефлегмацией существуют определенные соотношения нагрузок, при которых достигается максимальное обогащение дистиллята.
4.2.3. ДИНАМИЧЕСКИЕ ХАРАКТЕРИСТИКИ КОЛОНН
Динамические характеристики ректификационных колонн пытаются рассчи-; тывать, применяя различные математические модели. По Кёллеру и Шоберу [264] динамика колонн становится объектом изучения в тех случаях, когда нашей целью [, является: 1) исследование выходных параметров колонн во времени после простого или комбинированного возмущающего воздействия на процесс ректификации; ’ 2) моделирование процессов ввода и вывода колонн из рабочего режима, а также ^отклонений от него (предусмотренных или случайных); 3) поверочный расчет [нестационарных режимов промышленных установок; 4) расчет стационарных режимов как предельных случаев переходного процесса ректификации; 5) модели-?рование процессов управления установками; 6) улучшение динамических харак-
' теристик колонн с учетом существенных факторов, проявляющихся в неустано-
49
вившемся режиме (при изменении нагрузок по газу и жидкости); 7) теоретическое объяснение пульсационных явлений в ректификационных установках и проведение альтернативных расчетов в попытке устранить подобные явления.
Адольфи [11 ж] разработал графический метод расчета динамических характеристик колонн для ректификации бинарных смесей; в основе метода лежит диаграмма Мак-Кэба и Тиле. Вагнером с сотр. [162 а, б] исследована динамическая характеристика насадочных ректификационных колонн. Приводится информация о динамике колонн при разделении бинарных идеальных и неидеальных смесей, а также тройных систем. Исследована динамическая характеристика последовательно соединенных ректификационных колонн. Для расчета динамики колонн широко применяются ЭВМ (см. разд. 4.15).
4.3. РАЗДЕЛЕНИЕ СМЕСЕЙ ВЗАИМНО
РАСТВОРИМЫХ КОМПОНЕНТОВ
Взаимная растворимость жидкостей тем выше, чем ближе их химическое сродство (например, принадлежность к одному гомологическому ряду). Данные о закономерностях, наблюдаемых при смешении органических соединений, приведены в работе Штау-дингера [12]. В большинстве случаев взаимная растворимость возрастает с повышением температуры; выше критической температуры смешения Ткр имеет место неограниченная растворимость. На рис. 28 приведена диаграмма растворимости для смеси вода—фенол.
Перегонке подвергают, как правило, гомогенные растворы; при расслоении смеси ее сначала разделяют декантацией. Разу-
Рис. 28.
Диаграмма состояния системы вода—фенол.
50
Рис. 29.
Диаграммы равновесия пар—жидкость для различных бинарных смесей: . а — бензол—вода, вода — хлорбензол, вода—ксилол; б — вода—фурфурол; в — вода—н-бутанол, вода —изобутанол; г — emop-бутанол—вода; а — уксусная кислота—н-октан, этанол—вода, сероуглерод—ацетон; е — * н-гексан—бензол, н-гептан —бензол, метанол—вода; ж — бензол—толуол, j этан—бутан, циклогексан—н-октан; з — ацетальдегид—фурфурол, аце-
у той—уксусная кислота, ;. азотная кислота—вода,
метилацетилкетон — пропионовая кислота; и — соляная кислота—вода, циклогексанол—фенол.
имеется, следует учитывать, что полностью взаимнонерастворимых ^веществ не существует. Растворенные вещества нельзя разделить
(механическими способами, и эффективным методом разделения г в подобных случаях является перегонка.
Перегонку в токе носителя, главным образом перегонку
= с водяным паром, и азеотропную ректификацию применяют также
для разделения смесей совершенно нерастворимых или частично растворимых компонентов. К перегонке в токе пара-носителя прибегают для снижения температуры процесса. При азеотропной же ректификации специальной добавкой определенного (вещества вызывают образование азеотропа между этим веществом
•R одним из компонентов разделяемой смеси, отделяя его таким ^Образом от остальных компонентов; получаемый при этом азеотропный дистиллят должен легко поддаваться разделению на ^составные части (см. разд. 6.2.1). Следовательно, в таких случаях необходимо знать диаграмму растворимости для азеотропной
51
Таблица 6.
Особенности кипения бинарных смесей
Тип смеси (см. рис. 29) Степень взаимной Свойства кипящего раствора Сравнение сил притяжения между одинаковыми (^1/1 н <*2/2) и неодинаковыми (aj/2) молекулами
растворимости компонентов
а Полностью нерас- Имеет минимальную тем- О|/2 «1/!
творимы пературу кипения “1/2 °2/2
б Частично раство- То же а1/2 а1/1
в г римы с областью расслаивания «1/2 «2/2
д Полностью растворимы » а1/2 а1/1 а1/2 а2/2
е То же Н ей деа л ьн ый р а створ, азеотропа не образует а1/2 < а1/1 «1/2 < «2/2
ж » Идеальный раствор а1/2 ~ а1/1
«1/2 ~ а2/2
3 Полностью рас- Неидеальный раствор, а1/2 > а1/1
творимы азеотропа не образует а1/2 > а2/2
и То же Имеет максимальную температуру кипения а1/2 > «1/1 «1/2 > а2/2
к » Образуются новые химические соединения -н с-1 Q Q с-1 с-1 сГ о"
смеси, чтобы выбрать соответствующую температуру, при которой данная смесь расслаивается. Из рис. 28 можно, например, видеть, что при температуре выше 68,8 °C гомогенный раствор существует при любом соотношении компонентов, а ниже этой температуры наличие или отсутствие расслоения определяется составом смеси.
Взаимная растворимость двух компонентов может изменяться в пределах от почти полной нерастворимости до полной смешиваемости; для характера же кипения смесей имеется гораздо больше возможных вариантов. В идеальных смесях молекулярные силы притяжения, действующие между одинаковыми и неодинаковыми молекулами, равны. Если силы притяжения между неодинаковыми молекулами меньше, чем между одинаковыми, то следует ожидать образования азеотропной смеси с минимальной температурой кипения (положительный азеотроп); в противоположном случае образуется азеотроп с максимальной температурой кипения (отрицательный азеотроп). Между этими двумя крайними смесями расположены неидеальные смеси, для которых кривая равновесия на соответствующей диаграмме
52
сверху или снизу асимптотически приближается к диагонали диаграммы. Штаге [13] наглядно представил эти взаимосвязи (табл. 6), а рис. 29 иллюстрирует соответствующие типы кривых равновесия. Более подробно эти вопросы рассматриваются в разд. 4.6 и 6.2.
Гильдебранд и Ротариу [14], указывая на различия в энтальпии, энтропии и активности, подразделяют растворы на идеальные, правильные, атермальные*, ассоциированные и сольватированные. Как можно судить по классификации бинарных растворов, предложенной Маузером и Кортюмом [15], теория бинарных жидких смесей, несмотря на многочисленные фундаментальные исследования, до сих пор еще находится в неудовлетворительном состоянии. В монографиях Манхена [16] и Шуберта [17] подробно излагаются основы термодинамики растворов.
4.4. ЗАВИСИМОСТЬ МЕЖДУ ДАВЛЕНИЕМ
ПАРА И ТЕМПЕРАТУРОЙ рt
При решении задачи разделения необходимо прежде всего установить связь между давлением р и температурой t для перегоняемых смесей, которую изображают в виде кривых давления паров. Если на миллиметровой бумаге построить график зависимости давления насыщенных паров от температуры, то с его помощью можно определить, при каком давлении лучше проводить дистилляцию или ректификацию (см. рис. 39). При этом для температуры лучше использовать логарифмическую шкалу. Выбор давления разгонки зависит от того, какая из следующих операций должна быть проведена: а) аналитическая разгонка; б) препаративная ректификация; в) перегонка с целью накопления продукта; г) сравнительная ректификация с целью моделирования промышленной ректификации в лабораторных условиях. При этом необходимо учитывать, принимая во внимание гидравлическое сопротивление колонны, что ректификацию следует проводить под давлением, исключающим опасность термического разложения вещества и обеспечивающим такую температуру в конденсаторе, при которой имеющаяся в распоряжении охлаждающая среда будет пригодна для конденсации паров.
При аналитической разгонке следует добиваться максимальной эффективности разделения; поэтому выбирают такое рабочее давление, при котором кривые давления паров для разделяемых компонентов находятся как можно дальше одна от другой, в результате чего обеспечивается более высокая относительная летучесть компонентов (см. разд. 4.6.2). То же относится и к препара-
* Гильдебранд называет атермальными такие растворы, в которых теплота смешения компонентов равна нулю, причем компоненты имеют различные мольные объемы. — Прим. ред.
53
тивной ректификации. При перегонке же с целью накопления продукта следует иметь в виду, что с уменьшением давления значительно возрастает объем паров, и тем самым ограничивается пропускная способность колонны. При сравнительной ректификации рабочее давление процесса выбирают в зависимости от того, какой вакуум может быть гарантирован в промышленной установке.
Весьма подробные данные по давлению насыщенных паров приведены Драйсбахом [18], который собрал также значения физических констант для 38 гомологических рядов на диаграмме Кокса (разд. 4.4.2). В таблицах Драйсбаха [19] указаны температуры кипения примерно для 500 органических веществ при 1, 10, 100 и 760 мм рт. ст. Сталл [20] собрал данные по температурам кипения для 1200 органических веществ при 1, 5, 10, 20, 40, 60, 100, 200, 400 и 760 мм рт. ст. Кроме того, он привел аналогичные данные примерно для 300 неорганических веществ и около 100 значений температуры кипения для интервала давлений от 1 до 60 ат. Штаге опубликовал уточненные данные по давлениям насыщенных паров соединений для соединений различных классов, а именно, для насыщенных спиртов жирного ряда Cj—С18 с прямой углеродной цепью [21 ], для метиловых эфиров насыщенных жирных кислот Cj—С18 с прямой углеродной цепью [22], фенолов [23] и летучих компонентов спиртовой барды и спирта-сырца [24]. Большое число справочных таблиц подготовил /Кордан [25]. Риддик и Туппс (младший) [26] приводят данные по давлению паров и другие физические величины для 254 растворителей. Заге и Ласей [27] представили данные по давлению паров и равновесию для легких углеводородов, H2S и СО2. Глазер и Рюланд [28] впервые построили кривые давления паров в интервале от 1 до 60 ат для 33 веществ. Йекель [29] приводит список литературы, охватывающей данные по давлению паров в области среднего и высокого вакуума. Дополнительные новые материалы можно найти в «Успехах технологии» и реферативных сборниках ([16] к гл. 1).
Несмотря на обилие накопленных данных, в лабораторной практике нередко еще возникает необходимость в самостоятельном определении давления насыщенных паров. В таких случаях целесообразно проводить собственные исследования; однако можно также воспользоваться различными методами расчета, описанными в разд. 4.4.2.
4.4.1. ЭКСПЕРИМЕНТАЛЬНОЕ ОПРЕДЕЛЕНИЕ ДАВЛЕНИЯ НАСЫЩЕННЫХ ПАРОВ
Существуют два принципиально различающихся метода определения давления насыщенных паров чистого вещества: а) динамический метод — определение температуры кипения при различных давлениях; б) статический метод — определение давления паров при различных температурах. Методика проведения измерения подробно описана Киницем в сборнике Губен—Вейля [30]. Милаццо [31] приводит сведения о методах и приборах, применяемых для измерения 54
малых давлений. Он отмечает, что при измерении давления паров в интервале от 10"5 до 10 мм рт. ст. хорошие результаты дает манометр Мак-Леода.
Применение статического метода в лабораторных условиях, особенно при повышенных температурах, связано с экспериментальными трудностями и чаще всего используют динамический метод определения давления паров, тем более, что контролировать давление не сложно, благодаря наличию точно работающих регуляторов. Разумеется, при подобных измерениях важно заранее убедиться в достаточной чистоте исследуемого вещества.
Необходимым условием точного определения температуры кипения является хорошее перемешивание кипящей жидкости с образующимися паровыми пузырьками. Классическим прибором для определения температуры кипения является эбуллиометр Светославского [32]. На рис. 30 изображен дифференциальный эбуллиометр в полумикроисполнении. Его можно использовать не только для определения точки кипения, но также для контроля чистоты веществ и для изучения явления азеотропии в многокомпонентных системах.
Вещество в объеме 10—12 мл загружают в оба колена 1 и 2. На стенки колена 1 наплавлен стеклянный порошок, а снаружи это колено обмотано греющей спиралью. Образующиеся пары, захватывая за собой жидкость, поднимаются в тубус 7, в котором измеряется температура кипения вещества. Далее пары проходят через тубус 6, в котором измеряется температура конденсации, в хо-
Дифференциальный эбуллиометр Светославского в полумикроисполнеиии с рабочим объемом 10 —12 мл:
/ — колено с расширением; 2 — колено прямое; 3 — трубка; 4 — счетчик капель; 5 — холодильник; 6, 7 — тубусы.
Рис. 31.
Эбуллиометр Светославского (усовершенствованный Ольшевским) для ге-тероазеотропиых смесей.
Рис. 32.
Эбуллиоскоп Вебера с теплоизолирующим кожухом:
1 — штуцер для загрузки жидкости и присоединения к вакуумному насосу; 2 — конденсатор; 3 — кипятильная трубка; 4 — теплоизоляция из стекловолокна; 5 — спираль компенсационного электрообогрева; 6 — карман; 7 - - конец разбрызгивающей трубки; 8 — рубашка; 9 — разбрызгивающая трубка; 10 — центральное углубление; 11 — электрическая свеча; 12 — колпак.
55
Рис. 33.
Установка Вебера для определения давления насыщенных паров:
1 — укороченный манометр; 2 — форвакуумный (буферный) сосуд емкостью 3 л; 3 — адсорбционные ловушки; 4 — ртутный диффузионный насос; 5 — масляный насос; 6 — вспомогательный манометр; 7 — манометр Мозера; 8 — эбуллиоскоп; 9 — воздушная камера емкостью 10 л; 10 — ртутный манометр; 11 — масляный манометр.
лодильник5, где пары полностью конденсируются. Жидкость возвращается в обогреваемое колено 1, проходя через счетчик капель 4 и трубку 3. Счетчик капель служит для контроля скорости испарения жидкости.
Для определения азеотропного состава сильно гигроскопических веществ, например пиридиновых оснований, фенолов, применяют трехступенчатый эбуллиометр. В этом случае измеряют одну температуру кипения и две температуры конденсации. При работе с чистыми веществами или азеотропными все три температуры должны быть одинаковыми. Усовершенствованный прибор для определения температуры кипения гетероазеотропных смесей * разработан Ольшевским (рис. 31).
Аналогично работает эбуллиоскоп Вебера, изображенный на рис. 32. С помощью этого прибора можно измерять давление насыщенных паров в интервале от 10 до 760 мм рт. ст. и выполнять другие операции, такие как калибровка термометров, эбуллиоскопические измерения, изучение равновесия пар—жидкость, получение характеристик различных фракций дистиллятов, например в нефтяной и коксохимической промышленности [33].
Прибор имеет вертикальную кипятильную трубку 3 с внутренним диаметром 34 мм и длиной 500 мм. Регулируемое нагревание жидкости обеспечивается снизу электрической свечой И, размещенной в углублении 10. Наружные стенки углубления для усиления парообразования покрыты наплавленным стеклянным порошком. С помощью колпака 12, доходящего почти до дна, все образующиеся пузырьки пара собираются, смешиваются с жидкостью и направляются в разбрызгивающую трубку 9 диаметром 4 мм, где происходит дополнительное перемешивание в двух шарообразных расширениях. Затем смесь через конец 7 трубки 9 в виде брызг попадает на карман 6 термометра. Разбрызгивающая трубка вместе с шариками окружена вакуумированной рубашкой 8. Отделившаяся от пара жидкость стекает с кармана термометра и через узкую кольцевую щель между рубашкой 8 и кипятильной трубкой 3 возвращается в цикл. Кончик термометра погружен в небольшое количество ртути и защищен карманом 6 с припаянным
* Определение понятий гомогенный азеотроп, а также гетерогенный азеотроп см. в разд. 6.2. — Прим. ред.
56
козырьком, предохраняющим его от попадания капель стекающего сверху_кои-денсата. Столбик ртути окружен конденсирующимися парами.
Наблюдение с помощью лупы за подсвеченной сзади шкалой термометра и подсчет десятичных делений шкалы через пленку конденсата и не представляет трудностей, если верхнюю часть эбуллиоскопа предварительно протравить в течение 2 мин 1%-ной фтористоводородной кислотой и затем прокипятить в мыльной воде. Кипятильная трубка 3 до самого конденсатора 2 окружена изолирующим слоем стекловолокна 4, в котором оставлена узкая смотровая щель. Под теплоизоляцией 4 на трубку 3 намотана спираль компенсационного электрообогрева 5, выполненная из тонкой проволоки. Мощность обогрева можно рассчитывать, условно представляя спираль в виде охватывающей прибор бесконечно длинной цилиндрической оболочки с равномерно распределенными источниками тепла. Электрообогрев регулируют с помощью амперметров и калибровочной кривой таким образом, чтобы без включения системы подогрева кубовой жидкости приближенно устанавливалась ожидаемая температура. В этом случае даже пары труднолетучих веществ доходят до конденсатора, расположенного на 250 мм выше кармана термометра. Адиабатический режим в разбрызгивающей трубке обеспечивается четырехкратной защитной системой, включающей вакуумированную рубашку, слой нагретой до кипения жидкости, стекающей в кольцевой щели, спираль компенсационного электрообогрева и слой теплоизоляции. Через штуцер 1 обычно загружают жидкость, а при работе под вакуумом к нему присоединяют вакуумную линию.
Общая схема установки, включающей кольцевой трубопровод для присоединения насосов, эбуллиоскопа и манометра, приведена на рис. 33. Стабилизацию
Рис. 34.
Схема прибора Херрингтона—Мартина для определения температуры кипения (например, пиридиновых оснований):
1 — кипятильная колба для исследуемого вещества; 2 — кипятильная колба для чистой воды; 3 — вакуумный масляный насос; 4 — газосборник емкостью 30 л; 5 — регулятор давления; 6 — газосборник емкостью 30 л в водяной бане (400 л); 7 — высоковакуумный масляный насос; 8 — сосуд с оксидом фосфора; 9 — парортутный эжекторный иасос; 10 — манометр . Мак-Леода; 11 — баллон с азотом; 12 — сосуд с аммиаком и раствором карбоната аммоиня над металлической медью; 13 — промывная склянка ' с разбавленной серной кислотой; 14 — 19 — сосуды соответственно с 10%-* иым раствором едкого натра, концентрированной кислотой, безводным хлористым кальцием, с силикагелем, с пятиоксндом фосфора, со стекло-' ватой; 20 — охлаждаемая ловушка.
57
Рис. 35.
Вид по А
Прибор Хеиииига—Штока для измерения давления паров легколетучих веществ:
/ — колбаХдля жидкости с термометром; 2 — шлиф с крючком для термометра; 3 — кран; 4 — патрубок для присоединения к вакуумной линии;
5 — измерительное колено; 6 — барометр; 7 — чашка со ртутью.
Рис. 36.
Изотеиископ, усовершенствованный Шубертом:
1 — шарик с исследуемой жидкостью; 2 — вспомогательный манометр;
3 — охлаждаемый шарик; 4 — патрубок для присоединения к буферной емкости; 5 — термостатирующнй кожух.
давления при 760 мм рт. ст. или при любом вакууме можно проводить по методам, описанным в разд. 8.3. Аналогичная установка описана Эблином [34]. Для определения температуры кипения с точностью ±0,003 °C рекомендуется прибор Херрингтона и Мартина [35], который применяли для измерения давления паров пиридина и его гомологов в интервале от 50 до 157° С (рис. 34). Высокая точность измерения давления достигается тем, что параллельно при тех же условиях, что и в перегонной колбе, испаряют особо чистую воду, и искомое давление вычисляют по температуре паров кипящей воды.
Очень изящный метод, однако обеспечивающий точность измерения всего лишь ±0,5 мм рт. ст., описан Нательсоном и Цукерманом [36]. Он основан на том явлении, что капля жидкости, висящая на конце вертикального капилляра (трубки термометра), в результате постепенного вакуумирования окружающего пространства отрывается от капиллира. В момент отрыва капли считывают показание ртутного манометра, соответствующее достигнутому давлению.
Метод Элиша для работ в микромасштабе с загрузкой порядка нескольких миллиграммов вещества был усовершенствован таким образом, что стало возможным проводить измерения также и при остаточных давлениях ниже 100 мм рт. ст. [37]. На обогреваемой микроподложке конструкции Кофлера можно также определять точки кипения при незначительных количествах вещества [38].
Давление паров при низких температурах можно также определять, применяя радиоактивные изотопы. Несмеянов [39] подробно описывает методику, специально разработанную применительно к металлам и сплавам. Для трудно-летучих веществ, например иода, нафталина и фенола, пригоден эффузионный метод [40].
В дополнение к изложенному ниже рассмотрено еще несколько статических методов определения давления насыщенных паров, в соответствии с которыми 58
температуру поддерживают постоянной с помощью термостата и измеряют установившееся давление по манометру. При измерении давления паров легколетучих веществ соответствующего интервалу температур от —20 до +20° С можно воспользоваться методикой, описанной Хеннингом и Штоком [41]. Устройство прибора показано на рис. 35.
В колбу 1 через шлиф 2 загружают исследуемое вещество, на крючке шлифа 2 подвешивают термометр с ценой деления 1/10° С и колбу помещают в ванну с охлаждающей смесью. Патрубок 4 соединяют с вакуумным насосом; кран 3 служит для отключения прибора от вакуумной линии. Измерительное колено 5 погружают в чашку 7 со ртутью барометра 6.
Систему вакуумируют и закрывают кран 3, который следует заранее проверить на герметичность. Затем колбу 1 слегка нагревают, чтобы поднять давление паров, после чего нагревание прекращают и кран 3 открывают. Эту операцию повторяют до полного удаления воздуха из прибора. После того, как в колбе 1 установится постоянная температура, можно начинать измерение. С помощью зеркальной шкалы (а еще точнее с помощью катетометра) определяют разность уровней для менисков ртути в измерительном колене 5 и барометре 6, которая и представляет собой давление паров исследуемой жидкости (в мм рт. ст.). При каждой температуре необходимо выполнить несколько операций по измерению давления, а результат усреднить. Целесообразно предварительно проверить работоспособность этого прибора путем измерения давления паров какого-либо чистого вещества, для которого точно известны соответствующие данные.
Описанный Смитом и Мензисом [42] изотенископ благодаря применению вспомогательного манометра и буферной емкости позволяет определять давление насыщенных паров для высококипящих веществ, причем получаются относительно хорошие результаты. Рис. 36 иллюстрирует устройство прибора, усовершенствованного Шубертом. При работе этого прибора очень важно, чтобы во вспомогательный (U-образный) манометр 2 перегналось нужное количество исследуемого вещества. Кроме того, необходимо с помощью крана так отрегулировать вакуум в буферной емкости (на рисунке не показана), присоединенной к патрубку 4, чтобы показания вспомогательного манометра 2 установились на нулевой отметке. Тогда главный манометр (присоединенный к буферной емкости) покажет давление паров исследуемой жидкости, установившееся в левом колене вспомогательного манометра 2. Изотенископ размещен в термостатирующем кожухе 5, что обеспечивает равенство температур, при которых находятся манометр 2 и шарик 1 с исследуемой жидкостью.
Измерение давления насыщенных паров проводят следующим образом [43]. Шарик 1 трубки изотенископа на 3/4 объема заполняют исследуемым веществом. С помощью толстостенной вакуумной резиновой трубки прибор подсоединяют в точке 4 к буферной емкости, содержащей воздух. Прибор термостатируют, и всю систему медленно вакуумируют (с помощью вакуумного насоса, присоединенного к буферной емкости). Когда давление достаточно понизится, вещество в шарике 1 закипает, его пары конденсируются в верхнем шарике 3. Конденсат стекает в нижнюю часть вспомогательного манометра 2 и тем-самым полностью исключает контакт кипящего в шарике 1 вещества с воздухом, содержащимся в.буферной емкости. Когда убеждаются, что воздух из левой части изотенископа удален полностью, в термостатирующем кожухе 5 устанавливают постоянную температуру и регулируют вакуум, добиваясь того, чтобы мениски жидкости в U-образной трубке установились на одном и том же уровне. Сразу же после этого замеряют температуру термостатирующего кожуха и давление, показываемое ртутным (главным) манометром. Чтобы быть уверенным, что в изолированной части изотенископа (т. е. в шарике 1) больше нет воздуха, отсасывание воздуха и измерение давления повторяют несколько раз. После получения нескольких повторяющихся результатов начинают медленно повышать температуру. При этом одновременно изменяют давление, следя за тем, чтобы мениски жидкости в U-образной трубке постоянно находились почти на одном и том же уровне. Во избежание обратного проникновения воздуха в шарик 1 следует всякий раз избегать избыточного давления в буферной емкости.
Мениски жидкости в U-образной трубке окончательно устанавливают на одном и том'же уровне при постоянном давлении в буферной емкости путем не-
59
Рис. 37.
Прибор Кортюма с сотр. для измерения давления паров статическим методом:
1 — паровая камера; 2 — дроссель; 3 — сборник; 4 — платиновые контакты; 5 — поплавки; 6 — приемник жидкости; 7 — стандартный шлиф NS20; 8 — краны; 9 — токоподводящие провода для платиновых контактов; 10 — патрубки для присоединения главного манометра; 11 — конические шлифы; 12 — стандартный шлиф NS12; 13 — приемник конденсата;
14 — патрубок для присоединения промежуточного манометра; 15 — железный сердечник; 16 — магнитная катушка; 17 — пластина из пористой керамики.
значительных изменений температуры в термостатирующем кожухе.
Кортюм с сотр. [44] для получения данных по общему давлению паров в системе вода—диоксан при 35° С применил прибор (рис. 37), обеспечивающий более высокую точность измерения (до ±0,2 мм рт. ст.).' Шлюндер [45] описывает простой статический метод измерения давления паров над водными растворами солей LiCl, MgCl2 и Mg (NO3)2 при температуре 20—90° С.
Масс-спектрометрический метод измерения низких давлений паров позво-ляет определить парциальные давления определенного вещества в присутствии ' загрязняющих примесей.
Рек [46] применил три различных статических метода для измерения очень низких давлений (0,056—17,9 мм рт. ст.) паров анилина. В качестве измерительных средств наиболее пригодными оказались ртутный манометр и манометр, заполненный анилином. Для определения давления насыщенных паров при значениях до 60 ат и при температурах до 500° С Глазер и Рюланд применили автоклав из стали V4A.
4.4.2. РАСЧЕТ И СПОСОБЫ ПРЕДСТАВЛЕНИЯ
ДАННЫХ ПО ДАВЛЕНИЮ НАСЫЩЕННЫХ ПАРОВ
Если нельзя непосредственно измерить давление паров какого-либо вещества или рассчитать его на основе уже имеющихся литературных данных, то это давление можно найти, исходя из атомных и структурных составляющих. Как чисто расчетные, так и графические методы определения являются всегда приближенными.
Петер [47 ] исследовал связь между давлением паров и моле-, кулярной структурой вещества, применив уравнение Ван-дер-Ваальса:
1g (1/П) = а (1/0 — 1) (16)
где П = р/ркр — приведенное давление; 0 = 'Г/Ткр — приведенная температура; Ркр и fKp — критические давление и температура; а — константа.
При расчете констант а по правилу аддитивности, исходя из соответствующих атомных и структурных составляющих, получаются значения, хорошо согласующиеся с эксперименталь-60
В'-
I ними данными. Примерно в 50% исследованных случаев разность I между расчетными и экспериментальными значениями «составила I менее 1 %. Эти расхождения никогда не превышали отклоне-г ний а, обусловленных влиянием температуры.
| Путем обработки результатов 73 опытов и сравнительных |г- вычислений были получены следующие значения атомных и ! структурных составляющих для определения а:
С —2,154 СООН 2,247
н 1,139 СО2 (сложный эфир) 0,507
сн2 0,124 Двойная связь 2,295
О 0,266 Тройная связь 4,605
ОН (спирт) 2,327 Разветвление в цепи С—С 0,056
с6н5 1,708
N —0,765
С1 1,356
к Исходя из структурной формулы соединения, критической | температуры и критического давления, по уравнению (16) можно к рассчитать давление насыщенных паров р для любой темпера-| туры Т. Аддитивность констант а сравнима с аддитивностью к мольной рефракции и парахора. Коллар и Наги [47а ] описывают ? способ, основанный на методе Киннея, позволяющий рассчиты-£ вать кривые давления паров по молекулярным структурам для интервала температуры кипения от 0 до 400 °C. При этом поляр-ность молекулы, конечно, не должна быть слишком большой.
| Аналогичный метод для оценки температур кипения органи-Р ческих жидкостей разработал Пирсон [48], исходя из размеров Г и формы молекул и учитывая их ассоциацию введением в расчет-। ную формулу поправочного коэффициента. Уравнение Эглоффа Г для расчета температуры кипения также дает возможность обой-| тись без экспериментальных данных [49]. Если для какого-либо I вещества известны температуры кипения при двух различных К- давлениях и имеется полная кривая давления паров какого-либо г’ эталонного вещества, то с помощью правила Дюринга [50] можно | рассчитать полную кривую давления паров для данного вещества. | Правило Дюринга выражается следующим уравнением:
к. Окип, 1—Анп>г)/(01 ®г) = ? (17)
В. где /Кип>1 и /к11п,2 — температуры кипения рассматриваемого вещества А при ₽ давлениях pt и р2; 0х и 02 — температуры кипения эталонного вещества Б при Давлениях рг и р2; <7 — константа.
| Из этого правила следует, что для двух веществ А и Б отноше-Е. ние разностей температур кипения при двух различных давлениях Е остается постоянным.
К Применительно к большому числу веществ Рехенберг [51 ] Е выбрал в качестве эталона воду, для которой имеются точные К данные по давлению паров.
61
Пример расчета температуры кипения уксусной кислоты; в качестве эталона выбрана вода.
1. Известные величины:
= 760 мм рт.ст. р2 = Ю мм рт. ст.
Уксусная кислота /Кип, 1 = 118,1 °C /кип, 2 = 63, 0 °C Вода 0j= 100,0 °C 62 = 51,5 °C
По этим исходным величинам в первую очередь вычисляем константу q, значение которой необходимо для дальнейших расчетов:
<7 = (118,1 — 63,0)/(100,0 —51,5) = 1,14
2. Расчет температуры кипения уксусной кислоты при 200 мм рт. ст.: pi = 760 мм рт. ст. р2 = 200 мм рт. ст.
Уксусная кислота /КИп,1= 118,1 °C /кип.г -? Вода 0Х - 100,0 °C 02 = 66,4 °C
/кип, 2 = /кип, 1 - q (91 - 02), /2 = 118,1 - 1,14 (100,0 - 66,4) = 80 °C
Полученное расчетное значение хорошо согласуется с измеренной величиной. По такой схеме могут быть рассчитаны и другие данные, однако необходимо иметь в виду, что для других эталонных веществ опытные значения температур кипения в области низких давлений часто бывают неточными. Разумеется, йри этом и разности температур, рассчитанные по правилу Дюринга, будут отклоняться от истинных значений. Следует стремиться к тому, чтобы температуры кипения эталонного вещества были установлены с погрешностью не более ±0,05 °C. В качестве эталона рекомендуется выбирать вещества, имеющие аналогичную структуру с исследуемым веществом, например соединения из одного гомологического ряда. Янцен и Эрдманн [52] при вычислении кривой давления паров для насыщенных жирных кислот с длинной прямой углеродной цепью использовали в качестве эталона миристиновую кислоту. Как показала практика, для полярных жидкостей в качестве эталона пригодна вода, а для неполярных или слабо полярных жидкостей — и-гексан. Еще один способ подбора эталонного вещества описан Киреевым [53].
Весьма часто зависимость между давлением насыщенных паров чистого вещества и температурой описывают уравнением Антуана [54]:
lg р=- a — b/(i + с) (18)
где а, Ь, и с — константы, зависящие от природы данного вещества; константа р является групповой константой и для алифатических углеводородов ее можно принять равной 230.
Использование этого уравнения возможно при условии, что константы а и b для соответствующего вещества известны из опыта. Публикуемые данные по давлению паров очень часто представляют в виде констант а я Ь. Примером являются таблицы Драйсбаха [19]. Вихтерле и Линек [58] опубликовали значения констант уравнения Антуана, которые можно использовать в памяти ЭВМ, управляющей производственными процессами. От-62
Дельные константы рассчитаны итерационным способом, причем Г указаны погрешности их определения.
Детальные исследования Томсона [551 показали, что по i уравнению Антуана получаются значения, наиболее близкие | к экспериментальным данным. Константы а и Ь уравнения Ан-I туана можно вычислить, если известны температуры кипения К вещества при двух давлениях р и рх.
I Константу Ь в уравнении Антуана можно вычислить по фор-I муле:
I ь = (1g р- + 230) (/„ +230)/(/ -/„) (19)
f Подставляя найденное значение b и с = 230 в уравнение Ь Антуана (18), находим константу а:
а = Ig р + b/(t + 230) (20)
к Значения для р№ и можно найти в табл. 7 для соединений | каждого гомологического ряда на диаграмме Кокса.
| Если известны точка кипения при атмосферном давлении и | критическая температура вещества, то с помощью универсальной К, формулы Риделя [56 ] для расчета давления паров можно получить Ь всю кривую давлений. Если давления измерены для двух любых К температур, то можно вычислить критическую температуру. Г Формула Риделя отличается особой точностью и позволяет уста-новить, являются ли вещества ассоциированными или нет. Раз-
В работанный Рекхардом [57] метод особенно пригоден для опре-Г деления температур кипения при вакуумной перегонке таких Ц многокомпонентных смесей, как смолы, дегтярные масла. Расчет jg облегчается применением двух номограмм.
К Обзор методов расчета давления паров дан в монографии И [59]. Отмер и Пу [60] приводят обзор методов расчета, основан-И ных на использовании свойств эталонных веществ. Универсаль-Е' ное уравнение для расчета давления насыщенных паров угле-водородов предлагают Зиа и Тодос [60а]. Среднее отклонение
L вычисленных по этому уравнению значений давления паров № от экспериментальных составляет 0,38%.
Ц Графические методы определения давления паров по сравнению К с расчетными методами обычно проще и требуют меньшей затраты р времени. По правилу Дюринга кривую давления паров получают К' следующим образом. Температуры кипения данного вещества А В* и эталона Б, соответствующие одному и тому же давлению, пред-К ставляют в прямоугольной системе координат в виде точки, К абсцисса которой равна температуре кипения вещества Б, а Ор-H. Дината — температуре кипения А. Точки, нанесенные для раз-Н личных давлений, лежат все без исключения на одной и той же HL прямой. На рис. 38 показана диаграмма Дюринга, характери-В' зующая давление паров уксусной кислоты; она построена с ис-ИЕ.пользованием воды в качестве эталонного вещества. Давление И Насыщенных паров уксусной кислоты для какой-либо определенна 63
tкип воды(5)t°C
Рис. 38.
Прямая, характеризующая давление паров уксусной кислоты, построенная по правилу Дюринга.
Эталонное вещество — вода.
Рис. 39.
Кривые давления паров воды (/), бензола (2) и суммарная кривая для Паров воды и бензола (3).
ной температуры находят следующим образом: эту температуру откладывают на оси ординат диаграммы Дюринга, затем отсчитывают на оси абсцисс соответствующую температуру воды и по кривой давления водяных паров (рис. 39) определяют соответствующее этой температуре давление. На диаграмме, представленной на рис. 38, можно, конечно, предусмотреть шкалу для давлений водяного пара, но в этом случае трудно отсчитывать промежуточные значения давления (ввиду неравномерности шкалы).
[5S На рис. 39 показаны обычные кривые давления паров; совершенно ясно, что для получения надежных кривых требуется сравнительно много данных. Поэтому стали искать способы выражения этих зависимостей в виде прямых линий. В прямоугольной системе координат зависимость логарифма давления паров от обратной абсолютной температуры кипения 1/Т носит линейный характер в соответствии с уравнением Клаузиуса—Клапейрона:
lgp = _Z^L ’ +с (21)
К 1
где: ЛНу — энтальпия испарения; R — универсальная газовая постоянная; с — константа интегрирования [61].
64
На рис. 40 указанная зависимость построена для нескольких Веществ с применением специальной бумаги *; по оси ординат которой отложены значения 1g р, а по оси абсцисс — 1/71; причем наноси абсцисс также отложены значения температуры по шкале Цельсия в интервале от —23 до -ф 127 °C [62]. Очевидно, по наклону этих прямых можно определить энтальпию испарения. В соответствии с другой формой уравнения Клаузиуса—Клапейрона (21)
lg₽2 = lgPi(A#V. 1/д#У.г) + с (22)
можно построить графическую зависимость логарифма давления паров вещества 2 от логарифма давления паров вещества 1; при этом также получается прямая линия. Тангенс угла наклона этой прямой равен отношению энтальпий испарения веществ. Чтобы угол наклона прямой был по возможности ближе к 45 °, в качестве эталонного вещества для углеводородов следует принять «-гексан, а для ассоциированных веществ, например кислот и спиртов — воду, поскольку кислоты и спирты подобно воде, обладают большой энтальпией испарения. На основе урав-
* Бумага фирмы «Шефере Файнпапир», Плауэн, ФРГ; заказ № 602 для интервала температур от —24 до 726 °C; заказ № 603 для интервала температур от —23 до 127 °C. — Прим. ред.
: Зависимость давления паров некоторых веществ от температуры в системе ( координат igр — 1/7:
к 1 — пропионовая кислота; 2 — вода; 3 — этанол; 4 — «-гексан; G — диэтнловый эфир.
\Р«с. 41.
Диаграмма Кокса для гомологического ряда алкилбензола.
3 Э. Крель
65
Нения (22) Отмер с сотр. [63] разработал номограмму и таблицы, с помощью которых можно определять давление паров, энтальпию испарения и другие физические величины.
Если для какого-либо вещества известны температура кипения и энтальпия испарения, то можно пользоваться приведенной у Виттенбергера [64] диаграммой Бергхольма и Фишера [65], в которой кроме специальной сетки в координатах 1/Т — 1g р имеется также верхняя шкала в килокалориях. Для расчета давления насыщенных паров Хоффман и Флорин [66] приводят метод, состоящий в том, что логарифм давления паров откладывают на так называемой «оси веществ», ведущей к полюсным лучам. Этот метод аналогичен способу с применением известной диаграммы Кокса [67]. На этих диаграммах, построенных для соединений отдельных гомологических рядов («семейств» на диаграмме Кокса) все прямые, характеризующие давление паров, соединяются в точке (Полюсе) с координатами tx, которые для веществ каждого гомологического ряда имеют определенные значения. Таким образом, достаточно знать одну температуру кипения при каком-либо давлении, чтобы путем соединения соответствующей точки на диаграмме Кокса с полюсом можно было получить прямую, выражающую зависимость давления паров от температуры. В табл. 7 приведены систематизированные Драйсбахом координаты полюсов /«, и рж для 21 гомологического ряда на диаграмме Кокса. На рис. 41 показана диаграмма Кокса для алкилбензолов.
Кокс строил график зависимости 1g р от 1// и в качестве направляющей линии проводил прямую, образующую угол с ординатой. В соответствии со значениями р, отложенными на оси ординат, на ось абсцисс были нанесены экспериментальные значения температур кипения для воды. Дэвис [68], а также Калингарт и Дэвис [69] усовершенствовали диаграмму Кокса, предложив откладывать на оси ординат 1g р, а на оси абсцисс 1/(/ + 230). В таком виде диаграмму Кокса можно рассматривать как графическую иллюстрацию уравнения Антуана (18) при групповой константе с = 230. Однако эта константа применима лишь к алифатическим углеводородам [70]. Для удобства пользования диаграммой Кокса очень важно, чтобы она была построена в оптимальном масштабе. Чтобы можно было откладывать значения температур до 200 °C с точностью до 1 °C, следует применять диаграмму Кокса размерами примерно 1,5 м х 0,95 м. Драйс-бах [19] приводит точные инструкции для построения диаграммы Кокса, позволяющей определять значения давления и температуры с достаточной для лабораторной практики точностью.
На диаграмме Кокса по оси ординат отложена логарифмическая шкала давлений от 0,1 до 107 мм рт. ст., разделенная на восемь интервалов, суммарная длина которых составляет 1,422 м. Участок оси абсцисс, на котором откладывают значения температуры в интервале от —100 до 10 000 °C, имеет длину 0,984 м.
66
Таблица 7.
Гомологические ряды и соответствующие им координаты полюсов t и рк. иа диаграмме Кокса
X §
Первый член и характеристика гомологического ряда Гомологический ряд <0». °C Ро»-ю», мм рт. ст. Множитель (/„+230) Xlg(p„/7
Бензол, насыщенные боковые цепи Галогенпроизводный бензола, насыщенные боковые цепи 1670 2,320 6 621
Бензол, ненасыщенные боковые цепи Галогенпроизводный бензола, ненасыщенные боко-,, вые цепи СпН2П_8 СцН2П_(8+т)Х щ. 1063 0,916 3 985
Нафталин Галогенпроизводный нафталина СпН2п-12 QiH2n-(i2+m)Xm 921,5 0,530 3 274
Ароматический амин Галогенпроизводный ароматического амина CnH2n-5 CnH2rt_(5+mjN Хщ 1750 3,800 7 324
Фенол Галогенпроизводный алкилфенола СпН2п-бО СпН2П-(6+ГП)ОХт 2337 11,170 10 697
Нитробензол Галогенпроизводный нитробензола CnH2rt_7NO2 CrtH2rt_(7+m)NO2X/n 2022 3,701 8 304
Ароматический альдегид Галогенпроизводный ароматического альдегида CnH2rt_8O CnH2n-(8+m)^X/n 600 0,150 1 905
Фенилэтиловый спирт Галогенпроизводный фенилэтилового спирта ОгНгп-вО СцН2п-(в+т)^Х^2 3300 20,000 15 550
Ароматический кетон Галогенпроизводный ароматического кетона QiH2n-ieO СпН2п-(1б+т)ОХт 2186 5,186 9 263
Ароматическо-алифатический кетон Галогенпроизводный ароматическо-алифатического кетона с„н2П_80 СпН2п(8-т)ОХт 2186 5,186 9 263
Ароматический эфир Смешанный ароматическо-алифатический эфир Циклогексиловый эфир, насыщенные боковые цепи СпН2П-14О СпН2п-вО СпН2дО 1850 3,400 7 597
3*
67
Продолжение табл. 7
Первый член и характеристика гомологического ряда Гомологический ряд + °C 0СО-Ю6. мм рт. ст. Множитель Ц„+230)х Xlg(p„/760)
Циклогексиловый эфир, ненасыщенные боковые цепи СпН2п_2О 1850 3,400 7 597
Циклопентан c„H2rt 1450 1,550 5 560
Циклогексан CnH2n 2076 2,076 8 104.
Алифатический углеводород (насыщенный) СпН2п+2 1240 1,300 4 753
Галогенпроизводный алифатический углеводород (насыщенный) 1450 1,860 5 693
Ненасыщенный алифатический углеводород 1725 1,6980 6 548
Первичный алкиламин (тем. кип. до 190 °C при 760 мм рт. ст.) ^•пН2п+з^ 1125 1,441 4 424
Первичный алкиламин (тем. кип. 190—350 °C при 760 мм рт. ст.) QiH2n+3N 1270 1,709 5 029
Алифатический эфир QiH2n+2O 1579 2,525 6 370
Алифатический кетон Сп^2ПО 3293 6,929 13 950
Нитропарафин QiH2n+1NO2 1350 2,000 5 404
Алифатический нитрил CnH2n+iN 2025 3,461 8 250
Алифатический сложный эфир СПН2ПО2 1639 3,800 6 913
Алифатический спирт QiH2n_20 12150 128,200 64 718
Алифатическая кислота (темп. кип. до 141 °C) Алифатическая кислота (темп. кип. выше 145 °C) Галогенпроизводная алифатическая кислота cnH2rto2 QiH2nO2 ^п^2П-т^2^-т 364 1458 0,060 9,060 1 125 6 881
Расположение делений на температурной шкале можно рассчитать по следующим формулам:
t =230(0/(37,17 — /) (23)
или
/ = 37,17^/(230 + /) (24)
где: t — температура, °C; I — длина в дюймах (1 дюйм = 24,5 мм).
Нулевая точка в средней части шкалы удалена от левого края диаграммы, соответствующего —100 °C на расстояние 0,438 м; длина I обозначает расстояние на шкале, соответствующее положительному или отрицательному отклонению откладываемого значения температуры от нуля. Рекомендуется применять температурные интервалы, приведенные в табл. 8.
Полученные для интервалов I деления подразделяют на еще более мелкие.
68
Таблица 8 А
Температурная шкала для диаграммы Кокса
Температурный интервал I, °C Цена деления, °C
—100—200 10
200—600 50
600—2000 100
2000—4000 500
4000—10000 1000
Таблица 8Б
Температурный интервал 11, °C Цена деления
первая разбивка вторая разбивка
—100—0 10 10X1
0—200 10 5X2
200—600 50 5X10
600—1000 100 2X50
Более 1000 100 Более мелкие деления не повышают точность отсчета.
В заключение рассмотрим еще один метод, специально предназначенный для расчета давления насыщенных паров веществ гомологического ряда. Согласно этому методу по оси ординат откладывают значения температуры кипения в линейном масштабе, а по оси абсцисс — число углеродных атомов в молекулах компонентов в логарифмическом масштабе; затем на диаграмме строят изобары для различных давлений, как это показано на рис. 42 применительно к парафинам С10—С20 с прямой углеродной цепью для остаточных давлений 5, 10 и 30 мм рт. ст. Таким способом можно успешно интерполировать неизмеренные значения и экстраполировать данные для соединений с числом атомов углерода в молекуле менее 10 и более 20. Кроме того, преимущество этого метода состоит в том, что в ходе
сю ci2 ci6 Cte сго Рас. 42.
Изобары предельных углеводородов С10—С20,
69
ректификации при определенном давлении в любое время можно ориентировочно оценить, из какого числа атомов углерода состоит молекула вещества в дистилляте.
Имеются еще более точные методы для расчета давления паров. В работах Хоффмана и Флорина [66], Фальдикса и Штаге [70], а также Штаге и Джуилфса [71 ] и [59] содержатся подробные обзоры этих методов. Очень важно для работы выбирать метод, отвечающий поставленным целям наилучшим образом, но всегда следует иметь в виду, что все эти методы являются приближенными и лишь путем тщательно проведенных экспериментальных измерений получают максимально точные данные.
4.5. УРАВНЕНИЕ СОСТОЯНИЯ
И ФАЗОВАЯ ДИАГРАММА р—х
Жидкость состоит из молекул, движущихся с различными скоростями, из которых самые быстрые наиболее легко пересекают и покидают поверхность жидкости, переходя в виде паров в пространство, находящееся над жидкостью (испарение). Для перевода частиц жидкости в парообразное состояние необходимо затратить определенное количество тепла, которое при этом отнимается от жидкости, в результате чего она охлаждается. При внешнем подводе тепла достигается более интенсивное испарение, и жидкость закипает.
Таким образом, пар является газом вблизи температуры кипения или конденсации. При перегонке образуются насыщенные пары, которые находятся в контакте с жидкой фазой, причем в состоянии равновесия число молекул, переходящих из жидкой фазы в газообразную, равно числу молекул, возвращающихся из газообразной фазы в жидкую. Ненасыщенный пар образуется лишь в том случае, если первоначально насыщенный пар не имеет больше контакта с жидкостью и нагревается выше температуры кипения или расширяется (при переходе в область более низкого давления). В этих условиях пар не имеет возможности принимать из жидкости дополнительное число молекул в соответствии со своей более высокой температурой.
Общее уравнение состояния (26) для газов вблизи точки сжижения при высоких давлениях и при ассоциации молекул можно рассматривать лишь как приближенное и предельное соотношение, т. е. оно справедливо только для идеальных газов, а не для насыщенных паров, с которыми обычно имеют дело при перегонке.
Вышеуказанное обстоятельство учтено в уравнении Ван-дер-Ваальса:
(р+«Мол)(^МОл-0 = ^ (25)
где а и b — константы, константа b учитывает собственный объем молекул газа, я/Умол —силы притяжения, действующие между молекулами; р — давление газа на ограничивающую его поверхность; Рмол — мольный объем газа.
70
Однако и уравнение состояния Ван-дер-Ваальса ни для какого вещества не является точным, так как межмолекулярные силы не подчиняются такому простому закону, который Ван-дер-Ваальс положил в основу своего уравнения.
Планк в качестве уравнения состояния предложил уравнение пятого порядка, которое лучше всего описывает реальное поведение газов и паров. Однако проводить расчеты по уравнениям третьего и пятого порядка очень неудобно, и поэтому в практике перегонки применяют общее уравнение состояния, в основу которого положены газовые законы Гей-Люссака и Бойля-Мариотта
pV = p0V07/273,15
pV = RT (26)
При этом делают допущение, что поведение паров подчиняется общему уравнению газового состояния с достаточной точностью. Газ тем больше приближается к идеальному состоянию, чем меньше его плотность (чем меньше давление) л чем выше температура.
Если в выражение р0 УО/273,15 = R подставить параметры для нормальных условий, а именно р0 = 1 атм (что соответствует 760 мм рт. ст. или 1,033 кгс/см2 или 1,013 бар) и Vo = 22,414 л (объем 1 моль при 0 °C), то получим мольную газовую постоянную R = 1,0-22,414/273,15 = 0,082 л-атм/К.
Для любого числа молей N газа или пара справедливо следующее уравнение состояния:
pV = N-0,082T (27)
При лабораторной перегонке имеют дело обычно с малыми количествами вещества, поэтому расчеты проводят в молях, а не в килограмм-молях, как это принято для промышленных процессов.
Если нужно рассчитать объем паров испаренного вещества при определенном давлении, то пользуются формулой
V = /V-0,0827 -760/р (28)
где V измеряется в л; р — в мм рт. ст.; N — число молей.
Для 1 моль при атмосферном давлении 760 мм рт. ст. (или 1 атм) формула (28) упрощается:
V = 7-0,082 (29)
Плотность паров рп при нормальных условиях рассчитывают, исходя из массы 1 моль, по формуле
рп = .47/22,4 (г/л) (30)
При пересчете на другое давление и другую температуру ( получаем ' _ М р 273,15
/ Рп~ 22,4 ‘ 760 ' 273,15 -|- / (3
: где р измеряется в мм рт. ст.; t — в °C.
71
Если числовые константы объединить в один множитель, то из (31) получим
рп = 0,01605/Ир/Т (32)
Масса газа вычисляется по простой формуле
G = Ирп (33)
Как было показано выше, именно межмолекулярные силы влияют на взаимную растворимость компонентов. Этим обусловлены и другие свойства бинарных смесей, представленные по Торманну [72] на рис. 43.
Если компоненты практически взаимно нерастворимы, то в смеси оба компонента и образующиеся пары ведут себя так, как будто жидкие фазы компонентов не соприкасаются между собой (см. рис. 43, тип 1). Эти условия имеют место при перегонке с паром—носителем (см. разд. 6.1).
По закону Дальтона общее давление газовой смеси складывается из давлений, которые имели бы отдельные компоненты при той же температуре и в том же объеме:
Р — Р1 + Ра (34)
Отсюда содержание легколетучего в парах смеси взаимнонерастворимых компонентов при той же температуре можно вычислить по формуле:
Pi = (Pi/P) Ю0% (мол.) (35)
Чтобы определить парциальные давления (см. рис. 43, ряд I), используемые при расчете состава паров для бинарных смесей нерастворимых компонентов, строят обе кривые давления паров и получают с их помощью суммарную кривую обоих парциальных давлений подобно тому, как это показано для смеси вода—бензол на рис. 39. При 760 мм рт. ст. температура кипения этой смеси (в том числе и температура дистиллята) составляет 69,0 °C. Содержание легколетучего компонента в парах вычисляют по формуле (35):
у* = (534/760) 100 = 70,2% (мол.) (36)
По номограмме на рис. 22 проводят пересчет и получают 91,0% (масс.).
Если рассмотреть горизонтальные ряды I и II на рис. 43, для которых на диаграммах по оси абсцисс указана концентрация в мольных процентах в расчете на легколетучий компонент (см. также рис. 29), то можно выявить следующие соотношения.
Ряд I. Парциальные давления при постоянной температуре для легколетучего компонента А и труднолетучего компонента В (диаграмма р — х).
При постоянной температуре давление паров смеси зависит только от ее состава. Если силы притяжения между одинаковыми молекулами больше, чем между неодинаковыми, то наблюдается весьма слабая тенденция к взаимному смешению компонентов. Процесс растворения протекает с поглощением тепла, что приводит к уменьшению энтальпии испарения. Но при этом увеличивается летучесть смеси, и давление паров будет выше, чем это соответствует идеальной смеси (типы 1—3). Отклонения от идеального поведения можно выразить через 72
Рис. 43.
Зависимость свойств бинарных смесей от содержания легколетучего компонента в жидкой фазе по данным Торманна [72]:
ряд I — парциальные давления легколетучего А и труднолетучего В компонентов при постоянной температуре; ряд II — общее давление прн постоянной температуре; ряд III — содержание легколетучего в парах при постоянном давлении (кривая равновесия); ряд IV — температура при постоянном давлении (кривая кипення);ряд V — температура прн постоянном давлении (пунктирная кривая—линия конденсации).
Тип I — отсутствие или незначительная взаимная растворимость компонентов; тип 2 — частичная растворимость; тнп 3 — полная растворимость с наличием азеотропной точки (максимальное давление паров и минимальная температура кипения); тип 4 — полная растворимость с приближением к идеальному раствору; тнп 5 — полная растворимость с наличием азеотропной точки (минимальное давление паров и максимальная температура кипения).
в широком интервале концентраций заметно ниже температур кипения обоих чистых компонентов (минимальная температура кипения). Для смесей типа 5, наоборот, в некотором интервале концентраций температура кипения лежит выше температур кипения обоих чистых компонентов (максимальная температура кипения). Точка соприкосновения изобарных кривых кипения и конденсации соответствует азеотропной точке смеси.
Кривая равновесия у—х показывает связь между концентрацией жидкости х и соответствующей концентрацией пара у, находящегося в состоянии равновесия с жидкостью. Следовательно, кривая равновесия является основой для расчета числа теоретических ступеней разделения по графическому методу Мак-Кэба и Тиле [77], который успешно и широко применяется благодаря своей простоте. На рис. 43 в ряду III представлены кривые равновесия для смесей различных типов. Для смесей взаимно нерастворимых компонентов кривая равновесия представляет собой прямую линию (тип 1), которая пересекает диагональ в одной точке, называемой азеотропной. В этой точке составы пара и жидкости одинаковы; обогащение паров легколетучим компонентом при более высокой концентрации жидкости х уже невозможно; напротив, в этой области концентраций пар содержит меньше легколетучего компонента, чем жидкость. При перегонке смесей взаимно нерастворимых компонентов (тип 1) или смесей только частично растворимых компонентов (тип 2) дистиллят имеет один и тот же состав в широком интервале изменения концентрации легколетучего компонента в кипящей жидкости и только в непосредственной близости от концентраций 0 и 100% появляются промежуточные составы дистиллята. Для смесей с максимумом на кривой давления паров при концентрации жидкости выше азеотропной (тип 3), а для смесей с минимумом на кривой давления паров при концентрации жидкости меньше азеотропной (тип 5) пары содержат меньше легколетучего компонента, чем исходная жидкость состава х. Для смесей типа 4 характерна форма кривой равновесия у*—х, свойственная идеальным смесям, для которых у* всегда больше х.
4.6.1. РАСЧЕТ КРИВОЙ РАВНОВЕСИЯ
Построение кривой равновесия расчетным путем возможно для смесей почти нерастворимых компонентов (т. е. в высшей степени неидеальных смесей) и идеальных смесей, а также для смесей частично растворимых компонентов. Для смесей же с максимумом или с минимумом на кривой давления паров экспериментальное определение является более предпочтительным, поскольку в данном случае для расчета кривой равновесия требуются другие, зачастую малодоступные данные по физическим свойствам, причем объем вычислений возрастает.
В последние годы опубликовано очень много статей, посвященных расчету кривых фазового равновесия. Подробный разбор 76
всех этих работ занял бы слишком много места; к тому же эта тема исчерпывающе рассмотрена Шубертом [17], а также Хала с сотр. [78]. В монографиях этих авторов приведен и проанализирован обширный материал, опубликованный ранее.
Намечается тенденция рассчитывать параметры равновесия в многокомпонентных смесях, исходя из экспериментальных данных для бинарных смесей и применяя цифровые вычислительные машины. Это вызвано тем, что данных по равновесию для бинарных смесей накоплено уже достаточно много [105, 106], в то время как исследование многокомпонентных смесей весьма трудоемко. На международном симпозиуме «Перегонка—1969» в Брайтоне [61 семь докладов были посвящены расчету кривых фазового равновесия на основе физико-химических свойств чистых компонентов. На симпозиуме обсуждались данные по равновесию для электролитов и неэлектролитов, вопросы термодинамической проверки данных по равновесию для неэлектролитов и методы расчета коэффициентов активности. Во многих случаях оправдано применение уравнения Вильсона [80]. Как сообщают Донт и Вайхманн [81 ], для расчета данных по равновесию бинарных растворов на основе зависимостей р—х (или t—х) можно использовать методы последовательных приближений. Круг, Хабер-ланд и Биттрих [82 ] дали оценку основным положениям по теории растворов, выдвинутым Маргулесом, Редлихом, Кистером, Ван-Нессом, Ван-Лааром, Скэтчардом, Хеймером, Вильсоном, Хейлем и Реноном, Праусницем (NRTL—Non—Random—Two— Liquid). Шуберт [17 ] полагает, что теоретический подход Ренона— Праусница [83] весьма перспективен. Он лишен недостатков метода Вильсона, основанного на использовании трехпараметрического уравнения. Применение метода Вильсона ограничивается нерасслаивающимися жидкими системами. Соотношение Ренона—Праусница связывает физическую модель Вильсона с теорией Скотта.
Для расчета кривой равновесия идеальных смесей, например смесей химически родственных веществ, особенно относящихся к одному гомологическому ряду, можно использовать различные методы. Чтобы дать представление о ходе расчета в целом, рассмотрим некоторые из них.
Для расчета равновесного состава паров на основе данных по давлению паров чистых компонентов следует применять закон Рауля. Этот закон устанавливает, что парциальное давление компонента равно произведению мольной доли этого компонента и давления паров компонента при температуре, равной температуре жидкости:
(40) (41) (42) (43)
77
Pi = Pixi (Х1 в мол. долях)
Pi = Pixi/100 (xj в мол. %)
Pi РзО ~ *1) р2=-р2(100 —Х1)/100
Таблица 9.
Давление паров бензола и толуола при различных температурах
i, °C Давление паров, мм рт. ст.
pj (бензол) р2 (толуол)
80 748 294 2,54
90 1013 408 2,46
100 1335 555 2,41
ПО 1739 747 2,33
Общее давление р складывается из парциальных давлений рг и р2: Р = Pi + Рч = Р?Х1 + pg (1 — х,) (Xj в мол. долях) (44)
Р= Pi+Р2 = IP?Xi + Р§(ЮО —х,)]/100 (Xj в мол. %) (45)
Так как равновесный состав паров равен отношению парциального давления компонента 1 к общему давлению, то находим Pi - P?xi____________P?*i
У1 Р1 + Р2 Р1Хг4-р^(1 — х2) р 1 >
Поясним сказанное на примере. Допустим, нужно, исходя из данных по давлению паров чистых компонентов, расчетным путем построить кривую равновесия у*—х при 760 мм рт. ст. для смеси бензол—толуол, подчиняющейся закону Рауля. Вначале в возможно более крупном масштабе строим график зависимости давления паров чистых компонентов от температуры,, затем по графику определяем давления паров обоих компонентов для различных температур (табл. 9).
Преобразуя формулу (44), получим выражение для концентрации жидкости
Х1 = -£Р^-100%(МОЛ-) (47)
Подставляя в (47) значения давления паров при 90 °C, получим
Х1 = шТз — 408 '100 = 58118% (мол-) (48)
Подставив в (46), найдем соответствующую равновесную концентрацию у*
1013-58,18/760 = 77,55% (мол.) (49)
Аналогичным образом можно вычислить и остальные точки кривой равновесия у*—х. Чем больше имеется данных, тем точнее будет построена кривая равновесия. Разумеется, такой расчет возможен не только для атмосферного давления, но и для любого другого давления, например 20 мм рт. ст. В этом случае р = 20, а температуры варьируют в интервале между температурой кипения компонента 1 при 20 мм рт. ст. и температурой кипения 78
компонента 2 при 20 мм рт. ст. Результаты таких расчетов особенно удобно представлять в виде таблицы (табл. 10).
Кривую равновесия можно найти также и графическим способом. Этот способ гораздо сложнее и отнимает больше времени, чем чисто расчетный метод, поэтому ограничимся только упоминанием о нем. Более подробное его описание приводят Торманн [72] и Розенгарт [84].
Для идеальных бинарных смесей кривую равновесия можно также рассчитать по уравнению, полученному на основе объединения законов Рауля и Дальтона:
У* = Pi xPi = х
1—У* Рг (1—*)Р2 1— X
(50)
где х — мольная доля.
Уравнение (50) можно представить в более удобной для использования форме:
у* = ах/(ах— хЦ-1) (в мол. долях) (51)
или
У* = ПхУа” ~Г(В % М0Л,) (52)
1/л -ф- ОС — 1
В эти уравнения входит величина а — рЧ/которую называют относительной летучестью компонентов смеси (см. разд. 4.6.2). Для идеальных смесей можно принять величину а постоянной в интервале между температурами кипения чистых компонентов (см. также табл. 10). Для смеси бензол—толуол в табл. 9 приведены значения а, рассчитанные по заданным р? и pl\ они лежат между 2,54 и 2,33, среднее арифметическое значение аср = 2,43.
Таблица 10.
Пример расчета кривой равновесия для жирных кислот С10—Сп нормального строения при общем давлении р = 20 мм рт. ст.
t, °C 0 Pl-мм рт. ст. 0 мм рт. ст. Xt по (47), % (мол.) Ух по (46), % (мол.) Относительная летучесть 0/ 0 « = pjp2
161,5 20 11,0 100 100 1,819
163 21 11,9 83,9 93,4 1,765
164 22 12,5 78,9 86,8 1,760
165 23 13,2 69,4 79,8 1,742
166 24,1 13,9 59,7 71,9 1,733
167 25,2 14,5 51,3 64,7 1,736
168 26,4 15,2 42,8 56,5 1,735
169 27,5 16,0 34,7 47,15 1,717
170 28,7 16,9 26,3 37,7 1,696
171 30,0 17,4 20,6 30,9 1,723
172 31,4 18 14,95 23,45 1,743
173 32,6 18,8 8,70 14,3 1,733
174,5 35,0 20 0 0 1,750
79
Таблица 11.
Пример определения равновесной концентрации у* для системы бензол—толуол при а = 2,43
X. % (мол.) Аналитический способ Графический способ Опытные данные Киршбаума [103]
у* вычисляют по (51) или (52), % (мол.) Д % (мол.) (по сравнению с опытными данными) у* находят по кривой. равновесия для идеальной смеси при а = 2,4 Д % (мол.) (по сравнению с экспериментальными данными)
10 21,2 —0,4 20,0 +0,8 20,8
20 37,8 —0,6 37,2 ±0,0 37,2
30 51,1 —0,4 51,0 —0,3 50,7
40 61,8 +0,1 61,0 +0,9 61,9
50 70,8 +0,5 70,8 +0,5 71,3
60 78,7 +0,4 78,4 +0,7 79,1
70 85,0 +0,7 84,8 +0,9 85,7
80 90,7 +0,5 91,0 +0,2 91,2
90 95,6 +0,3 95,6 +0,3 95,9
По двум значениям а, найденным при температурах кипения чистых компонентов, можно вычислить среднее геометрическое значение, которое практически совпадает со средним арифметическим
«ср = К (53)
или
1g «ср = (1g «1 + 1g аз)/2 (54)
Это значение а подставляют в уравнение (51) или (52) и для заданных концентраций х вычисляют состав равновесных паров у*. В табл. 11 приведены рассчитанные таким способом равновесные значения у* для системы бензол—толуол.
Для расчета равновесия в идеальных бинарных смесях можно также использовать номограмму Орличека и Пёлля [85] (см. рис. 21). Левая шкала соответствует концентрации жидкой фазы х (в мол. долях), а правая шкала показывает равновесную концентрацию паровой фазы у. Например, для смеси бензол—толуол при х = 50% (мол.) и а = 2,4 на правой шкале получим (штрих-пунктирные линии) равновесное значение у* = 70,8% (мол.); это значение совпадает с расчетным.
Номограмма на рис. 21 построена в мольных долях. При пользовании номб-граммой нужно иметь в виду, что не обведенные кружком значения на шкалах X и Z только тогда принимаются в расчет, когда величины а и Q превышают 1. Если же величины а или Q (средняя шкала) меньше 1, то на шкалах X и Z во внимание принимают только значения, обведенные кружком.
Данные по равновесию можно также очень быстро рассчитать с помощью круговой диаграммы Фурвийка [86], основанной на формуле (49).
80
Мы ограничимся изложением способов расчета бинарных смесей, поскольку многокомпонентную смесь при вычислениях можно рассматривать как совокупность бинарных смесей. Методы расчета тройных смесей частично или полностью взаимно растворимых компонентов подробно проанализированы Торманном [72] и Мацом [73]. Холло, а также Холло и Ленгьел [87] обсуждают вопросы равновесия между паром и жидкостью в трех-и четырехкомпонентных системах. Эти авторы исследовали три неидеальные четырехкомпонентные системы с применением новой аппаратуры. При этом они использовали новый метод расчета, позволивший уменьшить объем экспериментальных исследований. Равновесные данные для четырехкомпонентных систем можно представить с помощью двойных ортогональных проекций.
Специально для условий нефтяной промышленности Ум-штеттер и Флашка [88] разработали метод построения диаграмм равновесия по кривым давления паров для компонентов, которые в свою очередь могут представлять собой смеси различных веществ. Точность расчетов заметно повышается благодаря учету энтальпий испарения и растворения. Метод расчета пригоден также и для азеотропных смесей.
Для расчета паро-жидкостного равновесия в многокомпонентных смесях Вильямс и Хенли [89] разработали программу на алгоритмическом языке Фортран. Для учета реального поведения смесей предназначен ряд подпрограмм, которые позволяют вычислять фугитивности по вириальному уравнению, по соотношению Редлиха и Квонга или по способу Чао и Зидера. Коэффициенты активности можно учесть, используя рекомендации Вильсона, Ван-Лаара или Гильдебранда.
4.6.2. ЛЕТУЧЕСТЬ, ОТНОСИТЕЛЬНАЯ ЛЕТУЧЕСТЬ И КОЭФФИЦИЕНТ АКТИВНОСТИ
Летучесть F выражается в виде отношения парциального давления паров вещества к его мольной доле в жидкой смеси при определенной температуре, например для бинарной системы:
Fi = Pi/xi, F2 = р2/х2 (55)
Относительная летучесть компонентов бинарной смеси равна: «=-&- = -^т1- = (Fi > F2) (56)
F2 р2/х2 р2Х! 1 ’
Для идеальных смесей летучесть не зависит от соотношения компонентов и равна давлению паров чистого вещества. По закону Рауля для легколетучего компонента 1 идеальной смеси справедливо соотношение
Pi = P?*i (57)
а для малолетучего компонента 2
Р2 = Р^г или Pi == Ра 0 — xi) (58)
81
ОС
Рис. 45.
Зависимость относительной летучести компонентов смеси от разности температур кипения А*кип Для различных температур кипения £кИП смеси при атмосферном давлении.
Таким образом, парциальное давление компонента равно произведению давления паров чистого компонента и содержания этого же компонента в жидкости при той же температуре, выраженного в мольных долях.
По формуле (35) равновесная концентрация пара равна
У* = Pi/Р и yl = рг/р
Подставив уравнения (57) и (58) в эти два уравнения, получим:
У* = PiXjp и у* = р«х2/р (59)
При делении уравнений (59) друг на друга будем иметь
У*!у1 = Р‘.Х/(/’%'2) или У^/У-Р^ = Pl/P°2 = а (60)
Выражение (60) идентично (56), поэтому относительную летучесть а для идеальной смеси значительно проще выражать через соотношение давлений паров чистых компонентов, причем о о
Р1 > Pl.
Относительную летучесть можно также определить по точкам кипения компонентов. Применив правило Трутона при К. = = hHvIT = 20,5 кал/(моль-°С) и уравнение Клапейрона, Роуз [91] вывел соотношение между температурами кипения и относительной летучестью двух жидкостей, которое можно применять для ориентировочных расчетов:
lga = 8,2(T2-T1)/(7’2+Л) (61)
Мельпольдер и Хедингтон [90] вывели следующие более точные уравнения, которые дают погрешность от 0 до 6,2%, причем в подавляющем большинстве случаев погрешность не превышает 1%.
82
Для давления 760 мм рт. ст.:
lg а = (Z2 — /j) (3,99 + 0,0011939Т)/Т (62)
Для давлений выше и ниже 760 мм рт. ст. (от 10 до 1500 мм рт. ст.):
lg а = [(/2 - /j) (7,30 - 1,15 lg р + 77(179 1g р)]/Т (63)
где /2 — 6 — разность температур кипения компонентов, °C; Т — температура кипения смеси, К; р — рабочее давление перегонки, мм. рт. ст.
Уравнение (62) графически изображено на рис. 45 для интервала температур кипения смесей от —100 до -|-300 °C.
Роуз и Билес [901 применили ректификационную колонну для экспериментальной проверки данных по относительной летучести при конечных флегмовых числах в интервале v = 9,2—15,8. Для смеси к-гептан—метилциклогексан получено а == 1,074, что хорошо согласуется с опытными данными по фазовому равновесию. Значение а определяли с использованием опытных данных по формуле:
а = (у + xe/Xb)/[v + (1 - х£)/( 1 - х£)] (64)
Этот метод обеспечивает расчет значений а идеальных и неидеальных смесей при различных давлениях на основе экспериментальных данных, полученных при ректификации на насадочных колоннах. Метод применим также и к многокомпонентным смесям.
Разность температур кипения двух компонентов однозначно характеризует трудность разделения только идеальных смесей. Если смеси не идеальны, то решению задачи разделения может способствовать анализ зависимости относительной летучести а от давления.
Если а = 1 (1g а = 0), то кривые давления паров пересекаются, как например для смеси м-крезол — и-крезол, и в условиях, соответствующих точке пересечения, разделение двух компонентов ректификацией невозможно. При этом разделение может быть достигнуто только селективными методами перегонки (см. разд. 6.2). Чем больше величина а, тем более выпуклой становится идеальная кривая равновесия и тем легче осуществить разделение.
; Чаще всего, особенно, когда компоненты смеси принадлежат к одному гомологическому ряду, величина а увеличивается с по-V нижением температуры, поэтому разделение смеси легче прово-; дить под вакуумом. К такому выводу пришли, например, Хопкинс и Брент [92] на основании многочисленных опытов по ректификации. Колонны при пониженном давлении имеют точно
83
Рис. 46.
Зависимость коэффициентов активности компонентов системы метанол—вода от концентрации метанола [109].
Сплошные кривые построены по экспериментальным данным, пунктирные — расчетом по методу Маргулеса—Ван-Лаара.
такие же рабочие характеристики, как и при 760 мм рт. ст.*, и лишь увеличение относительной летучести в вакууме повышает степень разделения смеси. Так, Цыпкина [93] сообщает, что при разделении ректификацией смесей антрацен—карбазол и пирен—фторантен число теоретических ступеней разделения снизилось больше, чем на 50% при переходе к остаточному давлению 60—100 мм рт. ст. Есть смеси, для
которых величина а остается постоянной в широком интервале рабочих давлений, например система хлороформ — четыреххлори-стый углерод или «-гептан — метилциклогексан. Смеси подобного рода благодаря стабильным значениям а являются наиболее подходящими эталонными смесями (см. разд. 4.10.3). Однако встречаются
также смеси, относительная летучесть которых с повышением температуры также растет, например смесь 2,4-диметил пентан—2,2,3-триметилпентан. Такие смеси не имеет смысла разделять под вакуумом, скорее напротив, лучшее их разделение достигается ректификацией под давлением. Из теоретических соображений иногда целесообразнее проводить перегонку в изотермических условиях, т. е. поддерживать температуру куба постоянной за
счет понижения давления.
Для определения относительной летучести идеальной смеси можно использовать уравнение (60) после введения в него коэффициента активности у:
а = PiYi/(P2?2)
(65)
* Изменение давления иногда сопровождается изменением физико-химических свойств разделяемой смеси, а также гидродинамики потоков жидкости и пара. Например, при ректификации в кольцевом зазоре между вращающимся внутренним цилиндром и неподвижным внешним цилиндром применение вакуума приводит к ослаблению интенсивности или полному исчезновению вихрей Тейлора в паровой фазе, благоприятствующих массопереносу. Затухание вихрей Тейлора происходит вследствие повышения кинематической вязкости паров. В итоге эффективность колонны заметно снижается (см. Шафрановский А. В., Ручинский В. Р. Теор. основы хим. технол. 1971, т. V, № 1; Олевский В. М., Ручинский В. Р. Роторно-пленочные тепло- и массообменные аппараты. М., Химия, 1977. — Прим. ред.
84
Так как по формуле (35) у* ~ p-Jp (по закону Дальтона), то при р! = ру* получаем
Т? = PP?/(Pi^i) У2 = РУгЛФг) (66)
Отсюда для азеотропного состава, когда у* = х1г получим
71 = Р/Pi и ?а = Р/Р’
(67)
Если построить график зависимости у; от х1( то можно получить кривые, которые являются непосредственным наглядным и количественным показателем степени отклонения свойств реального раствора.. от закона Ра ул я_ (рис. 46).
Для идеальных смесей коэффициент активности равен 1. Отклонение значений у£ от 1 являются показателем неидеальности смеси. Если давление паров обоих компонентов больше, чем это соответствует закону Рауля (рх = Pi%i), то уг и у2 превышают 1 (1g V/ > 0); при этом говорят о положительном отклонении от закона Рауля. Если эти отклонения достаточно велики, то в смеси образуется азеотроп с максимальным давлением паров или с минимальной температурой кипения *. Наоборот, при больших' отрицательных отклонениях (1g у,- < 0) образуется азеотроп с минимальным давлением паров или с максимальной температурой кипения **. Очень большие отклонения от закона Рауля приводят к расслоению раствора на две отдельные жидкие фазы и тем самым к образованию так называемого гетероазеотропа, который ведет себя аналогично гомоазеотропу (см. разд. 6.2) [94].
Данные по коэффициентам активности и их зависимости от состава раствора нужны в первую очередь для того, чтобы уста-
новить, к какому типу относится та или иная жидкая смесь. С помощью этих данных можно также проверить надежность экспериментальных данных по равновесию. Определение коэффициентов активности приобретает большое значение также для расчета процессов азеотропной и экстрактивной ректификации, при которой имеют дело с тройными смесями. Методы расчета коэффициентов активности для подобных смесей подробно рассматриваются Кортюмом и Бухгольц-Майзенхаймером [74], а также Шубертом [17].
Коэффициенты активности уг компонентов в растворах опре-
деляют по данным экспериментального исследования парциальных
давлений в изотермических условиях (см. рис. 37). Если экспе-
риментально найденные значения р£ подставить в формулу (66),
то можно получить зависимость у£ от х. при постоянной температуре (см. разд. 4.4.1). Для систем, образующих азеотроп, коэф-
фициенты активности целесообразно определять по методу Карл-) сона и Кольбурна [95]; этот метод подробно рассматривается \ Орличеком и Пеллем [85]. Коэффициенты активности можно
* В советской научно-технической литературе такой азеотроп принято называть «положительным». — Прим. ред.
** Или «отрицательный» азеотроп. — Прим. ред.
85
точно рассчитать по кривой общего давления с помощью графического метода, предложенного Орличеком [96].
Данные по давлению паров и фазовому равновесию, найденные экспериментальным путем, поддаются термодинамической проверке с помощью коэффициентов активности. По уравнению (66) или (67) на основе опытных значений х и р или х и у* вычисляют коэффициенты активности и строят графическую зависимость 1g у, от х. Полученные кривые сравнивают с кривыми [17, 18], рассчитанными по уравнению Ван-Лаара [97] или Маргулеса [98]. Рис. 46 иллюстрирует этот способ на примере опытных данных Гельбина [109].
При определении коэффициентов активности, отвечающих требованиям термодинамической состоятельности, следует учитывать возможность ассоциации молекул в паровой фазе [98а].
4.6.3. ЭКСПЕРИМЕНТАЛЬНОЕ ОПРЕДЕЛЕНИЕ
КРИВЫХ РАВНОВЕСИЯ
Данные по равновесию пар—жидкость в бинарных и многокомпонентных смесях лежат в основе расчета необходимого числа теоретических ступеней разделения и других параметров процесса ректификации. Ландольт и Бернштайн [99] систематизировали данные по фазовому равновесию, опубликованные до 1933—1934 гг. включительно. Дополнительные данные по равновесию приведены в книгах Перри [100], Ю Чин Чу [101 ], а также Чу, Ванга, Леви и Пауля [102].
Штаге с сотр. опубликовал многочисленные сборники данных по фазовому равновесию; аналогичные сведения для легких углеводородов, H2S и СО2 можно найти в книге Заге и Ласея [27]. Якобс [104] приводит кривые равновесия для 50 смесей. Коган и Фридман [105] подготовили обширное собрание таблиц. Монография этих авторов, изданная в 1966 г., содержит данные но равновесию для 1765 бинарных смесей, 359 тройных смесей и 32 многокомпонентных смесей; в книге также дается введение в технику экспериментального исследования фазового равновесия в системе пар—жидкость и в методику термодинамической проверки данных по равновесию. Вопросы равновесия между паром и жидкостью подробно освещает также Хала с сотр. [78]. В первом разделе книги авторы рассматривают термодинамику растворов неэлектролитов, а во втором —технику лабораторного исследования фазовых равновесий, третий раздел содержит литературные ссылки на 1232 исследованные системы. Наиболее обширный сборник данных по фазовому равновесию при атмосферном давлении опубликован Хала с сотр. [106] в 1969 г. Долгосрочный проект PPDS* поможет накопить многочисленные данные, используемые при расчетах процессов дистилляции и ректификации [106а].
В практике лабораторной ректификации постоянно встречаются смеси, фазовое равновесие которых еще не исследовано; в таких случаях возникает вопрос о выборе наиболее целесообразной аппаратуры для получения искомых данных.
Исследователи фазового равновесия проводят различие между изотермическими и циркуляционными методами при постоянном давлении. Принцип циркуляционного метода состоит в том, что бинарную смесь известного состава испаряют и после установления фазового равновесия (т. е. состояния, при котором число молекул, покидающих жидкость, равно числу молекул, возвращающихся в жидкость) при определенном давлении измеряют концентрации кубовой жидкости и сконденсированных паров. Мюллер и Штаге ([39] к гл. 1) дают превосходное введение в технику экспериментальных измерений на примере дистилляции
* Информационная служба по физическим свойствам Британского института инженеров-химиков. — Прим. ред.
86
жирных кислот. Можно так же, как это предусмотрено в изотермическом методе, поддерживать температуру постоянной и измерять устанавливающееся при этом давление паров. Статические методы имеют то преимущество, что они позволяют работать с очень малыми количествами вещества. Кортюм и Файер [107] сообщают об очень простом статическом методе измерения давления паров при постоянной температуре в двойных и тройных жидких системах. Вебер [108] описывает универсальную дистилляционную установку для исследования фазового равновесия, состоящую из термостатов. Гельбин [109] дает сравнительную характеристику различных методов нахождения кривой равновесия.
Особенно важно точно знать ход кривой равновесия в ее начале и в конце, поэтому в интервалах концентраций хв от 0 до 10% (мол.) и от 90 до 100% (мол.) проводят как можно больше измерений. Обычно равновесную концентрацию у* определяют для следующих концентраций кубовой жидкости: 1, 3, 5, 10, 90, 95, 97, 99% (мол.). В интервале концентраций от 10 до 90% измерения проводят через каждые 5%. Разумеется, при этом учитывают характер смеси, так что при идеальном ходе кривой можно вполне обойтись без промежуточных точек в средней области.
При исследовании равновесия по циркуляционному методу в колбу 1 (рис. 47) загружают от 250 до 500 мл смеси и нагревают ее до кипения. Пары конденсируются в холодильнике 3, и при закрытом кране 4 конденсат стекает обратно в колбу /. По термометру 2 наблюдают за изменением температуры. Прибор должен быть тщательно теплоизолирован. После установления равновесия из патрубка 5 при открытом кране 4 отбирают пробу конденсата объемом 0,1—0,2 мл. Одновременно берут пробу жидкости из колбы /; отбор проводят при открытом кране 6 через патрубок 7. Измерения необходимо повторять несколько раз до повторения одной и той же концентрации проб три раза подряд.
При работе в условиях вакуума применяют пробоотборник, пригодный также и при работе под атмосферным давлением, который позволяет полностью исключить контакт пробы со смазкой крана. Концентрацию пробы определяют по показателю преломления с помощью калибровочной кривой п^ — % (масс.), которую строят заранее, составляя смеси из чистых исходных веществ. Для определения концентрации можно использовать и другие физические константы, например плотность или температуру замерзания. Серию опытов обычно начинают с наименьшей концентрации хв. После завершения опыта при одной какой-либо концентрации хв легколетучий компонент добавляют в куб в таком количестве, чтобы приблизительно получилось очередное значение концентрации;
биваться точного значения намеченной концентрации, достаточно, если она лежит вблизи желаемой точки измерения.
Чтобы получать точные данные при исследовании фазового равновесия, необходимо исключать такие источники погрешностей, как перегрев кипящей жидкости, возникновение градиента концентраций в кипящей жидкости, частичная конденсация паров над колбой, унос брызг жидкости парами, полное испарение брызг жидкости, нарушение стационарного режима вследствие чрезмерно большого объема проб и загрязнение проб при отборе. Делались попытки путем усовершенствования аппаратуры по возможности
при этом не следует
Рис. 47.
Усовершенствованный прибор Отмеря для исследования фазового равновесия;
1 — колба; 2 — термометр; 3 •— холодильник; 4, 6 — краны; 5 —- патрубок для отбора пробы пара в виде конденсата состава у*\ 7 — патрубок для отбора пробы кубовой жид-
кости состава х&.
87
Рис. 48.
Схема циркуляционного прибора Рёка и Зига с насосом Коттрелля:
1 — перегонная колба; 2 — нагревательная спираль; 3 — трубка Коттрелля; 4 — насос Коттрелля; 5 — участок измерения температуры; 6 — отражатель брызг; 7 — паровая камера; 8 —- холодильник; 9 — приемник дистиллята; 10 — трубка с расширением для отбора проб; 11 — стандартный шлиф NS19. Цифрами без кружков указаны размеры в мм.
Рис. 49.
Обогреваемый узел циркуляционного прибора с вакуумной рубашкой:
12 — вакуумированная рубашка; 13 — термостатирующая рубашка; 14 — стандартный шлиф NS14,5; 15 — стандартный шлиф NS9.
исключить эти источники погрешностей и обеспечить оптимальные рабочие условия. Идеальным было бы такое решение, которое обеспечивало бы измерение концентрации жидкости в колбе и конденсата пара без отбора пробы. В последнее время для этой цели стали использовать проточный рефрактометр (см. разд. 8.5). Благодаря применению такого рефрактометра Штаге с сотр. [НО] добился уменьшения времени выхода процесса на стационарный режим в циркуляционной аппаратуре до 10 мин и менее по сравнению с несколькими часами для обычного прибора Отмеря [111]. Следует отметить, что всегда выгоднее работать с возможно большим количеством Жидкости в колбе, благодаря чему периодический или непрерывный отбор проб жидкости для анализа не препятствует установлению фазового равновесия.
Если один из компонентов имеет очень высокое давление паров (например, в системе метанол — метиловый эфир муравьиной кислоты) или коэффициенты преломления компонентов очень близки, то для измерения концентрации с успехом можно применить газовый хроматографический анализ. Этот метод особенно удобен при исследовании равновесия в многокомпонентных смесях, так
88
как позволяет ограничиться очень малыми количествами жидкости для проведения анализа {112].
ЧтобыХизбежать брызгоуноса и перегрева жидкости, Джиллеспи [113] воспользовался насосом Коттрелля. Применение насоса Коттрелля в приборах для исследованиЯ-фазового равновесия весьма целесообразно, так как благодаря ему удается получать очень хорошие результаты,^выдерживающие термодинамическую проверку. Насос Коттрелля использован, например, в приборах Река и Зига [114] (рис. 48 и 49), в которых приемник дистиллята по емкости в 20 раз меньше колбы. Нагревательная спираль 2 (см. рис. 48), погруженная непосредственно в кубовую жидкость, обеспечивает равномерное кипение. Поднимающиеся пузырьки пара интенсивно перемешивают содержимое^колбы 1 и при прохождении через трубку Коттрелля увлекают за собой значительное количество жидкости. Образовавшаяся паро-жидкостная смесь выбрасывается на участок измерения температуры 5, защищенный отражателем брызг 6. В этом месте жидкая н паровая фазы разделяются. Жидкость стекает вниз, проходит через расширение [10] для отбора пробы и возвращается обратно в колбу 1. Пары поступают в паровую (расширительную) камеру 7 и полностью конденсируются в холодильнике 8. Конденсат стекает через приемник дистиллята 9 и затем возвращается в колбу 1.
Паровая камера 7 и насос Коттрелля 4 в усовершенствованном приборе, показанном на рис. 49, с целью исключения частичной конденсации паров, помещены в вакуумированную рубашку 12. Наружная стенка вакуумированной рубашки поддерживается при температуре кипения смеси с помощью циркуляционного термостата.
При такой конструкции прибора удается избежать искажения равновесной концентрации паров, вызванной их частичной конденсацией или полным испарением образовавшихся капель. Гидростатический напор насоса Коттрелля и уровень жидкости в колбе при правильном проведении опыта не оказывают никакого влияния на измерение давления и температуры. Исследуемое вещество ин на каком участке прибора не соприкасается с кранами или шлифами, поэтому загрязнение пробы смазкой исключено.
Прибор с насосом Коттрелля оказался пригодным также и для исследования систем с высокой относительной летучестью (А/Кипф> 100 °C) [115]. Усовершенствованный циркуляционный прибор «Лабодест» также оборудован насосом Коттрелля и снабжен электромагнитными клапанами с тефлоновыми наконечниками для отбора проб [112]. Аналогично выполнен прибор в микроисполнении, рабочий объем колбы составляет всего 100 мл жидкости (рис. 50). Указанные выше
Рис. 50.
Схема циркуляционного прибора «Лабодест» с насосом Коттрелля:
1 — стабилизатор вакуума; 2 — пульт управления; 3 — лабораторный плоский самописец; 4 — газовый хроматограф; 5 — электронный интегратор.
Изготовитель: фирма Фишера «Лабораторная и исследовательская техника», Бонн—курорт Годесберг.
89
Рис. 51.
Схема прибора Шуберта
для исследования равновесия (модель G15).
источники погрешностей в этих приборах сведены к минимуму. То же можно сказать и о приборе Шуберта [116], предназначенном для изучения равновесия, новейшая модель G15 которого показана на рис. 51.
В этом приборе соединительный узел между кипятильной колбой и паровым пространством выполнен в виде трубки Коттрелля. Конструкция трубки, соединяющей паровое пространство с холодильником, в основном такая же, как и в модели G13. Тепло, необходимое для испарения жидкости, поступает от обогревателя, обогреватель состоит из стеклянной дуги с внутренней нагревательной спиралью. На внешнюю поверхность дуги наплавлен стеклянный порошок для исключения перегрева жидкости при кипении. Эта дуга установлена на достаточном расстоянии от дна колбы так, что под нею может свободно вращаться магнитная мешалка в тефлоновой оболочке, обеспечивающая интенсивную турбулизацию жидкости. Форма дуги способствует образованию пузырьков пара преимущественно в центре кипящей жидкости, поскольку в месте наибольшего изгиба дуги наблюдается наибольший приток тепла.
Для пробоотбора служит электромагнитная маятниковая подвеска, вдоль которой стекает струя конденсата или охлажденной жидкости. Применение подобной маятниковой подвески вместо ранее использовавшейся качающейся воронки имеет то преимущество, что это позволяет исключить два стеклянных шлифа на каждом месте пробоотбора. Другой характерной особенностью при
90
бора G15 является совмещение капилляров (3 мм) для стока конденсата и отсе-парированной жидкости перед входом в среднюю часть прибора. Достигаемое при этом смешение обоих потоков жидкости способствует устранению значительных колебаний концентрации жидкости на входном участке зоны кипения, которые могут заметно нарушать равновесие. Все остальные узлы прибора не отличаются от узлов предшествующих моделей этого прибора.
Вихтерле и Хала [117] описывают микроприбор для изотермического исследования фазового равновесия в многокомпонентных системах при объеме проб всего лишь 2 мл. Нойманн и Вальх [118] разработали прибор для изучения фазового равновесия при низких температурах, который был применен для исследования системы СО2—СН4. Штаге и Фишер [118а] сообщают о дальнейшем усовершенствовании циркуляционного прибора «Лабодест», специально предназначенного для изучения смесей компонентов с ограниченной взаимной растворимостью.
Рис. 52.
Прибор Кумаркришны Рао с сотр. для исследования равновесия при давлениях до 15 кгс/см2:
1 — вентнль для отбора пробы жидкости; 2 — карман для термопары; 3 — продувочные вентили;. 4 — манометр; 5 — конденсатор; 6 — вентиль для отбора пробы пара; 7 — предохранительный клапан; 8 — куб;
9 — перфорированная медная трубка; 10 — дренажный вентнль.
Рис. 53.
Схема прибора Лидерсена—Хаммера для исследования фазового равновесия при остаточных давлениях до 1 мм рт. ст.:
1 — колбонагреватель; 2 — колба для исследуемой смеси (диаметр 6 см);
3 — колбы для отбора проб; 4, 5 — алюминиевые цилиндры с электрической спиралью; 6 — соединительная трубка; 7 — подвесной шарнир;
8 — воздушный холодильник; 9 — водяной холодильник; '10 — буферная емкость; 11 — вентнль; 12 — вакуумный насос; 13 — регулятор давления; 14, 15 — сосуды Дьюара, заполненные твердым диоксидом углерода;
16 — манометр Мак-Леода; 17 — приемник дистиллята; 18 — соединительный капилляр; 19 — вращающийся стол; 20 — ось вращения прибора.
91
Рис. 55.
Графический метод проверки термодинамической достоверности данных по равновесию в системе хлороформ—этанол по методу Редлнха—Кистера (t = 45 °C).
Рис. 54.
Схема прибора Юигханса—Вебера для исследования равновесия с применением инертного газа:
1 — сферический сосуд для исследуемой смеси; 2 — термометры с ценой деления шкалы 1/10 °C; 3 — изоляция нз стекловаты; 4 — пластина из пористой керамики.
Для исследования систем бензол —н-гексан и бензол—циклогексан при давлениях до 18 ат Кумаркришна с сотр. [119] использовал прибор, схема которого показана на рис. 52. Можно также применять обычную аппаратуру из стекла, заключенную в автоклав. Иост с сотр. [120] встроил в автоклав емкостью 20 л циркуляционный прибор из стекла с адиабатической рубашкой.
Для исследования равновесия при остаточном давлении вплоть до 1 мм рт. ст. Лидерсен и Хаммер [121] разработали прибор, который приводят во вращение, что обеспечивает периодическое обновление пленки жидкости в процессе испарения, а также интенсивное перемешивание кипящей жидкости и конденсата (рис. 53). Прибор был использован, например, для исследования фазового равновесия в системе н-дибутилфталат — н-дибутиловый эфир себациновой кислоты при остаточном давлении 1 мм рт. ст.
Метод изучения равновесия, основанный на разбавлении насыщенных паров потоком инертного газа, описан Юнгхансом и Вебером [122]. Этот метод был применен при исследовании бинарной смеси этилбензол—стирол, причем полимеризацию исключали путем добавки ингибитора. Метод, основанный на введении инертного газа в паровое пространство, имеет то важное преимущество, что он дает возможность проводить исследование в изобарных условиях; при этом изменяющиеся парциальные давления р± и р2 паров дополняются парциальным давлением инертного газа р3 до исходного наружного давления. Одновременно благодаря термостатированию обеспечивается изотермичность процесса (рис. 54).
Большое преимущество дает применение радиоактивных изотопов в исследованиях фазового равновесия [123]. Описано применение изотопа 36S [124].
Следует иметь в виду, что несмотря на тщательность измерений экспериментальные данные отдельных авторов более или менее различаются. В последнее время проводят много исследований для выявления источников погрешностей для уточнения и подтверждения экспериментальных данных.
Экспериментальные данные по равновесию можно проверить термодинамически по способам Кортюма с сотр. [44], Карлссона и Кольборна [95], Ридлиха и Кистера [125] и других исследователей [17, 74].
92
В Редлих и Кистер [125] показали, что условием термодинамической досто-
В верности данных по равновесию является соблюдение равенства
I j 1g (Ti/Ts) dXi = 0 (67a)
К
К На основе данных по равновесию вычисляют ?1/?2> ПРИ этом используют соотно-Н шение
1g (V1/V2) = 1g (//Л/Wi) — 1g (PxM) (676)
E где 4i — коэффициент активности соответствующего компонента; р, — концен-Е трация паровой фазы; х{ — концентрация компонентов в жидкой фазе; р° — дав-Е ление паров чистых Компонентов.
Е' На графике зависимости 1g Т1/Т2 от xt заштрихованные площади над и под Et осью абсцисс (рис. 55) должны быть равны в том случае, если изотермические данные надежны с термодинамической точки зрения. На рис. 55 в качестве М; примера показана зависимость подобного рода для системы хлороформ — этанол HI при 45 °C. Несовпадение площадей составляет всего 1,6%. Херингтон [127] рас-пространил этот метод на тройные системы и изобарные условия.
Вопросы термодинамической проверки равновесных данных, полученных в изобарных условиях, были рассмотрены Биттрихом [128]. Он дал критическую К оценку важнейшим известным методам. Херингтон [6] разработал новый графи-^Е ческий метод («тест на симметричность площадей»), который позволяет проверить Щ экспериментальные данные для ограниченного интервала концентраций бинар-ных и тройных смесей. Тао [129], исходя из уравнения Гиббса—Дюгема, выво-дит соотношение для проверки термодинамической достоверности данных по фа-зовому равновесию в многокомпонентных системах. В монографии Шуберта [17] ,^Е подробно обсуждаются прикладные методы измерения коэффициентов активно-сти и способы термодинамической проверки данных по фазовому равновесию би-нарных систем.
И 4.7. ЧИСЛО ТЕОРЕТИЧЕСКИХ СТУПЕНЕЙ К РАЗДЕЛЕНИЯ (ЧИСЛО ТЕОРЕТИЧЕСКИХ К ТАРЕЛОК)
[Иг' Как уже было отмечено в гл. 2, с разработкой большого числа И| различных ректификационных колонн возникла необходимость сравнительной оценки их разделяющей способности. Под разде-ляюще® способностью колонны подразумевают количество дистил-(^Е лята определенной концентрации, полученное в единицу времени из смеси определенного состава при заданных условиях ректи-Ик Фикации *•
^Е Один из первых методов определения разделяющей способности К, состоял в следующем. Результаты, полученные при ректификации ^Е в одних и тех же условиях, смеси, содержащей, например, 50% :^Е (масс.) толуола, оформляли в виде графиков зависимости темпера-i^E туры ректификации от количества отобранного дистиллята (кри-пИе вая разгонки). Сравнивали ход кривых, полученных для различ-
К * В советской литературе под разделяющей способностью колонны пони-У^Е мают эффективность ее работы, выраженную числом теоретических ступеней 1^Е( Разделения или единиц переноса. — Прим. ред.
93
Рис. 56.
Метод определения полюсного расстояния 5 для периодической ректификации по кривой разгонки (метод Боумена—Чичелли).
W — выход дистиллята.
Рис. 57.
Эволюция тарельчатой колонны:
а — простой перегонный аппарат, эквивалентный одной теоретической ступени разделения; б — тарельчатая колонна с тремя реальными тарелками.
ных разгонок, и делали качественный вывод, например о том, что разделяющая способность зависит от высоты колонны и ее нагрузки.
Более точные сведения о разделяющей способности получают путем измерения какой-либо физической характеристики дистиллята, например показателя преломления, и затем построения графика зависимости этого показателя от количества отогнанного дистиллята. Одна лишь температура кипения вещества не является однозначным критерием степени его чистоты. Эти методы оценки разделяющей способности вполне пригодны для сравнения различных колонн, но не дают возможности заранее рассчитать размеры колонны и параметры ректификации, обеспечивающие заданную степень разделения. Поэтому для характеристики разделяющей способности были предложены следующие показатели: выход дистиллята определенной степени чистоты [130]; количество промежуточной фракции, ограниченной двумя концентрациями дистиллята; наклон кривой разгонки на промежуточном участке [1311; число конечных ступеней [132]*.
* По определению [132] число конечных ступеней при периодической ректификации зависит от концентрационных пределов промежуточной фракции дистиллята, относительной летучести разделяемых компонентов и флегмового числа. — Прим. ред.
94
Оба последних показателя можно рекомендовать для оценки разделяющей способности при периодической ректификации бинарных смесей. Было введено понятие полюсного расстояния, определяемого по наклону кривой разгонки в точке, в которой дистиллят содержит точно 50% (мол.) легколетучего компонента (рис. 56).
С введением понятия теоретической тарелки или теоретической ступени разделения появилась возможность количественно оценивать как эффективность колонны, так и степень трудности разделения.
Теоретическая тарелка— это гипотетический участок колонны, соответствующий идеальной тарелке, на котором между паром, поднимающимся из нижележащего участка колонны (теоретической тарелки), и жидкостью, стекающей с вышележащего участка колонны (теоретической тарелки) наступает термодинамическое равновесие (см. рис. 24) *.
Поясним понятие теоретической тарелки более подробно на примере эволюции тарельчатой колонны. Самый простой аппарат для перегонки состоит из перегонной колбы для испарения жид-
кости и приставки для конденсации паров и отвода конденсата. При этом, согласно определению, такой аппарат соответствует одной теоретической тарелке, поскольку пары, поднимающиеся из колбы, находятся в термодинамическом равновесии с жидкостью, загруженной в колбу ** (рис. 57д). Для достижения более
высокой степени разделения Адам предложил устанавливать после-
довательно несколько перегонных колб и каждую последующую колбу нагревать парами, выходящими из предыдущей колбы. В результате частичной конденсации паров в соединительных трубках, охлаждаемых воздухом, образуется некоторое количество флегмы. Если последовательно расположенные перегонные колбы
разместить одну над другой, то получится уже известная тарельчатая колонна (рис. 576).
Для дополнительного пояснения понятия теоретической тарелки рядом со схематическим изображением тарельчатой колонны (рис. 58) приведена кривая равновесия, которая иллюстрирует процессы, протекающие в колонне. Допустим, что как и при исследовании фазового равновесия, колонна работает с бесконечным флегмовым числом, т. е. без отбора дистиллята. В колбу загружена смесь, содержащая — 10% (мол.) компонента 1 (легколетучий). Пары, образующиеся при испарении кубовой жидкости, после установления равновесия имеют концентрацию у\ = 25% (мол.). При конденсации этих паров образуется жидкость состава х2 на тарелке 2 (колбу рассматриваем как тарелку 1). Пары, поднимающиеся из перегонной колбы, за счет теплообмена с жид-
* При этом равновесными являются концентрации паров и жидкости,
' удаляющихся из рассматриваемого участка колонны. — Прим. ред.
** По ряду причин указанное равновесие полностью достигается в редких
случаях. — Прим.
ред.
95
Рис. 58.
Взаимосвязь между идеальной колонной с идеально работающими тарелками и кривой равновесия.
костью, находящейся на тарелке 2, вызывают испарение из нее легколетучих фракций. Концентрация легколетучего компонента во вновь образующихся парах у\ составляет уже 50,4% (мол.). Эти пары конденсируются на тарелке 3, в результате чего жидкость на тарелке также имеет концентрацию х3 — 50,4% (мол.). Поднимающиеся с тарелки 3 пары имеют концентрацию //з == 76% (мол.) и т. д. При отводе в холодильник паров у\, поднимающихся с тарелки 4, в результате конденсации получают дистиллят концентрацией = 91% (мол.).
Для обогащения смеси, начиная с концентрации 10% (мол.), которую имеет жидкость в колбе, до концентрации 91 %, которую имеет дистиллят, требуется в общей сложности четыре теоретических ступени разделения. Для этого нужна колонна с тремя идеально работающими тарелками и куб (перегонная колба), который соответствует одной теоретической ступени разделения *. Вместо термина «теоретическая тарелка» в дальнейшем будет применяться выражение «теоретическая ступень разделения», поскольку оно более точно отражает сущность метода ступенчатых построений, который применяютдля оценки обогащения, достигаемого как в тарельчатой, так и в насадочной колонне.
Понятие теоретической ступени разделения было использовано при характеристике эффективности насадочных колонн и ряда методов разделения. Оно позволяет количественно оценить эффективность работы применяемой аппаратуры. Длину участка колонны, который необходим для перехода от концентрации жидкости х к равновесной концентрации пара у *, называют высотой,
* При этом следует учитывать, что куб не всегда эквивалентен целой теоретической ступени. — Прим. ред.
96
эквивалентной теоретической ступени (ВЭТС). В литературе на английском языке используют термин НЕТР (height eguivalent to a theoretical plate).
Если принять за ось координат г ось колонны, получим
(^l)z+B3TC (xl)z = (68)
или
(//1/1/2 )z+ВЭТс/(^1/i's) =а (69)
Для разделяющей колонны длиной I получим
ВЭТС = (/пт
Число теоретических ступеней разделения, достигаемое в колонне, является функцией большого числа параметров как аппаратурного, так и технологического характера. Путем одних только математических построений до сих пор еще не удалось разработать метод расчета ВЭТС. На основе многочисленных экспериментальных результатов получены только эмпирические соотношения (см. разд. 4.2 и 4.8).
На реальных тарелках практически никогда не достигается к. п. д. 100%, что возможно для идеальных тарелок; обычно к. п. д. составляет 50—90% *. Это вызвано, во-первых, тем, что перемешивание пара и жидкости в большинстве случаев не является совершенным, и, во-вторых, тем, что пар, особенно при больших скоростях, увлекает брызги жидкости на вышележащую тарелку. Кроме того, колонны, как правило, работают не с бесконечным флегмовым числом, а с конечным, так как целью любой ректификации является получение дистиллята. Как показал Аншюц [133], коэффициент полезного действия тарелок может быть учтен при графическом построении теоретических ступеней разделения по методу Мак-Кэба и Тиле.
Под флегмовым числом v понимают отношение количества жидкости R, возвращаемой после конденсации паров в верхнюю часть колонны, к количеству жидкости Е, отбираемой в виде дистиллята:
v = R/E
Если дистиллят не отбирают, го флегмовое число является бесконечным:
v = lim R/E — оо £->0
Количество паров D, выходящих из колонны, складывается таким образом из R и Е:
D = R Е (70)
* Известны случаи, когда для ситчатых тарелок с переливами при работе с сильно пенящимися жидкостями удавалось в определенной области нагрузок получить к. п. д. выше 100%. — Прим. ред.
4 Э. Крель 97
Рис. 59.
Кривая равновесия смеси бензол—толуол прн 760 мм рт. ст. (построение рабочих лнкик и ступеней разделения).
Это справедливо по отношению к каждому компоненту, что выражается уравнением
Dy—RxB-\-ExE (71)
При подстановке (70) в (71) получим
у = RxB!(R + Е) 4- ExeKR 4- Е) (72)
Разделив на Е числители и знаменатели в правой части уравнения (72), получим
RxB Ехе
У = £(/?,'£ -]-£/£) + £(£/£+£/£) (73)
Подставив вместо RIE условное обозначение для флегмового числа v, получим уравнение рабочей линии укрепляющей части колонны
У = Vxb№ + !) + хе№ +
(74)
Это линейное уравнение выражает зависимость состава пара от состава жидкости для любого поперечного сечения колонны (при условии адиабатичности процесса) и представляет собой рабочую линию а—с (рис. 59). Если мольные энтальпии испарения компонентов сильно различаются, то это необходимо учесть при расчете числа теоретических ступеней по методу Мак-Кэба и Тиле.
Различия в мольных энтальпиях испарения могут оказывать заметное влияние на число теоретических ступеней разделения особенно при малых флегмовых числах или при малой относительной летучести компонентов и высокой разделительной способности колонны. Графический метод Мак-Кэба и Тиле в этом случае заметно усложняется, так как при этом рабочие линии процесса ректификации не являются прямыми. Однако видоизменение метода Мак-Кэба и Тиле, предложенное Фишером [134], относительно упрощает графические построения. Биллет [135] вывел уравнения для расчета рабочих линий, соответствующих процессу ректификации бинарных смесей при различных мольных энтальпиях испарения компонентов. Тум [136] разработал метод прямого расчета числа теоретических ступеней разделения при ректификации идеальных бинарных смесей с конечным флегмовым числом, в котором учтены различия в энтальпиях испарения.
Неретник с сотр. [137] пришел к выводу, что для энтальпий испарения различных веществ можно найти такие поправочные коэффициенты N, что все вещества в полученной таким образом 98
новой системе измерения будут иметь одинаковые скорректированные энтальпии испарения. Тем самым рабочие линии для систем, исходные компоненты которых имеют весьма различающиеся энтальпии испарения, в результате простого преобразования можно было бы получать на диаграмме Мак-Кэба и Тиле в виде прямых линий. Если относительная летучесть компонентов не зависит от состава смеси, указанное преобразование достаточно выполнить всего для нескольких концентраций.
В системе координат с осью абсцисс х и осью ординат у рабочая линия укрепляющей части колонны пересекает ось ординат в точке
y = xE/(v+l) (75)
При бесконечном флегмовом числе (у = со) ордината этой точки становится равной 0, так как
lim(/ = 0 (76)
V->oo
Это означает, что рабочая линия при v = оо проходит через начало координат и совпадает с диагональю диаграммы равновесия пар— жидкость. Угол наклона рабочей линии к оси абсцисс определяется зависимостью
tga = y/(o+l) (77)
Сделаем упрощающее допущение, что мольные энтальпии компонентов, а следовательно и смеси, одинаковы. Тогда с помощью графического метода Мак-Кэба и Тиле [77] можно рассчитать число теоретических ступеней разделения, необходимое для разделения смеси при определенном флегмовом числе, которое превышает минимальное флегмовое число. Вычисления можно проводить как для периодического, так и для непрерывного режима работы.
1 Следует подчеркнуть, что все вышеизложенные рассуждения относились к тарельчатым колоннам. В колоннах этого типа массо-t обмен происходит при барботировании потока паров через слой жидкости на тарелке, при этом между паром и жидкостью дости-[ гается более или менее полное термодинамическое равновесие. [ Процесс изменения температуры и состава паров и жидкости повто-t ряется от тарелки к тарелке; таким образом, обогащение паров t происходит по ступенчатому закону. В насадочной колонне при I прохождении потока паров над пленкой жидкости, орошающей I насадку, имеет место непрерывный процесс массообмена. Однако Г ни в каком месте насадки пары не остаются настолько долго, чтобы [ между ними и пленкой жидкости могло установиться термодина-I мическое равновесие. Непрерывное обогащение достигается в ре-К зультате многих элементарных актов разделения. Поэтому для I характеристики насадочных колонн ввели термин «высота единицы I переноса» (heigt of transfer unit — HTU).
I Простые приемы вычисления числа ступеней, разработанные I для тарельчатых колонн, можно использовать также для наса-I- 4* .99
дочных колонн. Однако следует учитывать, что такие расчеты дают лишь хорошее приближение без строгого теоретического обоснования.
О пользе концепции теоретической ступени можно получить определенное представление, если сравнить процессы разделения в тарельчатой и насадочной колоннах по возможности одинаковых размеров, работающих в идентичных условиях. Если колонны имеют одинаковую разделяющую способность,то можно уверенно сказать, что обе колонны эквивалентны определенному числу теоретических ступеней разделения.
4.7.1. РАСЧЕТ ЧИСЛА ТЕОРЕТИЧЕСКИХ
СТУПЕНЕЙ РАЗДЕЛЕНИЯ ПО МЕТОДУ МАК-КЭБА И ТИЛЕ ДЛЯ ПЕРИОДИЧЕСКОЙ РЕКТИФИКАЦИИ
В качестве примера рассмотрим разделение смеси бензол-толуол. Поставлена следующая задача: смесь, содержащую 40% (мол.) бензола, необходимо обогатить до концентрации 98% путем периодической ректификации.
В координатах х—у строим кривую равновесия (см. рис. 59) и через точку хв = 40% (мол.) на оси абсцисс проводим прямую, параллельную оси у, до пересечения с равновесной кривой в точке Ь. Прямая, проведенная через точку хБ = 98% (мол.) параллельно оси ординат, пересекает диагональ в точке а. Соединив точки а и Ь, получим рабочую линию, которая пересекает при продолжении ее за точку b ось ординат в точке с, соответствующей у = 37% (мол.). Из уравнения (75) находим:
"мин = (МО-1 <78>
сМин = 98/37 — 1 = 1,65 (79)
Значение 1,65 представляет собой минимальное флегмовое число, при котором для разделения смеси бензол—толуол требуется бесконечно большое число ступеней разделения. Так как в этом случае точки хЕ, хв и у* лежат на рабочей линии, то минимальное флегмовое число может быть также выражено уравнением [103]
°мин = (хе-^Ж-хв) <8°)
в котором у * соответствует точке пересечения b рабочей линии а—с с кривой равновесия. Находим
%ин = (98-62)/(62-40)~ 1,65 (81)
При флегмовом числе меньше 1,65 обогащение до концентрации 98% (мол.) вообще невозможно. Если увеличить флегмовое число (у > 1,65), то потребуется конечное число ступеней разделения и оно будет тем меньше, чем больше флегмовое число. Примем, что флегмовое число v = 1,85; тогда длина отрезка, отсекаемого на оси ординат рабочей линией, будет равна:
«/ = 98/(1,85 4-1) = 34,4 (82)
100
Таблица 12.
Число теоретических ступеней разделения, требуемое для разделения смеси бензол—толуол при хв= 40%, хе — 98% (мол.), 1'мин ~ 1,65
Рабочее флегмовое число Отрезок, отсекаемый иа оси ординат, ХЕ у- о + 1 Число теоретических ступеней разделения, п
1,85 3,44 10,0
3,00 24,5 7,0
5,00 16,4 6,0
10,00 8,9 5,5
20,00 4,7 5,0
50,00 1,9 5,0
оо 0,0 4,5
Таблица 13.
Флегмовые числа, обеспечивающие разделение смеси бензол—толуол при хв от 80 до 5% (мол.), п — 10 и хе = 98% (мол.)
Хв, % (мол.) Флегмовое число
минимальное рабочее
80 0,58 0,65
60 0,94 1,09
40 1,65 1,85
20 3,46 3,90
10 7,10 8,80
5 13,00 25,00
Точку пересечения с, соответствующую у — 34,4, (см. рис. 59) соединяем с точкой а и получаем рабочую линию, характеризующую выбранные условия ректификации. Начиная от точки Ь,
наносим теоретические ступени разделения, вписывая их между равновесной кривой и рабочей линией; получим 10 теоретических ступеней разделения.
Следует выяснить, насколько уменьшается число теоретических ступеней разделения с увеличением флегмового числа. В табл. 12 приведены числа теоретических ступеней, необходимые для разделения смеси бензол—толуол при разных флегмовых числах. На рис. 60 показана кривая равновесия этой смеси с соответствующими рабочими линиями.
Очевидно в данном случае разделения нет смысла увеличивать флегмовое число выше 5; увеличение числа ступеней становится таким незначительным, что дальнейшее повышение флегмового числа себя не оправдывает.
Следует учитывать, что при периодической ректификации содержание легколетучего компонента в кубовой жидкости постепенно уменьшается в результате отбора дистиллята в головке колонны концентрация хв стремится к нулю. Предположим, что в результате разгонки концентрация кубовой жидкости xs уменьшилась до 10% (мол.). Еще раз, как вначале, проведем расчет. Ниже дано сравнение конечных рабочих параметров раз-
гонки с исходными данными, соответствующими началу разгонки:
'Начало разгонки...............
Сравнительный расчет ..........
ХВ
40
10
ХЕ
98
98
°мин °раб
1,65 1,85
7,10 8,80
п
10
10
101
Рабочие линии процесса ректификации смеси бензол—толуол в обогаща ющей части колонны для различных флегмовых чисел.
х^ — 98% (мол.). Линии построены по данным табл. 12.
Рис. 61.
Зависимость флегмового числа от концентрации кубовой жидкости xg.
Xg = 98% (мол.), число теоретических ступеней разделения п = 10.
Для того, чтобы установить, до каких пределов нужно повысить флегмовое число (при изменившемся составе кубовой жидкости) для получения дистиллята прежнего состава расчетное число теоретических ступеней разделения для начала и конца разгонки было намеренно выбрано одинаковым. Если оставить флегмовое число v = 1,85, то концентрация дистиллята хЕ с уменьшением концентрации жидкости в перегонной колбе снизилась бы.
Таким образом, при периодической ректификации недостаточно установить условия процесса для начального момента. Следует также решить, до какой концентрации кубовой жидкости целесообразно проводить разгонку или какое максимальное флегмовое число необходимо выбрать, чтобы длительность разгонки не была бы слишком большой. Если принять предельное флегмовое число и = 25 и хв = 80% (мол.), оставив постоянными хЕ ~ 98% (мол.) и число теоретических ступеней разделения п = 10, то для снижения концентрации легколетучего компонента в кубовой жидкости с 80 до 5% (мол.) необходимы флегмовые числа, указанные в табл. 13.
Из графика зависимости ораб от хв (рис. 61) легко увидеть, что в области концентрации низкокипящего компонента ниже 10% (мол.) уменьшение его концентрации на каждый мольный процент потребует значительного увеличения количества орошающей жидкости. На практике разгонку обычно начинают при флегмовом числе v = 2, а при повышении температуры в верху колонны 102
флегмовое число увеличивают до 5, и затем еще раз до 10. Целесообразность дальнейшего увеличения флегмового числа следует проверять в каждом отдельном случае. Необходимо только иметь в виду, что чем меньше соблюдаются требуемые условия ректификации, тем больше будет объем промежуточной (нецелевой) фракции.
Основываясь на уравнениях Релея, Вебер [1381 разработал диаграмму, с помощью которой для идеальных смесей при известном значении относительной летучести компонентов можно определить, какой состав имеет кубовая жидкость, если первоначальная загрузка куба равна Во, а количество кубового остатка после разгонки составляет В{. Фогельпол 1139] разработал метод расчета ректификации идеальных тройных смесей в тарельчатых колоннах. Доменех с сотр. [139а] создал математическую модель процесса ректификации. Биллет [140] представил уравнения, с помощью которых для периодической ректификации бинарных смесей при постоянной концентрации дистиллята можно рассчитать, применяя смешанные графоаналитические способы, такие параметры ректификации, как флегмовое число, количество наработанного продукта, состав кубовой жидкости и объем проходящих в колонне паров смеси для различных моментов разгонки. Применительно к глубокой очистке жидкостей путем периодической ректификации Вилкокс [140 а] вывел специальные уравнения для расчета процессов разделения, направленных на получение дистиллята весьма высокой степени чистоты.
При расчете процесса периодической ректификации следует учитывать, что по мере изменения состава кубовой жидкости одновременно повсюду в колонне (в парах и в жидкости) уменьшается доля легколетучего компонента (см. разд. 4.10.5).
4.7.2. РАСЧЕТ ЧИСЛА ТЕОРЕТИЧЕСКИХ СТУПЕНЕЙ РАЗДЕЛЕНИЯ ПО МЕТОДУ МАК-КЭБА И ТИЛЕ ДЛЯ НЕПРЕРЫВНОЙ РЕКТИФИКАЦИИ
Лабораторную ректификацию большей частью проводят в периоди-' ческом режиме, однако возможны такие процессы разделения, для которых непрерывный способ является предпочтительным или даже ) обязательным (см. разд. 5.2.2). В противоположность периодиче-( ской ректификации, при которой концентрации кубовой жидкости,
дистиллята и всего содержимого колонны непрерывно изменяются, при непрерывной ректификации эти параметры остаются неизменными. При непрерывной ректификации исходную жидкую смесь
постоянного состава предварительно подогревают до температуры, установившейся в колонне в месте ввода смеси; ее пары обога-
щаются в укрепляющей части колонны 1 (рис. 62). Участок колонны 3 между местом ввода исходной смеси 2 и кубом (перегонная колба 4) называют исчерпывающей частью. Обозначим коли-
.чество дистиллята (головной продукт) через Е, а количество про-
дукта, отбираемого из нижнего конца колонны (над кубом) или
103
Рис. 62.
Установка (модель IIIv) для непрерывной вакуумной ректификации по нормалям «Дестииорм»:
1 — укрепляющая часть колонны; 2 — место ввода исходной смеси; 3 — исчерпывающая часть колонны; 4 — перегонная колба.
Изготовитель: народное предприятие
«Комбинат технического стекла», Ильменау).
из самого куба (кубовый остаток или кубовый продукт) через А.
При работе установки] непрерывного действия следят за тем, чтобы постоянно сохранялся материальный баланс:
Z = Е + Л (83) а также
x_Z = хРЕ + х.А (84) где Z, Е, А — соответственно количество питающей жидкости, головного (целевого) продукта и кубового продукта (отходы).
Рассмотрим процесс разделения смеси жирных кислот нормального строения Св—С7, содержащей 81% (мол.) кислоты С6. Остаточное давление при разгонке должно составлять 20 мм рт. ст. Рассчитаем число теоретических ступеней, необходимое для разделения
этой смеси по непрерывному способу, если требуется получить головной продукт, содержащий 96% (мол.) низкокипя-щего компонента, а кубовый остаток — 0,5%.Кривая равновесия для этой смеси экспериментально не изучена, она была рассчитана по формуле (52) при а = 2,10 (рис. 63).
Через точки а (хЕ = 96%) и b (xz = 81%) проводят рабочую линию процесса ректификации для укрепляющей части колонны. Эта линия пересекает ось ординат в точке с, которой соответствует у = 59,8% (мол.). По уравнению (78) вычисляем минимальное флегмовое число:
ОмИа=(^)- 1 = 96/59-8 - 1 = 0’6
(85)
Чтобы не получить слишком большого числа теоретических ступеней разделения, примем флегмовое число v = 1, тогда отре-
104
зок, отсекаемый рабочей линией на оси ординат, будет равен у = 96/(1 + 1) =48 (86)
Рабочая линия а—с', соответствующая конечной стадии разгонки, пересекает ось ординат в точке 48% (мол.), а вертикальную линию, проведенную через xz = 81% —в точке d. Через ; точку хА = 0,5% проводят линию, параллельную оси ординат, ! до пересечения с диагональю в точке е. Соединяют точки d и е и j получают прямую, которая является рабочей линией процесса ( ректификации для исчерпывающей части колонны. Начиная от ;* точки d, вычерчивают ступени разделения для укрепляющей части колонны. Эти ступени располагаются между кривой равновесия
S и рабочей линией d—а\ для укрепляющей части колонны до-г статочно иметь 4 ступени разделения. Затем откладывают ступени разделения от точки d вниз и между равновесной кривой и рабочей ^линией процесса для исчерпывающей части колонны d — е получают «' всего 13 ступеней. Если кривая равновесия построена в мелком v масштабе (примерно 25 X 25 см) или подходит очень близко к диаго-
нали, то рекомендуется участок диаграммы, лежащей между 10 и 0%, отдельно вычертить в большем масштабе, как это показано
в левой части рис. 63; такой прием облегчает построение ступеней
(разделения.
1 Для заданного разделения необходимо в общей сложности (17 теоретических ступеней, из которых 13 приходятся на исчер-; пывающую часть колонны, а 4 — на укрепляющую часть. В разд. ( 4.10.9. и 7.3 показано, как можно практически получить эти (17 теоретических ступеней.
X, 7о(мОЛ.)
UC. 63.
клетная кривая равновесия смеси жирных кислот Св—С7 нормального роеиия при 20 мм рт. ст. и пример графического способа расчета числа Юретических ступеней разделения для непрерывной ректификации.
105
т. . °/о( мал.)
лонны увеличится на
Рис. 64.
Влияние температуры исходной смеси при непрерывной ректификации на положение рабочей линии процесса в обогащающей части колонны.
Описанный прием определения минимального флегмового числа для непрерывной ректификации основан на допущении, что разделяемая смесь поступает в колонну при температуре кипения. В этом случае количество жидкости в исчерпывающей части ко-
количество исходной смеси, т. е.
R' = R + Z
(87)
Если температура исходной смеси ниже температуры кипения, то происходит частичная конденсация паров; количество конденсирующихся паров определяется количеством тепла, необходимым для нагревания смеси до точки кипения. Это обстоятельство учитывают поправочным коэффициентом е, вследствие чего уравнение (87) принимает вид
R' = R^-eZ (88)
e==1+(^кип-^)/А77г (89)
где QzKIIII — энтальпия смеси при температуре кипения; Qz — энтальпия смеси при фактической температуре смеси в точке питания; &HV — мольная энтальпия испарения.
При этом получают значение абсциссы в точке g (рис. 64):
x'z = xz/e (90)
Прямая g — d пересекает рабочую линию в точке в, вследствие чего минимальное флегмовое число дополнительно уменьшается. Если исходная смесь поступает при температуре кипения, то е = 1, а точкам d и f соответствует одна и та же абсцисса xz [ 103 ]. Если часть смеси поступает в виде паров, то можно воспользоваться рекомендациями Каверса [142], которые позволяют и в этом случае проводить расчет числа теоретических ступеней с помощью диаграммы Мак-Кэба и Тиле.
Поль [143] описал метод расчета параметров исчерпывающей части колонны для смесей с очень низкой концентрацией легколетучего компонента. Этот метод основан на использовании диаграммы Мак-Кэба и Тиле. При этом способе расчета кривую равновесия можно приблизительно представить в виде прямой. Норват и Шуберт [144] разработали метод расчета, который можно ис-106
пользовать при высоких концентрациях легколетучего компонента в головном продукте [около 99,99% (мол.)] и при низких концентрациях этого компонента в кубовом остатке. Если диаграмму Мак-Кэба и Тиле построить в логарифмических координатах, то точность расчета увеличится. Аналитические методы определения числа теоретических ступеней и рабочих параметров для непрерывной ректификации идеальных смесей подробно изложены в гл. 4.
4.7.3. ОПРЕДЕЛЕНИЕ ЧИСЛА ТЕОРЕТИЧЕСКИХ СТУПЕНЕЙ РАЗДЕЛЕНИЯ ПО МЕТОДУ МАК-КЭБА И ТИЛЕ ДЛЯ СМЕСЕЙ С КРИВЫМИ РАВНОВЕСИЯ, ИМЕЮЩИМИ ТОЧКУ ПЕРЕГИБА И АЗЕОТРОПНУЮ ТОЧКУ
Если на кривой равновесия для какой-либо смеси имеется точка перегиба, например для смеси этанол—вода, то число теоретических ступеней разделения определяют методами, описанными в разд. 4.7.1 и 4.7.2. Однако при этом вводятся следующие ограничения, а именно: концентрация легколетучего компонента в головном продукте хЕ должна быть меньше концентрации в азеотропной точке, а при определении минимального флегмового числа < рабочую линию процесса ректификации для укрепляющей части : колонны следует 'проводить по касательной к кривой равновесия I (рис. 65). Если провести рабочую линию а—с1; как обычно, от г точки а через точку Ь2, то на кривой равновесия получатся три I точки пересечения blt b2 и Ь3. Даже при некотором увеличении I флегмового числа, начиная с его минимального значения, этой f рабочей линии соответствует бесконечно большое число теорети-I ческих ступеней разделения. Чтобы получить конечное число тео-I ретических ступеней, необходимо перейти от минимального флег-I мового числа, соответствующего касательной к кривой равнове-I сия (рабочая линия а—с), к несколько большему флегмовому числу, определяемому рабочей линией а—с'; после этого можно строить ступени разделения в обычном порядке. Касательная а—с пересекает кривую равновесия только в одной точке &4 и касается ее в точке Ьъ. Отсюда ясно, что минимальное флегмовое число остается постоянным в широком интервале концентраций. Рис. 66 иллюстрирует
Рис. 65.
Графическое определение минимального флегмового числа для разделения смесей, кривая равновесия которых имеет точку перегиба.
107
о 10 20 зо Чо 50 во
X- , Чо(мОЛ.)
Рис. 66.
Зависимость минимального фле; .OBrtr'j числа от при непрерывной решифи* кации смеси этанол—вода.
760 мм рт. ст.; = 81,6% (мол.).
зависимость умин от состава исходной смеси xz при ректи-фикации смеси этанол—вода, когда требуется получить дистиллят концентрацией 81,6% (мол.), или 92% (мол.). Как видно из рис. 66, в пределах значений xz от 7,5 до 65,3% (мол.) минимальное флегмовое число остается постоянным и равным 1,3.
Если кривая равновесия, начиная с нулевой концентрации, проходит ниже диагонали, а после пересечения с диагональю выше нее, то это означает, что данная смесь является азеотропной с максимумом на изобарной кривой кипения или минимумом на изотермической кривой давления паров. При этом точка кипения азеотропной смеси лежит выше точек кипения обоих чистых компонентов. В качестве примера можно привести смесь азотная кислота — вода (см. рис. 29 и). Температура кипения Кртео азотной кислоты 86,0° С, воды 100,0° С, азеотропа, содержащего 37,81% (мол.) кислоты, 122° С. Для этой системы Флатт [145] приводит метод графического расчета рабочих условий ректификации.
4.7.4. ОПРЕДЕЛЕНИЕ ЧИСЛА ТЕОРЕТИЧЕСКИХ
СТУПЕНЕЙ РАЗДЕЛЕНИЯ ДЛЯ СМЕСЕЙ СО СЛАБО
ВЫПУКЛЫМИ КРИВЫМИ РАВНОВЕСИЯ
И ПРИ НЕБОЛЬШОМ РАССТОЯНИИ МЕЖДУ КРИВОЙ
РАВНОВЕСИЯ И РАБОЧЕЙ ЛИНИЕЙ
Если кривая равновесия смеси, например л/-крезол — п-крезол, незначительно отклоняется от диагонали (рис. 67), то очень трудно точно построить ступени разделения в интервале низких значений хв и высоких значений хЕ. В этом случае для облегчения графических построений кривую равновесия следует вычертить в большом масштабе (порядка 2x2 м), а также дополнительно укрупнить масштаб в интервалах концентраций 0—10% и 90— 100% (мол.). При этом кривая равновесия практически принимает вид прямой (ср. с рис. 63).
Аналогичный прием можно использовать и в том случае, когда кривая равновесия достаточно выпукла, но область между кривой равновесия и рабочей линией слишком узка для точного по-
* Условное обозначение температуры кипения прн 760 мм рт. ст., принятое в немецкой литературе; в советской литературе применяется термин н. т. к. (нормальная температура кипения). — Прим. ред.
108
X , °!о(мОЛ.}
Рис. 67.
Кривая равновесия смеси л-крезол—л-крезол
Остаточное давление 30 мм рт. ст.
X, °/о(мОЛ.')
Рис. 68.
Кривая равновесия смеси толуол—н-октаи с асимптотическим приближением к диагонали.
760 мм рт. ст.
строения ступеней разделения (рис. 68). При этом число наносимых ступеней вследствие асимптотического приближения равновесной кривой к диагонали или рабочей линии является столь большим, что точность метода становится ограниченной.
Для ректификации с бесконечным флегмовым числом Штаге и Шульце [146] предлагают метод расчета числа теоретических ступеней, который связан с построением так называемой дифференциальной кривой. Согласно этому методу по возможности в большем масштабе (ось абсцисс примерно 1 м) строят график зависимости разности у* — хв от хв. Таким образом получают дифференциальную кривую выпуклой формы (рис. 69). С помощью кривой строят график зависимости «число теоретических ступеней разделения — концентрация» (рис. 70). Построение начинают с очень низкой концентрации, например 0,16% (мол.), которой на дифференциальной кривой (см. рис. 69) соответствует обогащение (у*—хв) = 0,28%. Это значение прибавляют к 0,16% и получают концентрацию жидкости на второй тарелке, равную 0,44%. Для этого значения на диаграмме снова определяют обогащение, равное 0,70%, которое прибавляют к 0,44% и получают концентрацию на следующей
20
О.2В
Я к 10
Рис. 69.
Дифференциальная кривая равновесия смеси бензол —к-гептан по данным Штаге-Шульце.
Xq — концентрация бензола в смеси.
109
35
О 20 ‘tO 60 80 100
П
Рис. 70.
Зависимость числа теоретических ступеней разделения от концентрации для смеси бензол—н-гептан (по данным Штаге—Шульце).
Рис., 71.
Зависимость числа теоретических ступеней разделения от показателя преломления для эталонной смеси н-гептан—метилциклогексан.
тарелке, равную 1,14%. Эти графические построения продолжают до достижения концентрации 99,8%, получая в результате кривую зависимости числа теоретических ступеней разделения от концентрации (см. рис. 70). Необходимое число теоретических ступеней разделения при бесконечном флегмовом числе находят по кривой следующим образом: из точек хв и хЕ на абсциссе проводят вертикальные линии до пересечения с кривой, и разность чисел по ординате дает искомое число теоретических ступеней разделения. Например, для обогащения смеси бензолом с 25% (мол.) в кубе до 85% в дистилляте потребуется 11,2—6,6 = 4,6 теоретических ступеней разделения при бесконечном флегмовом числе.
Вместо значения абсциссы хв можно также использовать и другие показатели, например плотность или показатель преломления, и аналогичным способом определять число теоретических ступеней разделения; при этом на график наносят соответствующие значения показателей для загрузки куба и дистиллята и находят разность соответствующих чисел на оси ординат. Рис. 71 иллюстрирует зависимость числа теоретических ступеней от коэффициента преломления для модельной смеси н-гептан — метилциклогексан 1147].
Для ректификации с конечным флегмовым числом Шефер разработал метод расчета теоретических ступеней с использованием двух номограмм и двух диаграмм. Этот метод, особенно пригодный для многократно повторяющихся расчетов числа теоретических ступеней разделения п, основан на приближенной гиперболической зависимости между п и флегмовым числом v, применявшейся еще Фишером:
(« - «мин) (V - ‘'мин) = («' - «мин) (°' - "мин) (91)
110
Фишер [149] предлагает метод, который в отличие от метода Шефера основан на использовании только одной номограммы. Другие графические и графоаналитические методы расчета изложены в обзоре Штаге и Джуилфса [71 ].
«Метод относительной летучести» Маца [1501 позволяет рассчитывать параметры периодической и непрерывной ректификации идеальных смесей также и в тех случаях, когда значения хЕ находятся в интервале 99—99,8%.
4.7.5. АНАЛИТИЧЕСКИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ
ЧИСЛА ТЕОРЕТИЧЕСКИХ СТУПЕНЕЙ РАЗДЕЛЕНИЯ
для ПЕРИОДИЧЕСКОЙ РЕКТИФИКАЦИИ
До сих пор мы рассматривали, главным образом, графические и графоаналитические методы определения числа теоретических ступеней, так как они более наглядно передают понятие ступени разделения. Наряду с этим разработаны многочисленные методы и уравнения, позволяющие чисто расчетным путем определить условия ректификации для заданной степени разделения.
Графические методы имеют то преимущество, что они базируются на кривой равновесия и тем самым учитывают также и неидеальное поведение смеси. Применяя чисто расчетный метод, обычно допускают, что по свойствам смесь приближается к идеа-альной.
4.7.5.1. Определение числа теоретических ступеней разделения по разности температур кипения
О взаимосвязи между разностью температур кипения и необходимым числом теоретических ступеней разделения можно судить по приведенным ниже данным для бинарных смесей углеводородов при атмосферном давлении, позаимствованным у Хильберата [1511.
Необходимое число тео-
Разность температур кипения, °C ретических ступеней разделения
1,5
3,0
5,0
7,0
100
55
30
20
Расчет облегчается, если исходить из степени очистки легколетучего компонента. Диаграмма Брэгга—Льюиса [152] позволяет сразу же найти минимальное число теоретических ступеней разделения в зависимости от разности температур кипения для бинарных эквимольных идеальных смесей (рис. 72).
111
Рис. 72.
Диаграмма Брэгга- Льюиса.
А^кип — разность температур кипения компонентов; пмнн — минимальное число теоретических ступеней разделения при полном возврате флегмы.
Для любой разности температур кипения необходимое число теоретических ступеней разделения можно также рассчитать по следующему уравнению:
п=(Т2 + 7’1)/Л(7’2-7’1) (92)
где 7\ и Та — температура кипения легколетучего и труднолетучего ком-
понентов соответственно, К; К — константа, причем К = 2,5 соответствует максимальному числу теоретических ступеней разделения, К = 3 — оптимальному, а К = 4— минимальному числу теоретических ступеней разделения.
Однако в этом уравнении не учтены остальные условия ректификации такие, как флегмовое число, рабочее давление, общая удерживающая способность колонны.
По уравнениям Странджио и Трейбала [152а] можно ориентировочно рассчитать некоторые параметры ректификации (о и п) также и для многокомпонентной смеси при условии постоянства относительной летучести и при вводе исходной смеси либо в виде жидкости, либо в виде паров.
4.7.5.2. Расчет числа теоретических ступеней разделения с помощью фактора обогащения и формул Роуза
Формула (93) предназначена для расчета числа теоретических ступеней разделения по относительной летучести а и коэффициенту обогащения Fr*. Последний представляет собой эмпирическую величину, выведенную Куком на основе многочисленных опытов по ректификации:
Fr = an = xEixBJ (хЕхв^ (93)
где хв и — содержание легколетучего и труднолетучего компонентов в кубовой жидкости соответственно; хЕ^ и хЕг — содержание легколетучего и труднолетучего компонентов в дистилляте соответственно.
Величина Fr достигает 10000 при четком разделении, когда легколетучего компонента в дистилляте содержится 99% (мол.), а в кубе — 1 %:
Fr~ 99-99/1-1 =9801 » 10 000 (94)
* Коэффициент обогащения часто называют также коэффициентом разделения. — Прим. ред.
112
^В При обычном разделении результаты считаются удовлетво-рительными, если дистиллят содержит 90 %, а кубовая жидкость —
^В 1% легколетучего компонента; в этом случае величина Fr равна S 1000:
V Fr =90-99/10-1 = 891 « 1000 (95)
Отсюда получают для четкого разделения
п = 4,0/lg а теоретических ступеней (96)
Щ а для обычного разделения
п = 3,0/lg а теоретических ступеней (97)
Разумеется, коэффициент обогащения можно рассчитать также ^В и для других условий, например для концентрации дистиллята ^В 95% (мол.) и концентрации кубового остатка 1%:
Fr = 95-99/5-1 = 1850 (98)
а" = 1850 (99)
^В Тогда
К; п lg а = 1g 1850 (100)
и
в^Вг п = 3,3/lg <х теоретических ступеней разделения (101)
^В Результаты вычислений по уравнениям (96) и (97) приведены ^В в табл. 14, они использованы при построении диаграммы, пока-^В занной на рис. 73.
^В Роуз [154] усовершенствовал этот метод, исходя из того, что для большинства процессов разделения результаты очистки вполне удовлетворительны, если первые порции дистиллята, со-ставляющие 40% от всего его количества, содержат выше 95% (мол.) легколетучего компонента при условии, что компоненты исходной идеальной бинарной смеси находятся в равных коли-чествах. Дополнительно предполагается, что удерживающая спо-
М| Таблица 14.
^^В Число теоретических ступеней разделения, рассчитанное
^^В по эмпирическому коэффициенту обогащения
Относительная летучесть Разность температур кипения, °C Требуемое число теоретических ступеней разделения
при обычном разделении по кривой / на рнс. 73 и по формуле (97) при четком разделении по кривой 2, на рис. 73 н по формуле (96)
'3,0 30 5 8
(2,0 20 9 13
И ,5 10 17 22
|1,3 7 25 35
1,25 6 30 42
1,20 5 38 50
fl, 15 4 50 65
У,12 3 70 80
И ,07 2 100 150
113
Рис. 73.
Зависимость относительной летучести от разности температур кипения компонентов и зависимость необходимого числа теоретических ступеней разделения от а при различных коэффициентах обогащения (см. табл. 14): П1 — рассчитывается по формуле (97), Fr = 1000; п2 — по формуле (96), Fr = 10 000.
Рис. 74.
Зависимость числа теоретических ступеней разделения лмакс, «опт и лмин от относительной летучести данных по методу Роуза при обычной степени очистки (см. табл. 15).
собность колонны по жидкости настолько мала, что ею можно пренебречь, в силу чего справедливо равенство п — V. Кроме того, должно быть известно среднее значение относительной летучести, на основе которой можно получить максимальное пмакс, оптимальное пОПт и минимальное пмин числа теоретических ступеней разделения (рис. 74):
«макс = 3,6/1g а (102)
«опт =2,85/lga (103)
«мин = 2,3/1g а (104)
Таблица 15.
Числа теоретических ступеней разделения, определенные по методу Роуза
а 1g а пмнн попт пмакс
2,0 0,3010 8 9 12
1,5 0,1761 13 16 20
1,25 0,0696 24 29 37
1,10 0,0414 56 69 87
1,05 0,0212 108 134 170
1,03 0,0140 203
1,02 0,0085 335
114
использование формул (102—104),
Рис. 75.
Зависимость числа теоретических ступеней разделения от флегмового числа при обычной ректификации по данным Роуза.
1 — v = 3/2п [см. уравнение (105)1; n = 73/lg а, [см. (104)]; К=4 [см. (92)1; 2 — о = п [см. (106)]; п = = 2,85/lg а [см. (103)]; К = 3; 3 — о = 2/з [см. (107)]; п = 3,6/lg а [см. (102)]; К = 2,5.
Значения указанных выше величин приведены в табл. 15, которая поясняет а также графика на рис. 74.
При обычных условиях разделения принято исходить из
того, что низкое значение п можно компенсировать соответствующим приростом v, а более высокие значения п допускают меньшее значение v. Однако очень важно не выходить при этом из определенных пределов, так как слишком низкие значения п невозможно неограниченно компенсировать увеличением флегмы, а слишком высокие значения п не допускают неограниченного уменьшения флегмового числа v. В соответствии с этим по данным Роуза получаются следующие выражения для флегмовых чисел (рис. 75):
^макс = 3/2пмнн (Ю5)
^ОПТ — ^оПТ
^мин = 2/ЗпМаКс
(106)
(107)
4.7.5.3. Расчет минимального числа теоретических ступеней разделения по уравнению Фенске для идеальных смесей при v = оо
Относительная летучесть компонентов идеальной бинарной смеси может быть рассчитана по уравнению:
а = (у11у1)1(хх1х2) (108)
где у* и у*, — концентрация легколетучего и труднолетучего компонентов в парах соответственно; и х2 — концентрация легколетучего и труднолетучего компонентов в жидкости соответственно.
Если элементарное обогащение, достигнутое на одной теоретической ступени разделения, многократно увеличивается на п последовательно расположенных ступенях разделения, то суммарный эффект разделения можно выразить следующим образом:
ап = хе/^~хе) xb/Q хв)
Если прологарифмировать равенство (108а), то можно прийти к уравнению Фенске [155] для определения минимального числа
(108а)
115
Рис. 76.
Номограмма Мельпольдера—Хедингтона для определения минимального числа теоретических ступеней разделения идеальных смесей при v = оо.
теоретических ступеней разделения (см. разд. 5.1.4.2) при периодической ректификации идеальных смесей (концентрация указана в мольных процентах):
1М* 1 * * * V — хв)/хв(* — хв)1 ] П08(й
Линн------------1 (ЮоО)
или (см. разд. 5.1.4):
1 Г хе хв
«мин= ~'sT' In 1 — In “Т----
О I — Хс 1 — Хп с о
где 6 = а — 1.
По сравнению с методом Роуза расчет по этому уравнению можно проводить для различных значений хв и хЕ при условии
V = ОО.
Мельпольдер и Хедингтон [901 на основе уравнения (1086) разработали номограмму, по которой можно быстро и просто определить искомые величины (рис. 76).
На номограмме приведен пример расчета п при условии: хв = 15% (мол.), хЕ = 90%, а = 1,2. Находят точку пересечения вертикальной линии, соответствующей хв= 15% (мол.), с кривой хЕ = 90%. От точки пересечения проводят горизонтальную линию до линии а — 1,2. От новой точки пересечения опу-
116
число
(Ю9)
(ИО)
(1И)
скают перпендикуляр на нижнюю шкалу, указывающую теоретических ступеней разделения, и находят п = 22.
Используя обозначение
хЕ (ЮР — хв) хв (100 x£)
Рихтер [156] преобразовал уравнение Фенске к виду
п = (A71g а) — 1 или
К = (л+ l)lga
Методы, описанные в разд. 4.7.5.2. и 4.7.5.3, основаны на зависимости:
п— c/lga (112)
в которой величина с зависит от начальных условий разгонки. Однако применение этой формулы ограничено идеальными смесями.
Минимальное флегмовое число, необходимое для разгонки идеальной бинарной смеси, вычисляют по следующей формуле °мин = [(МХВ1) -a(X£2/XB,)l/(a- ’> (113>
Соответствующая этому уравнению номограмма Смоукера приведена в книге Харбанда [238].
4.7.5.4. Другие методы расчета числа теоретических ступеней разделения и их сравнительная оценка
Приведенными выше методами, очевидно, не исчерпывается все многообразие расчетных способов определения числа теоретических ступеней разделения. Пол [157] опубликовал относительно простые методы расчета числа теоретических ступеней разделения для периодической и непрерывной ректификации идеальных смесей при бесконечном и конечном флегмовом числе. Кроме того, следует сослаться на работу Штаге и Джуильфа [71 ], в которой, как и в книге Роуза с сотр. [1531, приведены другие точные и приближенные методы расчета. Цуидервег [158] предполагает метод, учитывающий общую удерживающую способность колонны (см. разд. 4.10.5) и размер промежуточной фракции в условиях периодической разгонки.
Все методы расчета числа теоретических ступеней разделения, описанные в разд. 4.7.5, сопоставлены между собой и с графическим методом Мак-Кэба и Тиле в табл. 16 на примере следующей задачи разделения: хв = 40% (мол.), хЕ = 98%, a = 2,4, А/кнп = = 30° С.
Из данных табл. 16 видно, что результаты расчетов по методам II и V, I и VII, а также VI при и = оо хорошо согласуются между собой.
117
Таблица 16.
Сравнение различных методов определения числа теоретических ступеней разделения
Метод расчета и п Примечание
I. Графический, Мак-Кэба и Тиле (данные табл. 12) 1,85 3,00 5,00 10,00 20,00 50,00 оо 10,0 7,0 6,0 5,5 5,0 5,0 4,5 При проведении периодической разгонки параметры хв и v изменяются, как показано ниже: хв, % (мол.) v 80 0,65 60 1,09 40 1,85 20 3,90 10 8,80 5 25,00
II. Диаграмма на рис. 72 5,0 8,0 (х£= 99%) (хЕ= 99,9%) хв = 50% (мол.)
III. Формула (92) 9,7 8,0 6,0 (макс.) (опт.) (мин.) хв и хЕ не учитываются
IV. Формула (96) Формула (97) 10,0 7,5 [х£ = 99% (мол.)] ]х£ = 90% (мол.)] Хв = 1% (мол.)
V. Формула (102) Формула (103) Формула (104) 8,8 7,1 6,0 9,0 7,1 5,8 Хв = 50% (мол.), первые порции дистиллята (40% от всего количества) имеют концентрацию выше 95% (мол.)
VI. Формула (108в) оо 3,7
VII. По Цуидер-вегу (11584 к гл. 4) 26 10 Соответствует концу разгонки при хв = = 5 % (мол.) по данным табл. 13
Методы II и IV дают лишь ориентировочное значение необходимого числа теоретических ступеней разделения. За основу берут максимальное значение п, а флегмовое число повышают по мере возрастания температуры в головке колонны.
4.8. ТЕОРИЯ ПРОЦЕССОВ, ПРОТЕКАЮЩИХ В НАСАДОЧНОЙ КОЛОННЕ
4.8.1. ПРОЦЕСС РАЗДЕЛЕНИЯ В НАСАДОЧНОЙ КОЛОННЕ
От реальной тарелки в тарельчатой колонне мы перешли к понятию теоретической ступени разделения. Что же происходит при ректификации в насадочных колоннах, содержащих упорядоченную
118
или неупорядоченную насадку? Вследствие противоточного движения орошающей жидкости и паров между'кубом и конденсатором и при наличии двух противоположных тенденций, а именно к установлению равновесия за счет диффузии в горизонтальном направлении и к смещению равновесия за счет извне созданного противотока, становится возможным разделение компонентов и обогащение паров легколетучим компонентом. Различия в траекториях движения паров и жидкости в насадочной и тарельчатой колоннах схематически показаны на рис. 77. Еще в разд. 4.2 мы познакомились с тем, что хаотически загруженный слой насадки очень сильно влияет на характер аэродинамического и гидродинамического режима в колонне.
Следует указать, что невозможно достаточно полно описать основные закономерности процесса разделения в насадочной колонне, если оперировать только такими величинами, как высота, эквивалентная теоретической ступени или единице переноса. Зицман [159] показал, что массообмен в насадочной колонне протекает тем интенсивнее, чем легче проникают компоненты из ядра одной фазы к границе раздела жидкость — газ и оттуда далее в ядро другой фазы. Поэтому необходимо принять во внимание два диффузионных сопротивления, а именно: 1ЕП при массопере-носе внутри паровой фазы и 1ЕЖ при массопереносе внутри жидкой фазы. Диффузионные сопротивления зависят от среднего пути переносимого вещества в соответствующей фазе, от степени перемешивания фазы в точках контакта между насадочными телами, от турбулентных завихрений и других факторов, которые уже были обсуждены в разд. 4.2. Соотношение между диффузионными сопротивлениями в газовой и жидкой фазах, экспериментально измеренные Зицманом для семи различных типов насадки, указаны в табл. 17. Из данных табл. 17 следует вывод, что вклад диффузионного сопротивления газовой фазы в общее сопротивление массопереносу при ректификации может составлять от 9 до 96%.
Оптимальную поверхность фазового контакта обеспечивают правильным подбором насадки, наиболее подходя щей’для заданного процесса разделения. Каналообразование J f можно ограничить, выбирая колонну с отноше-
\J нием диаметра к поперечному размеру насадоч-Д ного тела, превышающим 10 : 1, применяя < > / аэродинамически выгодные насадочные тела, например седловидную насадку, и секционируя
„ 2 А Рис. 77.
* *, * * / у Схематическое изображение процесса массо- и теплообмена в на-
* 1/1 садочной колонне (а) и в тарельчатой колонне с двумя реальными
а б тарелками (^).
фф Траектории движения паров н жидкости; -> направление мас-__ сопереноса.
119
Таблица 17. Соотношение диффузионных сопротивлений в паровой Wn и жидкой №ж фазах прн различных типах насадки по данным Зицмана £159] Опыты проведены в колонне высотой 1 м и диаметром 30 мм. Эталонная смесь хлорбензол—этилбензол; наименьшая концентрация смеси — 20% (мол.) хлорбензола; относительная летучесть — 1,11. Температура дистиллята около 110 °C; турбулентное движение паров Наличие или отсутствие стадии предварительного затопления насадки <75 CU <75 <и <75 <75 Ф <75 W СХЗ СХЗ CU CU гад ЧДЧ гтжп с[ с(с[ ж ж
+ в ь + 10 1ГЭ СО ИЗ СО ИЗ СО 04 ь- lON 05 из 05 С"- -'t-ФСО 1ГЭСО1Л А А О О ООО 000 о о о* о о*
к h с Ю О О О U5 г-^ооиз счиэ—сч - — <о -Ф со о* о" о* —" о —• о" ь ь о* о 04 —
ВЭТС, см -ф СО О -Ф О ИЗ ИЗ 04 СО 00 СО СОСО 0 — 0 10 Т-Г10 coco 04 СО
Нагрузка, мл/(см2-ч) OO ООО OOiO О О У2 ю 0 r-. r-. rococo t*--ф 04 b- —00 05 b- —« — — — 04 — — — — 04 — — —
£ * ВСЮ > * О О Я я 04 04 000 Tf -ф 50 OO 00 10 t"- io ic co 05^05 А A о" о* о* о о" oo"o* о” о" o' 0 0
Материал насадки 1 Стекло » » » I Керамика 1 » » Стекло матированное Сталь V4A » им.
Размер насадки, мм о со ОС0 Тр rf тр тр тр 1Л1Л тр тр а XX XXX XXX x-х XX XX сх C0-C0 тр тр тр xt ’Cf Ю 1Л тр rf 5 S
Тип насадки Кольца Рашига Кольца Рашига Седла Берля Шары Проволочные кольца од-новитковые То же Проволочные кольца двухвитковые ♦ Измерено в трубе диа
насадку, благодаря чему орошающая жидкость чаще собирается и перераспределяется на нижележащих слоях насадки. Влияние каналообразования на эффективность работы колонны наглядно показано на рис. 78. Опыты проводили со смесью н-гептан—метилциклогексан. Очевидно, незначительное канало-образование в колоннах с 10—20 теоретическими ступенями разделения не очень заметно сказывается на эффективности процесса разделения; в колоннах же с 100 теоретическими ступенями'разде-ления каналообразование сильно ухудшает эффективность колонны. Для смесей с высокой относительной летучестью компонентов (например, при а = 1,07) эффект каналообразования незначителен; если же а < 1,07, то эффективность разделения заметно ухудшается [1601.
Несмотря на то, что чаще всего первоначальное’распределение жидкости является равномерным, нередко бывает, что насадка смачивается неравномерно и, кроме того, отдельные участки пленки стягиваются в струйки. Штуке [161] тщательно проанализировал вопросы смачивания насадки и исследовал, каким образом тип насадки, свойства жидкости, нагрузка колонны и рабочее давление процесса ректификации (и тем самым температура процесса) оказывают влияние на неравномерность образования пленки. В результате он пришел к следующим выводам (см. также разд. 4.2).
1. Струйчатое обтекание насадки имеет место главным образом при малых нагрузках; большее количество жидкости относительно лучше распределяется на поверхности насадки. 2) Чем больше плотность жидкости и чем меньше ее вязкость, тем с большей вероятностью даже на полностью смоченной насадке (под действием силы тяжести) формируется струйное течение. 3) Чем меньше поверхностное натяжение, тем легче при малых нагрузках смачивается твердое тело. 4) Высокая разделяющая способность, которую следует ожидать при использовании мелкой насадки, может быть достигнута только при достаточно равномерном смачивании всей насадки. При более высоких нагрузках на разделяющую способность отрицательно влияет возрастающая неравномерность пленки. 5) Основные закономерности, связанные с влиянием
Рис. 78. Влияние каналообразования на разделяющую способность колонны.
, F — коэффициент уменьшения разделяющей спо-
Каналоооразооание, °fo собности; п — число теоретических ступеней разделения; у — концентрация жидкости в головной ___ части колонны.
121
рабочего давления при ректификации, до сих пор не установлены. Некоторые исследователи обнаружили наличие оптимального давления при ректификации, другие же отмечали отсутствие зависимости разделяющей способности колонны от давления.
Интерес представляет также динамическое поведение насадочных колонн. Хайсе, Хиллер и Вагнер [162] исследовали поведение насадочной колонны во времени при ректификации тройной почти идеальной смеси этилацетат — бензол — трихлорэтилен при ступенчатом изменении соотношения расходов жидкости и паров.
Однако этими факторами не исчерпывается сложность процессов, протекающих в насадочной колонне. Крель (см. разд. 4.2) указал на важность состояния межфазной поверхности и перечислил факторы, определяющие процесс ректификации в насадочной колонне:
1) свойства разделяемой смеси; это — относительная летучесть, характер фазового равновесия, состав смеси, взаимная растворимость компонентов в жидкой смеси, плотность паров и орошающей жидкости, поверхностное натяжение на границе раздела паров и жидкости, вязкость паров и жидкости, удельная теплоемкость компонентов, энтальпия испарения компонентов, смачиваемость; 2) характеристика колонны (диаметр, высота слоя насадки, способ укладки насадочных тел); 3) характеристика насадки (высота, диаметр, толщина стенок насадочных тел, свойства поверхности, свободный объем, конструкционный материал и форма насадочных тел); 4) факторы, обусловленные режимом работы: это — способ и характер подачи и распределения орошающей жидкости, удерживающая способность колонны или отношение количества удерживаемой жидкости к количеству загруженной жидкости, рабочее давление, потеря напора (перепад давления), предельная скорость.
Из этих данных следует, что ректификация в насадочной колонне представляет собой сложный процесс и исследование влияния отдельных факторов на разделение часто сопряжено с большими экспериментальными трудностями. Возможно, это и является объяснением того, почему в литературе часто встречаются противоречивые сведения по одним и тем же вопросам.
4.8.2. ОПРЕДЕЛЕНИЕ ЧИСЛА ЕДИНИЦ ПЕРЕНОСА
Количественная характеристика процессов, протекающих в насадочной колонне, по- указанным выше причинам может быть получена лишь полуэмпирически на основе теории подобия. Чильтон и Кольборн [163] для оценки эффективности массообмена в насадочных колоннах ввели понятие числа единиц переноса (ЧЕП). Это понятие учитывает тот факт, что в насадочной колонне в противоположность тарельчатой колонне массо- и теплообмен осуществляется в виде бесконечно малых элементарных ступеней
122
Рис. 79-
Сравнительная характеристика теоретической ступени разделения и единицы переноса
— — — — единица переноса, теоретическая ступень разделения; а — рабочая линия (2) круче, чем кривая равновесия (/) (Ду уменьшается); б — рабочая линия (2) параллельна кривой равновесия (?) (Ду не изменяется).
разделения. Для теплопередачи движущей силой является температурный перепад, а для массопередачи — разность парциальных давлений или концентраций переносимого вещества. ЧЕП — это безразмерная величина, которая определяется по разности концентраций между соответствующими точками кривой равновесия и рабочей линии [73].
Рассмотрим часть диаграммы для графического определения числа теоретических ступеней разделения по методу Мак-Кэба и Тиле (рис. 79). В тарельчатой колонне между жидкостью состава ylt находящейся на тарелке, и поднимающимися парами устанавливается термодинамическое равновесие *. Концентрация паров, покидающих тарелку, равна у*. Такую же концентрацию (г/2) имеет жидкость, находящаяся на вышележащей тарелке **. В паровом пространстве между тарелками (а следовательно, между точками у± и у2) массообмен практически не происходит.
Иначе протекает процесс в насадочной колонне. Изменение
концентрации здесь в каждом слое между сечениями г/х и у2 пропорционально у* — у. Только когда кривая равновесия и рабочая линии расположены параллельно (см. рис. 796), имеет место случай, при котором ЧЕП и число теоретических ступеней разделения п совпадают, поскольку в рассматриваемой области концентраций разность у* — у остается постоянной. Такая зависимость наблюдается при ректификации идеальных смесей, компоненты которых имеют мало различающиеся температуры кипения. По-> добные смеси используют главным образом при испытании колонн.
Вообще,’ по'обогащающему эффекту единица переноса идентична теоретической ступени разделения, рассчитанной для разности концентраций у*—у, являющейся средней между значением у*—у\ и соответствующей разностью концентраций уь—уь в конце единицы переноса [71].
Если движущая сила \у увеличивается по мере продвижения
потока паров в колонне, то для одного и того же процесса разделения единиц переноса требуется меньше, чем теоретических ступеней разделения. При уменьшении \у (см. рис. 79а) имеет место другая зависимость. Если смеси идеальны или почти идеальны,
* Термодинамическое равновесие имеет место между жидкостью, стекающей с тарелки, и парами, уходящими с тарелки (конечно, при идеальной работе тарелки). — Прим. ред.
' ** Т. е. жидкость, стекающая с вышележащей тарелки. — Прим. ред.
123
Таблица 18.
Сравнение числа теоретических ступеней разделений и числа единиц переноса
а Уи % (мол.) (соответству-ет хв) У г. % (МОЛ.) (соответству-те л-£) ЧЕП по урав нению (120) м по уравнению (1086)
1,08 10 99 119,5 . 119,5
1,08 50 99 61,0 59,8
2,50 10 99 10,7 10,0
2,50 50 99 6,9 5,0
то между ЧЕП и числом теоретических ступеней разделения нет существенных различий. Наоборот, эти различия весьма значительны и должньГучитываться для неидеальных смесей и смесей с высокой относительной летучестью, как это следует из данных, приведенных в табл. 18 [164].
При условии *, что все сопротивление массопереносу сосредоточено в паровой фазе, пол чи ; согласно определению 1163] уравнение
у* л
ЧЕП=| -Д- (114)
Vi аналогично уравнению ке л
НА <"5>
*в
По аналогии с высотой, эквивалентной теоретической ступени разделения
Высота колонны РЭГС =--------------- см
п
(ПЕТР—height equivalent to a theoretical plate) (116)
получают высоту единицы переноса
ВЕП =-------гГпТт----см (HTU—height of a transfer unit) (117)
4hl 1
Высота единицы переноса является мерой сопротивления массо-перепосу. Она слабо зависит от скорости газа **.
Мац [73] отмечает, что процессы массо- и теплообмена в отличие от термодинамически обратимых процессов протекают не
* Формулы (114) и (115) справедливы и без принятия такого допущения. — Прим. ред.
** Иногда влиянге скорости газа на ВЕП (HTU) довольно ощутимо (см., например, Шафрановсхий А. В., Ручинский В. Р. Теор. основы хим. техи., 1967, т. I, № 1, с. 111 или 1970, т. IV, № 1, с. 106). — Прим. ред.
121
бесконечно медленно, а в измеримое время и с конечной скоростью. Он предлагает использовать для решения интеграла (114) выражение
(118)
где аН — характеристическая величина для поверхности массообмена; а — удельная поверхность массообмена, м2/м3; И — высота колонны, м; K/w — коэффициент скорости; К — скорость массопередачи, м/ч; ш — скорость потока паров, м/с.
Исходя из этого выражения, Мац вывел уравнения, с помощью которых легко рассчитать ЧЕП при бесконечном флегмовом числе
ЧЕП = —Ц- 1п -* ~У1 + In (119)
т — 1 1 — т — 1 г/1
; где т — тангенс угла, который образует с осью абсцисс касательную к кривой равновесия в рассматриваемой точке.
В книге Маца 173 ] даны примеры расчета ЧЕП как для v = оо, так и для конечного флегмового числа, превышающего минималь-[ ное флегмовое число.
1 Для идеальных смесей и бесконечного флегмового числа урав-I нение (114) поддается интегрированию; при этом получают
I 2 303 хР (1 — xR) 1— Хд
I ЧЕП = 1g -Ц-.---------Ц + 2,303 1g --® (120)
I а —1 xB(l-%V Х~хе
| Для конечного флегмового числа рассчитать ЧЕП по такому I же методу значительно сложнее [71]. Чильтон и Кольборн [163] I описали приближенный графический метод расчета ЧЕП, который К представляет собой дальнейшее развитие метода Мак-Кэба и | Тиле. Он пригоден для всех смесей, для которых известна кривая К равновесия. При этом в рассматриваемом интервале концентраций рабочая линия процесса ректификации для укрепляющей части Е колонны не должна подходить слишком близко к кривой равно-Г весия. Задача сводится к тому, чтобы подобрать подходящее среднее значение движущей силы, совпадающее с величиной обогащения, соответствующей единице переноса. На диаграмме равновесия в обычном порядке вычерчивают рабочую линию и Е через точку хв проводят вертикальную “линию, которая пересе-К кает рабочую линию в точке ylt а кривую равновесия — в точке у’. На рис. 79 такое построение схематически пояснено для отдель-К ного участка диаграммы равновесия; при этом на рис. 79а кривая К равновесия проходит менее круто, чем рабочая линия, а на рис. К 796 линии параллельны между собой.
К Как показано на рис. 79а, одна единица переноса дает обога-Вщение у'ь — yi, которое является средним значением для разно-Встей уь—уь и у’—уг. Одна теоретическая ступень разделения дает В обогащение t/2—уг.
к Единицы переноса можно строить в виде ступенек на диаграмме В-Мак-Кэба и Тиле, но при этом надо следить за тем, чтобы степень
125
Рис. 80.
Энтальпийная диаграмма смеси этанол—вода при 760 мм рт. ст.
обогащения уь—уъ соответствующая единице переноса, была равна среднему значению для у!—у\ и у*ъ—уъ-Точку уь следует наметить на рабочей линии таким образом, чтобы соблюдалось условие
у'ь - Ух = 0,5 [(^ - j/i) + (уь - у&)]
(121)
Построения ступенек, соответствующих единицам переноса, прово-
дят методом проб и ошибок, затраты времени на это не очень велики. Если кривая равновесия и рабочая линия являются прямыми (хотя и не параллельными), то точность метода можно повысить, используя среднее логарифмическое значение для у*—у\ и уь—уь-Однако в большинстве случаев погрешность, вносимая спрямлением одной или обеих кривых или использованием среднего арифметического вместо среднего логарифмического, не существенна. Чтобы наглядно представить взаимосвязь между числом теоретических ступеней разделения и числом единиц переноса, выше был намеренно рассмотрен наиболее простой из графических методов. Другие точные и приближенные методы описаны в работах Штате и Джуилфса [71 ]. В весьма интересной статье, содержащей математическое описание понятий п и ЧЕП, Аркенбут и Смит [166] показали, что согласно их расчетам нередко ВЭТС оказывается более удобной единицей длины колонны, чем ВЕП.
Укажем еще на два метода расчета числа теоретических ступеней, которые не зависят от конструктивных особенностей ко-
лонны и поэтому могут применяться как для тарельчатых, так и для насадочных колонн, а также для колонн с другими видами насадок. Меркель [167] разработал метод, в соответствии с которым процессы противоточного массообмена представляют в энтальпийной диаграмме Н—х—у. По ней находят изменение состава жидкости и пара, их количества, а также подводимую и отводимую теплоту (рис. 80). К сожалению, получено незначительное число энтальпийных диаграмм, и применение этого метода ограничивается небольшим числом смесей. Некоторые сведения по этому методу можно найти в литературе [73, 75, 103]. Биттер [261 ] дал сводку различных приемов вычислений для определения числа теоретических ступеней разделения при ректификации бинарных смесей; эти приемы основаны только на уравнениях рабочих линий и служат основой для графических методов решения с применением энтальпийной диаграммы.
126
4.8.3. ИНТЕНСИВНОСТЬ ПРОТИВОТОЧНОГО
МАССООБМЕНА И ВРЕМЯ, НЕОБХОДИМОЕ
ДЛЯ РАЗГОНКИ
Удерживающая способность (УС) колонны, т. е. удерживание некоторого количества жидкости в колонне, является необходимым условием для проведения ректификации (см. разд. 4.10.5). УС определяет время, требуемое для проведения процесса (см. разд. 4.10.5). Если число теоретических ступеней разделения и флегмовое число увеличиваются пропорционально 1/lg а, то время разгонки, обусловленное соотношением количеств удерживаемой и загруженной жидкости, возрастает в квадрате. Таким образом, время разгонки однозначно характеризует степень трудности разделения. Если составить отношение
Число теоретических ступеней • Расход флегмы
УС колонны (122)
для колонн 1 и 2, исходя из условий их работы, то отношение времен tn : /2, необходимых для проведения одного и того же процесса разделения в этих колоннах, будет соответствовать отношению факторов интенсивности противоточного массообмена /2 : /j при условии, что во всем остальном колонны в одинаковой мере отвечают поставленной задаче ректификации. Величина I выражается соотношением [168]:
/ = tiRjH (с'1) (123)
Фактор / по единицам измерения является обратной величиной времени; эти же единицы измерения имеет частота. Фактор интенсивности не зависит от размеров аппаратуры и может быть вычислен для колонны любого объема, любой высоты и любого поперечного сечения.
Составной частью времени разгонки является также пусковое время tn, в течение которого не следует еще отбирать дистиллят. Отбор начинают после достижения достаточного обогащения легколетучим компонентом жидкости в головке колонны. Пусковое время рассчитывают по уравнению Вебера [170):
(ra4-l)2/(2Z) (124)
Время, необходимое для введения колонны в режим, увеличивается примерно пропорционально квадрату числа теоретических ступеней разделения, т. е. высоты колонны [152, 169]. Пусковое время колонны будет более подробно рассмотрено в разделе, посвященном получению стабильных изотопов (см. разд. 5.1.4); пусковому времени придается особое значение при п > 100.
Приняв за основу противоточного устройства модель Куна [3], состоящую иЗ двух плоскопараллельных вертикальных стенок, £ебер [170] рассчитал фактор интенсивности для эталонной смеси н-гептан — метилциклогексан; при этом он исходил из максимального числа теоретических ступеней (3,54 на 1 см высоты колонны) и оптимальной скорости паров 0,1525 см/с (табл. 19).
127
Таблица 19.
Значения фактора интенсивности при различных числах теоретических ступеней разделения и скоростях потока паров
Ректификации эталонной смеси м-гептан — метилциклогексан в противоточном устройстве согласно модели Куна
Скорость потока паров, см/с Число теоретических ступеней разделения иа 1 см высоты колонны Фактор интенсивности /•10», с“‘
0,1525 3,54 260
0,305 2,83 367
0,4575 2,12 381
0,610 1,66 375
1,525 0,70 322
7,62 0,14 217
Из данных табл. 19 видно, что максимальное число теоретических ступеней разделения (3,54 на 1 см) не соответствует максимальному фактору интенсивности, равному 381 при числе теоретических ступеней 2,12 на 1 см и при скорости паров 0,4575 см/с. Далее можно видеть, что после максимального значения 381 фактор интенсивности снова уменьшается, но намного медленнее, чем следовало бы ожидать в связи с сильно уменьшившимся числом теоретических ступеней разделения. Таким образом, для получения высоких значений фактора интенсивности не имеет смысла работать при низких скоростях паров. При повышенных нагрузках движение паров становится турбулентным, что оказывает благоприятное действие на массопередачу аналогично увеличению коэффициента диффузии. С возрастанием нагрузки колонны выше оптимального значения количество орошающей жидкости увеличивается, и под действием поднимающихся паров происходит подвисание жидкости в колонне. Удерживающая способность возрастает в степени, превышающей 2, вследствие чего фактор интенсивности снижается.
Сравнивая, по данным Вебера 1170], разделяющую способность колонн различного диаметра с насадкой в виде дисков из проволочной сетки при нагрузке, составляющей 2/3 от максимального значения, получим результаты, приведенные в табл. 20.
Таблица 20.
Условия ректификации в колонне с орошаемыми сетчатыми дисками при оптимальной нагрузке, составляющей а/3 от максимального значения
Внутренний диаметр, мм Нагрузка, г/с Число теоретических ступеней разделения на 1 см высоты колоииы УС, г/см Фактор интенсивности /•10», с-1
17,0 0,139 0,460 0,239 268
25,4 0,333 0,370 0,228 537
50,8 1,530 0,144 0,328 670
128
Таблица 21.
Условия ректификации для заданной степени разделения
при постоинном числе теоретических ступеней и постоянном отношении УС к количеству загруженной жидкости
Загрузка, кг Диаметр колонны, мм Высота колонны, м Время разгонки, ч
1,0 17,0 1,о 100
1,18 25,4 1,24 49
4,40 50,8 3,20 40
Если те же данные пересчитать на постоянное число теоретических ступеней и постоянное отношение УС к количеству загруженной жидкости, то для заданных условий разделения будут получены значения времени разгонки, приведенные в табл. 21.
Последняя колонна (см. табл. 21) является наилучшей, поскольку она работает наиболее интенсивно.
Чтобы показать порядок значения фактора интенсивности, приведем еще три максимальные значения этого фактора, полученные Вебером [170] при нагрузке колонны 2/3 от максимальной:
Насадочная колонна с насадкой из стеклянных спиралей диаметром 0,3 см..................165-10~3, с-1
Насадочная колонна с матированными кольцами
Рашига 2,4X2,4 мм....................... 370-10"3, с*1
Тарельчатая колонна Брууна ...............140-10~3, с-1
Таким образом, фактор интенсивности является параметром, который легко рассчитать. Его можно использовать для сравнения различных типов колонн, поскольку он представляет собой величину, которую можно определить даже для весьма сложных процессов ректификации.
Практически к тем же выводам пришел Коллинг [171], который исследовал производительность различных насадок и сформулировал определение производительности как количества дистил-
Таблица 22.
< Условия ректификации для получения дистиллята, содержащего 90% (мол.)
i я-гептана
Таблица 23.
Условия ректификации при постоянной скорости отбора дистиллята (40 мл/ч)
Флегмовое число Нагрузка колоииы, мл/(см*.ч) Концентрация дистиллята, % (мол.)
10 70 71,5
15 100 78,5
25 160 84,5
50 300 88
' 5 Э. Крель
129
Рис. 81.
Зависимость степени чистоты дистиллята от числа теоретических ступеней разделения и флегмового числа для исходной смеси м-гептан—метилцикло-гексаи [(50% (мол.)]
*£ — содержание «-гептана в дистилляте.
Рис. 82.
Зависимость степени чистоты дистиллята от скорости отбора дистиллята при различных флегмовых числах для исходной смеси «-гептан—метил-циклогексаи [50% (мол.)].
Цифры на кривых обозначают нагрузку в мл/(см2, ч).
лята определенной степени чистоты, полученного в единицу вре-мени. На рис. 81 показана зависимость степени чистоты дистиллята от числа теоретических ступеней и флегмового числа для смеси 50% (мол.) я-гептана и 50% метилциклогексана. Если нужно получить дистиллят концентрацией 90% (мол.), то рекомендуется проводить разделение в условиях, указанных в табл. 22.
Таким образом, можно получить в два раза больше дистиллята той же чистоты, если нагрузку увеличить в 4,6 раз, а флегмовое
число удвоить.
Если же исходить из постоянного количества отбираемого дистиллята, то в соответствии с рис. 82 можно получить значения, указанные в табл. 23.
Результаты исследования фактора интенсивности указали на необходимость пересмотра прежнего мнения о том, что скорость потока паров, а следовательно, и нагрузку ко-
Рис. 83.
Зависимость ВЭТС от скорости паров для иасадки из колец Рашига (/) и седел (2) размером 25 мм.
130
лонны следует поддерживать возможно более низкими для достижения максимальной разделяющей способности. Чем выше нагрузка колонны, тем лучше результаты разделения при одинаковой скорости отбора дистиллята. Не следует забывать и о том факте, что большее количество жидкости относительно легче распределить по насадке, в результате чего достигается лучшее ее смачивание. Это действительно до определенного предела, после чего разделяющая способность снова уменьшается. Такая зависимость становится особенно очевидной, если построить графическую зависимость ВЭТС от скорости потока паров [103]. Например, для колец Ра-шига диаметром 25 мм ВЭТС остается постоянной при скоростях парового потока от 0,2 до 1,0 м/с, т. е. при пятикратном увеличении нагрузки (рис. 83). Седловидная насадка имеет максимальную ВЭТС при линейных скоростях 0,08 и 1,6 м/с. Как видно из диаграммы, работать в интервале между этими двумя скоростями паровых потоков нецелесообразно, так как при этом разделяющая способность снижается.
4.9. ОПРЕДЕЛЕНИЕ ЧИСЛА ТЕОРЕТИЧЕСКИХ СТУПЕНЕЙ РАЗДЕЛЕНИЯ ПРИ ПЕРИОДИЧЕСКОЙ И НЕПРЕРЫВНОЙ
РЕКТИФИКАЦИИ МНОГОКОМПОНЕНТНЫХ СМЕСЕЙ
Под многокомпонентной смесью подразумевают систему с числом компонентов больше двух. При этом различают смеси с точно определенными компонентами и сложные смеси, состоящие из очень большого числа компонентов, часто с весьма близкими температурами кипения. Такими сложными смесями являются, например, нефть и бензин. Как правило, для подобных смесей определяют лишь кривую разгонки. В зависимости от требуемой точности для разделения применяют либо прибор Энглера (см. разд. 7.2), либо эффективную колонну, с помощью которой легко выделить близкокипящие фракции.
Для тройных смесей условия ректификации могут быть установлены с помощью треугольной системы координат [72—74 , 78]. Корт [172] предлагает метод расчета минимальных флегмовых чисел при непрерывной ректификации трех- и четырехкомпонентных смесей, как идеальных, так и азеотропных.
Хаузену [173] удалось решить дифференциальное уравнение для идеальной трехкомпонентной смеси, а Вирн и Тийссен [174] разработали графический метод определения числа теоретических ступеней, основанный на приближенном представлении многокомпонентных смесей в виде бинарных. На эту возможность указал еще ранее Львов [175 ], который в своей теоретической работе рассматривал процесс ректификации любой многокомпонентной смеси как процесс разделения, состоящий из совокупности параллельно
б* 131
Выход дисггщппята, г
Рис. 84.
Диаграмма ректификации смеси жирных кислот С4—Св нормального строения при остаточном давлении 20 мм рт. ст.;
---температура кипения при 20 мм рт. ст.;----кислотное число (к.ч.);
50
---------показатель преломления пр.
Горизонтальные отрезки у кривых указывают на теоретическое значение физических величин.
протекающих процессов разделения бинарных смесей. Таким образом, задача сводится к тому, чтобы многокомпонентную смесь представить в виде бинарной, содержащей «ключевые» компоненты, которые труднее всего разделить (следовательно, смесь с наименьшей относительной летучестью компонентов), и в основу расчетов положить кривую равновесия для этой смеси. Для лабораторной практики применительно к разделению идеальных смесей метод «ключевых» компонентов является наиболее рациональным, к тому же приближенные расчеты можно проверить с помощью сравнительно простых пробных разгонок.
Мостафа [175а] представляет многокомпонентную смесь в виде эквивалентных бинарных смесей, обладающих различной относительной летучестью, и определяет число теоретических ступеней разделения с помощью графического метода Мак-Кэба и Тиле. Если компонентов много, то можно выбрать один «ключевой» компонент, относительная летучесть которого является средним значением для Относительных летучестей всех компонентов.
Шьен [1756] приводит точный метод расчета минимального числа теоретических ступеней разделения для ректификации многокомпонентной смеси. Приближенный метод разработан Серовым с сотр. [175в].
132
Непрерывный способ ректификации имеет то преимущество, что условия процесса остаются неизменными. Кроме того, при этом имеется возможность в качестве головного или кубового продукта получать как отдельные компоненты, так и смесь нескольких компонентов. Если необходимо получить х различных фракций, то для разделения требуется (х—1) колонн. Возьмем в качестве примера предварительно отогнанную смесь жирных кислот С4—С8, для которой на рис. 84 показана диаграмма разгонки [176]. Очевидно, для непрерывного разделения этой смеси на отдельные фракции (по числу атомов углерода) потребовалось бы четыре колонны. Опыт показывает, что лучше работать, отбирая отдельные фракции по возможности в виде головного продукта (рис. 85); при этом фракции получаются в виде прозрачного и бесцветного дистиллята. Однако можно проводить ректификацию и по схеме, изображенной на рис. 86.
Сначала рассмотрим колонну IV на рис. 85. В этой колонне разделяется бинарная смесь С7—С8. Условия ректификации для этого случая рассчитать весьма просто. В колонну III вводят тройную смесь и получают один компонент в головном продукте и два компонента в кубовом продукте. Сравнивая кривые равновесия смесей С6—С7 и С,—С8, выбирают для расчетов ту кривую равновесия, которой соответствует более низкое значение относительной летучести компонентов а.
В колонну II поступает смесь из четырех компонентов. Снова находят относительную летучесть компонентов каждой из бинарных смесей С5—С6, С6—С7 и С,—С8 и для расчетов выбирают ту кривую равновесия, которой соответствует наименьшее значение а. Разумеется, следует иметь в виду, что в процессе массообмена компоненты С7 и С8 почти не содержатся в паровой фазе. Эти компоненты можно рассматривать скорее как балластное или
Рис. 85.
Схема непрерывной ректификации смеси жирных кислот С4—С8 нормального строения с отбором компонентов С4—С7 в дистилляте и компонента С8 в кубе.
Рис. 86.
Схема непрерывной ректификации смеси жирных кислот С4—С8 нормального строения с отбором компонентов С4, С6 и С7 в дистилляте н компонентов Св и С8 в кубе.
133
«вытесняющее» вещество. Поэтому рекомендуется внести поправку в значение а с учетом ожидаемой температуры, состава кубового продукта и его летучести. Если температура кубовой жидкости сравнительно высока, то из имеющихся значений а для трех бинарных смесей, выбирают наименьшее значение а. Если температура куба сравнительно низка, то за основу достаточно взять наименьшее значение а для двух первых из названных смесей. Ввиду постоянного присутствия в работающей колонне определенного количества жидкости и очень незначительного содержания компонента СБ рекомендуется исходить при расчете из смеси С6—С7, а остальные компоненты рассматривать как балластное вещество.
Аналогичные условия имеют место в колонне I. По отношению к температуре головного продукта температура в кубе является сравнительно высокой, поэтому из значений относительной летучести для смесей С4—С6 и С5—С6 выбирают наименьшее.
Если компоненты смеси принадлежат к одному гомологическому ряду, как в рассмотренном примере, то значения а снижаются по мере увеличения числа атомов в молекуле компонентов, образующих смесь:
2,66
2,50
2,10
2,13
Таким образом, с увеличением молекулярной массы компонентов разделение становится все более трудным.
В качестве головного и кубового продуктов можно отбирать и смеси различных компонентов, как показано на рис. 86 для колонны I. В дистилляте получают фракцию С4—С6, а в кубе — фракцию С7—С8; следовательно, граница раздела смеси лежит между компонентами С6 и С7. В этом случае прежде всего выбирают кривые равновесия для крайних пар компонентов, т. е. для С4—Сй и С,—С8, и рассчитывают число теоретических ступеней разделения и другие условия ректификации, необходимые для обогащения смеси до хЕ = 95% (мол.) при непрерывном режиме работы колонны. Для двух полученных чисел ступеней вычисляют среднее значение. За основу можно взять также кривую равновесия для смеси С6—С7, поскольку количественно фракция С4—С6 преобладает, а компонент С8 почти не участвует в массообмене.
Исследования Фальдикса и Штаге [176 а] позволили найти оптимальные схемы системы, состоящей из двух ректификационных колонн. Мюллер [1766] разработал различные схемы соединений колонн с боковым отбором продукта из колонн при разделении многокомпонентных смесей.
Биллет [176в] отмечает, что отклонения от оптимальных условий ректификации могут привести к значительному увеличению производственных затрат. Он рассматривает различные схемы 134
соединения основных и вспомогательных колонн, а также вопросы рационального использования обогащающего эффекта дефлегмации при дистилляции и ректификации. Эти данные следует учитывать также при работе в лаборатории и на опытных установках.
Разработаны многочисленные методы расчета параметров процесса ректификации для идеальных многокомпонентных смесей, которые подробно изложены Торманном [177], а также Эллисом и Фрешуотером [178]. Особо следует отметить приближенную формулу Кольборна [179] и Андервуда [180], позволяющую определять минимальные флегмовые числа. Простой приближенный метод расчета минимального числа теоретических ступеней разделения при v = оо принадлежит Фенске [181], который с целью упрощения рассматривает многокомпонентную смесь как бинарную. При этом условно принимается, что в смеси преимущественно содержатся «ключевые» компоненты, температуры кипения которых образуют постепенно возрастающую последовательность, а разности температур кипения для различных соседних компонентов смеси примерно одинаковы. Если через х1 обозначить содержание низкокипящего «ключевого» компонента, содержание которого в кубовом продукте невелико, а через х2 — содержание высоко-кипящего «ключевого» компонента, содержание которого невелико в головном продукте, то уравнение Андервуда—Фенске для расчета минимального числа теоретических ступеней разделения будет иметь вид:
(ДгАзг) (х2в/х1в)
«мин =--------------- (1 20)
jg а12
где а12 — отношение давлений паров «ключевых» компонентов.
Хаберт [182] разработал метод расчета минимального числа Г теоретических ступеней разделения, согласно которому давление паров каждого компонента смеси относят к давлению паров самого 1 летучего компонента. В своем превосходном обзоре Бруийн [1831 ! рассматривает теорию ректификации многокомпонентных смесей I при минимальном флегмовом числе.
| Для точного расчета параметров колонн и условий ректифика-| ции многокомпонентных смесей необходимо применение электрон-| ных вычислительных машин. При этом проблемы моделирования I и оптимизации поддаются решению с минимальными затратами I времени. В разд. 4.15 обсуждены возможности применения цифро- вой и аналоговой вычислительной техники для решения задач Е разделения.
I 4.10. АППАРАТУРНО-ТЕХНОЛОГИЧЕСКИЕ | ПАРАМЕТРЫ ТАРЕЛЬЧАТЫХ
Ви НАСАДОЧНЫХ КОЛОНН. ИСПЫТАНИЯ колонн
До сих пор были рассмотрены лишь методы, позволяющие рас-
считать необходимое для разделения число теоретических ступеней или число единиц переноса. Испытания колонны с применением В 135
определенной эталонной смеси проводят, чтобы установить, скольким теоретическим ступеням или единицам переноса эквивалентна колонна. Такие испытания необходимы, так как отсутствуют подходящие методы, позволяющие рассчитать эффективность разделения в лабораторной колонне. При испытаниях в куб загружают эталонную смесь известного состава и доводят ее до кипения. Обычно испытание проводят при бесконечном флегмовом числе, т. е. без отбора дистиллята, так как это условие наилучшим образом поддается воспроизведению. Из головки колонны и из куба пробы отбирают по каплям, благодаря чему стационарный режим в колонне не нарушается. Анализом проб устанавливают достигнутое обогащение, соответствующее разнице в концентрациях кубовой жидкости и дистиллята. Методические вопросы испытания колонн подробно рассмотрены Андервудом [ 184 ] в одной из его ранних работ.
Определив число теоретических ступеней разделения, обычно обнаруживают, что это число меньше числа реальных тарелок. Следовательно, реальная тарелка работает не идеально, и поэтому работу тарелки оценивают по отношению найденного числа теоретических ступеней разделения к числу реальных тарелок. Это отношение называют средним относительным обогащением или средним коэффициентом полезного действия тарелки (по Киршбауму) [103 ]
Sep = П/Пр (126)
где Sep—средний к. п. д. тарелки; п — число теоретических ступеней разделения; Лр — число реальных тарелок.
Для насадочной колонны определяют ВЭТС
Высота колонны
ВЭТС =-----------------см (127)
Исследования Хюбнера и Шлюндера [103а] показывают, что средний коэффициент полезного действия тарелки (или к. п. д. Мерфри) можно с достаточной точностью рассчитать, используя модель турбулентной диффузии, при условии, что известны коэффициенты массоотдачи или турбулентной диффузии в газовой и жидкой фазах. Фогельпол и Серетто [1036] приводят сведения о значениях к. п. д. тарелки при ректификации тройных смесей.
Если высота, эквивалентная теоретической ступени разделения, совпадает с расстоянием между соседними реальными тарелками колонны, то это свидетельствует об идеальной работе реальной тарелки. В насадочной колонне высота теоретической ступени разделения соответствует ВЭТС.
Число теоретических ступеней разделения, установленное при испытании колонны, определяется кривой равновесия эталонной смеси и зависит от соблюдаемых условий ректификации и, разумеется, от размеров колонны. Если завод-изготовитель указывает для ректификационной колонны лишь число теоретических ступеней разделения без каких-либо дополнительных пояснений, то подобная информация практически бесполезна. Только при 136
Таблица 24.
Аппаратурно-технологические параметры, необходимые для оценки разделяющей способности колонн (в дополнение к числу теоретических ступеней)
Насадочные колонны Тарельчатые колонны Колонны с другими насадками См. р аздел
Диаметр колонны мм Пункты 1,3—7 те же, что и для насадочных колонн Пункты 1,3—7 те же, что и для насадочных колонн 4.10.1
Длина разделяющего участка (высота насадки), мм Эталонная смесь [состав загруженной в куб смеси, % (мол.)] Флегмовое число и расход орошающей жидкости, отнесенный к свободному поперечному сечению, мл/(см2-ч) УС колонны Рабочее давление, мм рт. ст. Нагрузка (мл/ч) или скорость потока паров (м/с) Длина разделяющего участка — рабочая высота колонны Длина разделяющего участка — рабочая высота^колонны 4.10.2 4.10.3 4.10.4 4.10.5 4.10.6 4.10.7
Форма насадочных тел, материал насадки (состояние поверхности), размеры насадочного тела (высота, диаметр, толщина стенки), активная поверхность насадки Расстояние между тарелками Размеры реальных тарелок Режим работы (например, число оборотов ротора в колоннах роторного типа) Размеры насадки 7.8
наличии дополнительных сведений относительно аппаратурнотехнологических параметров (табл. 24) возможна однозначная оценка и опытная проверка разделяющей способности колонны.
4.10.1. ДИАМЕТР КОЛОННЫ
По экспериментальным данным Мерча [185] при постоянных размерах насадочных тел ВЭТС возрастает с увеличением диаметра колонны. Однако для насадок из проволочной сетки влияние диаметра колонны (по данным Стедмана и Мак-Магона) не так ощутимо, как, например, для насадки из колец Рашига или из седел. На основе систематических исследований пристеночного эффекта (растекание жидкости к стенкам колонны), проведенных Муллином [186], пришли к выводу, что наибольшая эффективность колонны достигается при соотношении: диаметр колонны/ .диаметр насадочного тела = 10—12. Если это соотношение не
137
выдерживается, то следует предусмотреть как можно больше пере-распределителей жидкости по высоте колонны (см. разд. 4.2 и 4.8.1).
4.10.2. РАБОЧАЯ ВЫСОТА КОЛОННЫ
И СПОСОБЫ УКЛАДКИ НАСАДОЧНЫХ ТЕЛ
Под рабочей высотой колонны подразумевают высоту участка колонны (любой конструкции), на котором непосредственно протекает процесс разделения. Следовательно, рабочая высота в насадочных колоннах представляет собой высоту слоя насадки. Вследствие каналообразования и пристеночного эффекта ректифицирующий участок необходимо секционировать по всей высоте колонны, иначе в ее нижней части не будет обеспечен противоточный массообмен ([39] к гл. 1).
Киршбаум [187] по результатам испытания промышленных колонн установил, что число теоретических ступеней разделения не увеличивается пропорционально высоте слоя насадки. Казанский [188] тщательно исследовал пристеночный эффект в лабораторных колоннах. В частности, он обнаружил, что эффективность несекционированной колонны высотой 149 см, составляющая при определенных условиях 18 теоретических ступеней разделения, увеличивается до 24 ступеней после секционирования колонны на три участка. Работы Бушмакина и Лызловой [189] подтвердили эти результаты. При использовании в качестве насадочных тел константановых спиралей диаметром 1,8 мм было показано, что секционирование колонны на участки длиной по 25 см с целью сбора и перераспределения орошающей жидкости обеспечивает ее максимальную разделяющую способность. При увеличении числа секций от 1 и до оптимального значения каждое перераспределительное устройство повышает эффективность на 1,5 теоретической ступени. Автором проведены испытания насадки из фарфоровых седловидных насадочных тел размером 4X4 мм при v = оо. Результаты испытаний приведены в табл. 25.
Таблица 25.
БЭТС в зависимости от рабочей высоты колоииы и числа секций
Эталонная смесь четыреххлорнстый углерод — бензол [40%
(мол.)], исходный объем смесн 300 мл, давление атмосферное
Нагрузка, мл/г ВЭТС (см) при рабочей высоте колонны']
450 мм 900 мм
1 секция 2 секции
200 4,73 6,93 6,93
600 6,00 11,25 7,83
138
Рис. 87,
Устройство Алленби— Лере для неупорядоченной загрузки мелкой насадки:
1 — трубка; 2 — корковая пробка с отверстием; 3 — выступ.
Эти опыты выявили следующие факты, которые до сих пор недостаточно учитывались при лабораторной ректификации: 1) ВЭТС при одной и той же нагрузке зависит от высоты ректифицирующего участка; 2) если при малых нагрузках секционирование колонны не дает эффекта, то при более высоких нагрузках с введением секционирования эффективность разделения увеличивается. Суммарная поверхность насадки в слое определенной высоты зависит от способа ее укладки, влияющего также и на распределение жидкости [190]. Влияние способа укладки на перепад давления в колонне и ее разделяющую способность весьма значительно [191]. Для обеспечения беспорядочной укладки насадки Майлс с сотр. [192] применил способ, в соответствии с которым колонну наполняют минеральным маслом и бросают в нее насадку по одному элементу. Проще заполнять колонну, опускдя в нее по три-четыре насадочных тела при постоянном постукивании деревянной палочкой по стенкам колонны. Небольшие насадочные тела можно очень быстро загрузить с помощью устройства, описанного Алленби и Лёре [193] (рис. 87). Каждое насадочное тело попадает в колонну отдельно, благодаря чему обусловливается неупорядоченность расположения насадки. Насадочные тела насыпают на дно колбы в виде слоя высотой примерно 1 см. В трубку 1 (см. рис. 87) с помощью газодувки или воздухопровода, присоединенного к напорному патрубку вакуумного насоса, вдувают воздух, при этом насадочные тела приподнимаются и начинают вращаться. Выступ 3 притормаживает насадочные тела, которые через отверстие в корковой пробке 2 проскакивают по одному в соединительную трубку, ведущую в колонну.
В тарельчатых колоннах и в колоннах с иными насадками длина разделяющего участка соответствует рабочей высоте колонны. В тарельчатых колоннах эта высота равна расстоянию от нижнего края нижней тарелки до нижнего края верхней тарелки плюс один шаг между тарелками. Для колонн с другими насадками в качестве рабочей высоты указывают общую высоту специальной насадки, например рабочей высотой для колонн с вращающейся насадкой является высота ротора.
4.10.3. ЭТАЛОННАЯ СМЕСЬ И СОСТАВ ЗАГРУЖЕННОЙ ЖИДКОСТИ
В принципе, при испытании ректификационной колонны следует использовать те смеси, для разделения которых предназначена колонна. При отсутствии достаточно чистых веществ можно
139
ограничиться применением эталонных смесей, отвечающих определенным требованиям. Эталонные смеси следует выбирать в первую очередь с таким расчетом, чтобы степень трудности их разделения находилась в полном соответствии с предполагаемой разделяющей способностью колонны.
Эталонную смесь с низким значением относительной летучести компонентов а можно применять для колонн как с большим, так и с малым числом теоретических ступеней разделения; эталонную же смесь с высоким значением а можно использовать только для колонн с малым числом теоретических ступеней разделения.
Хальденвангер [194] наиболее полно сформулировал требования, предъявляемые к эталонным смесям: 1) по свойствам эталонная смесь должна приближаться к идеальному раствору, т. е. практически без отклонений подчиняться закону Рауля и иметь относительную летучесть компонентов, постоянную для всех концентраций; 2) данные по равновесию пар— жидкость должны быть известны или их можно легко рассчитать; 3) смесь должна состоять только из двух компонентов во избежание трудностей при измерениях и расчетах; 4) относительная летучесть компонентов должна иметь такое значение, чтобы в испытуемой колонне достигалось достаточное, но не слишком большое разделение; 5) температуры кипения смеси должны лежать в интервале, для которого нетрудно подобрать надежную тепловую изоляцию колонны; 6) компоненты смеси должны быть термически стойкими в условиях ректификации; 7) вещества и их смеси не должны вызывать коррозии конструкционных материалов, использованных в аппаратуре; 8) исходные вещества должны быть легко доступными; 9) вещества не должны содержать примесей; их чистота должна поддаваться проверке доступными методами; 10) смеси с любой концентрацией компонентов должны легко поддаваться анализу.
Международное сотрудничество в рамках Европейской федерации химической технологии (European Federation of Chemical Engineering) дало возможность стандартизировать эталонные смеси благодаря обширному экспериментальному материалу, накопленному членами федерации. В брошюре, опубликованной Цуидервегом [195], приведены 11 систем с данными по равновесию и со всеми необходимыми физическими свойствами, а также данные по химической стойкости компонентов.
Чтобы можно было сравнивать различные насадки и колонны по их разделяющей способности, рекомендуется использовать эталонные смеси, указанные в табл. 26. Для испытания колонн при атмосферном давлении в соответствии с международными нормами рекомендуется применять в первую очередь смеси «-гептан— метилциклогексан—бензол—1,2-дихлорэтан и четыреххлористый углерод — бензол для числа теоретических ступеней разделения п=50—60, бензол—этиленхлорид для п = 50—60 и четыреххло-140
ристый углерод—бензол для п < 30*. Брандт и Рёк [196] исследовали степень отклонения смеси «-гептан—метилциклогексан от идеальной и показали,что эту смесь можно назвать псевдоиде-альной, поскольку по свойствам она не соответствует традиционной классификации. Данную смесь можно с успехом применять в качестве эталонной для работы при остаточном давлении до 300 мм рт. ст., тем более, что мольные энтальпии испарения компонентов практически одинаковы (7,575 ккал/моль). Для колонн с числом теоретических ступеней разделения от 100 до 500 в качестве эталонного вещества пригодна тяжелая вода. Система 16Н2О—18НгО позволяет проводить испытания, когда п достигает 1000; однако при этом время, в течение которого устанавливается стационарный режим, измеряется месяцами (см. разд. 5.1.4). Для проведения точных измерений п можно также применять эталонные вещества, меченые радиоактивными изотопами 14С, 35 S и 36С1.
В интервале давлений от 20 до 400 мм рт. ст. смесь циклогексилциклопентан — «-додекан ведет себя почти как идеальная, а смесь дициклогексил — w-тридекан можно приближенно считать идеальной только в среднем интервале концентраций, т. е. от 30 до 90% дициклогексила [197].
Мерчу [185] удалось количественно описать влияние физических свойств жидкой смеси на разделяющую способность колонны. Он установил, что ВЭТС прямо пропорциональна выражению ат]/р (где а — относительная летучесть; т] — динамическая вязкость в сП; р — плотность в г/мл). Результаты своих исследований Мерч обобщил следующей эмпирической формулой:
ВЭТС = ft1A4ft*dfc«AI/3aT)/p (128)
где М — удельная нагрузка по паровой фазе, кг/(м2-ч); d — диаметр колонны, м; h — высота слоя насадки (рабочая высота колонны), м; ku и k3 — константы, которые зависят от типа и размеров насадки (табл. 27).
Формула (128) справедлива только при следующих условиях: 1) ректификация при атмосферном давлении; 2) величина М должна находиться в интервале 25—80% от предельной нагрузки; 3) отношение диаметра колонны к диаметру насадочного тела должно превышать 8—10; 4) ВЭТС рассчитывают при бесконечном или очень большом флегмовом числе.
Погрешность расчетов по формуле (128) не превышает ±10%. Автор, однако, предлагает, что для повышения надежности вычисленную величину следует увеличивать на 20%. Формула (128), таким образом, дает только грубое приближение, которое нужно проверять опытным путем.
Так как ВЭТС зависит от состава исходной смеси в кубе [92, 199], то, как показывает опыт, лучше всего поддерживать кон-
* Здесь смесь бензол—четыреххлористый углерод рекомендована одновременно для двух интервалов теоретических ступеней (я = 50—60 и п 30). Эту смесь предпочтительнее использовать при п 30. — Прим. ред.
141
X Таблица 26.
Характеристика эталонных смесей, применяемых при испытаниях колонн
Рабочее давление ректификации Бниариая смесь А + В Температура кипения, °C Коэффициент 20 преломления /гр Разность коэффициентов преломления А«Ь° Относительная летучесть п для и ?пы-тания колонн с ожидаемой эф-1 фективио- 1 стью
<А ZB А/ А В
760 мм рт. ст. Бензол—толуол * 80,1 110,6 30,5 1,50122 1,49647 0,0048 2,36—2,61 2—7
Бензол-н-гептан 80,1 98,4 18,3 1,5011 1,3876 0,1135 2,12—1,11 5—20
Метилциклогексан—толуол 100,98 110,6 9,62 1,4232 1,4968 0,0736 1,306—1,328 5—25
Бензол-1,2-дихлор-этан 80,1 83,5 3,4 1,5011 1,4448 0,0563 1,162—1,107 15—20
н-Гептан—метилциклогексан 98,4 100,85 2,45 1,3878 1,4232 0,0354 1,076—1,074 20—80
п-Ксилол-л-кси-лол ** 139,10 138,35 0,75 1,4960 1,4973 0,0013 1,0203—1,0204 50—250
5—100 мм рт. ст. (возможно до 1 мм рт. ст.) 2-Метилнафталин— 1-метилнафталин * — — — — — 0,0050 (<Ч°) 1,1400—1,1417 (5 мм рт. ст.); 1,1312—1,1328 (10 мм рт. ст.); 1,1035—1,1047 (100 мм рт. ст.) 15—45
5—100 мм рт. ст. транс- Декали н— ^ис-декалин * — — — 1,46932 1,48098 0,01166 1,483—1,460 (5 мм рт. ст.); 1,440—1,420 (10 мм рт. ст.); 1,314—1,300 (100 мм рт. ст.) 4—20
50—760 мм рт. ст. (возможно 20 мм рт. ст.)
Бензол—этилбензол
1,5246 1,4958 0,0288 1,188—1,194 (50 мм рт. ст.); 1,140—1,143 10—30
(300 мм рт. ст.); 1,130 (760 мм рт.
ст.)
8—16 ат Изобутан—н-бутан — — — — — — 1,308—1,241 (8 ат); 1,261— 1,217 (12 ат); 1,220—1,195 (16 ат) 6—25
1 —10 ат Метанол—этанол * — — — — — — 1,644—1,866 (1 ат); 1,474— 1,559 (3 ат); 1,395—1,447 (5 ат); 1,289— 1,319 (10 ат) 4—20
• Для анализа состава смесей лучше применять газовую хроматографию; ** Для анализа состава смесей лучше применять ннфра-красную спектроскопию нлн газовую хроматографию.
Таблица 27.
Константы klt k2 и k3 в формуле Мерча [185]
Тип насадки Поперечный размер насадочного тела, мм &3
Кольца Рашига 9,5 13,58 —0,37 1,24
12,7 44,95 —0,24 1,24
Седловидные насадочные тела 12,7 25,61 —0,45 1,11
Насадка Мак-Магона 6,35 0,0114 0,5 1,0
9,5 0,199 0,25 1,0
12,7 0,356 0,20 1,0
центрацию легколетучего компонента в кубовой жидкости на уровне не ниже 40% (мол.); при этом количество загруженной в куб жидкости должно превышать общую удерживающую способность колонны по жидкости примерно в восемь раз. Эталонную смесь следует выбирать с таким расчетом, чтобы обогащение по легколетучему компоненту, достигаемое в колонне, не превышало 90% (мол.). По разности температур кипения компонентов эталонной смеси можно выделить следующие группы смесей:
Для большого числа теоретических ступеней (п > 40)...........................
Для среднего числа теоретических ступеней (п = 10—40)........................
Для малого числа теоретических ступеней
(п < 10)...............................
разность температур кипения компонентов эталонной смеси, °C
0,5—3,0
3,0—10,0
Более 10,0
Для важнейших эталонных смесей составлены таблицы [92, 146, 147, 195], позволяющие вычертить диаграмму зависимости nD—п, аналогичную показанной на рис. 71 (зависимость числа теоретических ступеней от показателя преломления смеси). По оси абсцисс отсчитывают показатели преломления дистиллята и кубового продукта и с помощью оси ординат определяют соответствующие им числа теоретических ступеней разделения; разность этих чисел равна числу теоретических ступеней испытуемой колонны (см. разд. 4.7.4).
144
4.10.4. ФЛЕГМОВОЕ ЧИСЛО И КОЛИЧЕСТВО ОРОШАЮЩЕЙ ЖИДКОСТИ
Как уже было указано, число теоретических ступеней разделения обычно определяют при бесконечном флегмовом числе, т. е. без непрерывного отбора дистиллята, поскольку это условие поддается наиболее точному воспроизведению.
На практике часто необходимо знать, как изменяется число теоретических ступеней разделения при конечном флегмовом числе или какой поправочный коэффициент следует ввести в найденное число п при ректификации с конечным флегмовым числом, если в основу расчета положены результаты испытания эффективности при v = оо.
Число теоретических ступеней при конечном флегмовом числе определяют следующим образом: работают при определенной нагрузке с конечным флегмовым числом, а непрерывно отбираемый дистиллят так же непрерывно возвращают в куб (рис. 88). Затем в определенные промежутки времени из головки колонны и куба одновременно отбирают пробы и определяют их состав ф164].
Рис. 88.
Установка для испытания эффективности колонны при конечном флегмовом числе.
Рис. 89.
Зависимость поправочного коэффициента F для насадки из спиралей 2 X Х2Х0.2 мм от флегмового числа при различных нагрузках колонны.
145
Выход дистиллята, °/о(од.)
Рис. 90.
Зависимость состава дистиллята и кубовой жидкости от выхода дистиллята при ректификации смеси хлорбензол—этилбензол.
Нагрузка колонны 44 мл/ч; v = 8; рабочее давление 760 мм рт. ст. (/) н 20 мм рт. ст. (2); х — содержание хлорбензола, начальная концентрация ХВ = 37 % <мол->-
Рис. 91.
Зависимость числа эквивалентных теоретических ступеней разделения от выхода дистиллята в интервале 0—50% (об.) для смеси хлорбензол—этилбензол.
Условия ректификации:
1 — v = 24, 20 мм рт. ст.; 2 — v = 24, 760 мм рт. ст.; 3 — v = 8, 20 мм рт. ст.; 4 — v = 8, 760 мм рт. ст.
В колонне постепенно устанавливается стационарный режим, который выражается в постоянстве составов дистиллята и кубовой жидкости и соответствует любому моменту периодической разгонки. Рекомендуется после отбора последней пробы в течение некоторого времени отбирать дистиллят обычным образом (не возвращая его в куб) и затем повторить процесс при измененном составе кубовой жидкости.
На рис. 89 представлены поправочные коэффициенты F для насадки из спиралей размером 2x2x0,2 мм, которые были экспериментально определены автором по указанной методике (колонна диаметром 30 мм с рабочей высотой 1,15 м). Эти результаты были подтверждены исследованиями Науманна и Лейбница [201 ].
Под величиной F понимают поправочный коэффициент, на который нужно умножить число теоретических ступеней разделения, определенное при v = оо, чтобы получить фактическую разделяющую способность колонны при заданном конечном флегмовом числе:
«оо-Р = «КОН (129)
Из рис. 89 следует, что при испытании колонны с 60—70 теоретическими ступенями нецелесообразно увеличивать флегмовое число свыше 70. Это согласуется с уравнением (106), полученным Роузом.
Рекомендуется проводить периодическую ректификацию с использованием эталонной смеси при определенной нагрузке и при определенном флегмовом числе с отбором небольших проб дистиллята (около 1% от загруженной смеси); одновременно с каждой второй пробой дистиллята следует проводить капельный отбор и анализ жидкости из куба. Строят график зависимости состава
146
дистиллята и кубовой жидкости от выхода дистиллята (рис. 90) [92]. Эти данные сопоставляют с результатами испытания при бесконечном флегмовом числе, причем для этой цели используют понятия «число эквивалентных теоретических ступеней разделения» и «коэффициент полезного действия колонны».
Число эквивалентных теоретических ступеней разделения — это минимальное число теоретических ступеней, которое требуется для того, чтобы при бесконечном флегмовом числе вызвать такое же обогащение (хв —-> хЕ), как и при ректификации с конечным флегмовым числом. Все условия разгонки за исключением флегмового
числа остаются при этом одинаковыми.
При построении ступеней разделения на диаграмме Мак-Кэба и Тиле по указанному методу рабочую линию не используют, а ступени вписывают между диагональю и кривой равновесия, как это делается для бесконечного флегмового числа. Этот прием можно проиллюстрировать с помощью рис. 90. Так, после отбора 30% от общего объема дистиллята (при 20 мм рт. ст. и v = 8) получается, что 17% (мол.) хлорбензола содержится в кубовой жидкости, а 60% хлорбензола — в дистилляте. Если обе эти концентрации [хв = 17% (мол.) и хЕ = 60% (мол.)] нанести на диаграмму Мак-Кэба и Тиле или подставить их в уравнение Фенске (108в), то для v = оо получим обогащение, эквивалентное 17,5 теоретическим ступеням разделения. Эти расчеты можно выполнить для ряда точек, получая в итоге кривые, показанные на рис. 91. Как видно из сравнения кривых, число эквивалентных теоретических ступеней при высоком флегмовом числе больше, причем в начале ректификации при низком флегмовом числе (и = 8) наблю-
дается значительное уменьшение числа эквивалентных теоретических ступеней разделения. Из рис. 91 также следует, что давление
не оказывает существенного влияния на число эквивалентных
теоретических ступеней.
Диаграмма, построенная по опытным данным Коллинза и Ланца [200] (рис. 92) иллюстрирует взаимосвязь между числом теоретических ступеней при v = оо и 27 и числом эквивалентных
теоретических тарелок. При ректификации эталонной смеси «-гептан — метилциклогексан колонна с 30 реальными тарелками при нагрузке 3000 мл/ч имела при v = оо 19 теоретических ступеней, при v = 27 — 17 теоретических ступеней, а при v =
Рис. 92.
Зависимость числа теоретических ступеней (/, 2) и числа эквивалентных теоретических ступеней разделения (3) от нагрузки колонны:
1 — п при v — оо; 2~то же при и=27; 3 — пэкв при v = 27; 4 — кривая зависимости удерживающей способности колонны от нагрузки.
147
Рис. 93.
Устройство для измерения количества жидкости, стекающей в куб, и динамической удерживающей способности колонны.
Устройство изготовлено из стандартных деталей «Дестинорм».
Рис. 94.
Измеритель расхода жидкости с сифоном конструкции Зигварта—Штаге: / — шлиф для соединения с колонной; 2 — сифон; 3 — камера для сбора жидкости; 4 — шлиф для соединения с колбой.
Рис. 95.
Мерник жидкости «Лабодест» с магнитным клапаном конструкции Штаге.
= 27—12,5 эквивалентных теоретических ступеней. Это означает, что обогащение, достигнутое при v = 27, можно получить, применяя 12,5 теоретических ступеней разделения при v = оо. И, наоборот, необходимо подбирать колонну, которая при нагрузке 3000 мл/ч дает обогащение, соответствующее 19 теоретическим ступеням при v = оо.
Если отнести число эквивалентных теоретических ступеней пэкв к числу теоретических ступеней ru при v = оо и при такой же нагрузке, то, согласно Оболонцеву и Фросту [202], получим коэффициент полезного действия колонны:
К. п. д. = пэкв/п^ (при одной и той же нагрузке) (130)
Максимальное значение этой величины равно единице, оно тем ближе к 1, чем выше рабочее флегмовое число. 1
Изложенный выше метод расчета числа эквивалентных теоретических ступеней разделения и к. п. д. колонны имеет то существенное преимущество, что он позволяет сравнивать колонны по их разделяющей способности при различных рабочих условиях, поскольку все найденные числа теоретических ступеней разделения можно пересчитать относительно бесконечного флегмового числа v = оо.
Количество орошающей жидкости в основном измеряют в головке колонны. При условии, что колонна работает адиабатически, т. е. без рассеяния и подвода тепла, и мольные энтальпии 148
испарения компонентов различаются незначительно, количество жидкости, измеренное непосредственно над кубом, должно совпадать с количеством флегмы, измеренным в головке. Однако на практике вследствие тепловых потерь количество жидкости, стекающей в куб, как правило, больше количества флегмы, возвращаемой в верхнюю часть колонны. Поэтому целесообразно для одновременной проверки адиабатичности процесса ректификации измерять также и количество жидкости, стекающей в куб. Для этого применяют устройство, показанное на рис. 93. При измерениях кран устанавливают в такое положение, чтобы жидкость не стекала сразу в куб, а сначала попадала в градуированную воронку. С помощью секундомера измеряют количество жидкости, поступающее в единицу времени в воронку (т. е. расход жидкости). Этот прибор имеет то преимущество, что измеряемый расход жидкости можно варьировать в зависимости от условий разгонки. Недостатком устройства является возможность растворения смазки крана.
Это исключается в устройствах, предложенных Зигвартом и Штаге. На рис. 94 показано устройство с сифоном, а на рис. 95 — более удачная конструкция с магнитным клапаном. Геммекер и Штаге [245 ] дают превосходный обзор устройств для измерения количества орошающей жидкости.
4.10.5. ОБЩАЯ, СТАТИЧЕСКАЯ И ДИНАМИЧЕСКАЯ УДЕРЖИВАЮЩАЯ СПОСОБНОСТЬ КОЛОННЫ
Под общей удерживающей способностью колонны — УС (total holdup*) подразумевают количество вещества, которое в виде жидкости и паров находится в ректификационной колонне между кубом и конденсатором. Общая УС складывается из статической УС («захват» жидкости), называемой также УС в состоянии покоя, и динамической УС колонны (рабочая задержка — operating holdup*).
Информация об общей УС колонны очень важна, поскольку, как уже было показано в разд. 4.7.1, влияние общей УС усиливается с увеличением числа теоретических ступеней разделения. Общая задержка при большом флегмовом числе ухудшает разделение, а при малом флегмовом числе, наоборот, благоприятствует разделению. При очень большой общей задержке флегмовое число почти не оказывает влияния на разделяющую способность колонны. Для насадки из колец Рашига возможен теоретический расчет динамической УС, предложенный Йилмазом и Брауэром [11 в] (см. разд. 4.2.2).
Статической УС колонны называют количество жидкости, которое остается в колонне после предварительного затопления насадки или окончания разгонки и охлаждения. При определе
* Термины total holdup, operating holdup приняты в научно-технической литературе на английском языке. — Прим. ред.
149
нии статической УС в куб загружают жидкость в пятикратном количестве по сравнению с предполагаемой УС колонны и в течение I ч проводят ректификацию с бесконечным флегмовым числом. После охлаждения колонны измеряют количество жидкости, оставшееся в кубе. Разница между первоначально загруженным количеством и оставшимся количеством и представляет собой статическую УС. В насадочных колоннах статическая УС складывается из капель жидкости, оставшихся на насадочных телах и между ними, а также на стенках колонны, приставки и конденсатора *. В тарельчатых колоннах основную часть статической задержки составляют слои жидкости, оставшиеся на отдельных тарелках.
В соответствии с более простой методикой определения статической УС через верхний конец конденсатора в установку заливают отмеренный объем разделяемой смеси и по количеству собранной внизу жидкости устанавливают, сколько жидкости задержится в колонне. Измерение следует повторять несколько раз, чтобы достигнуть полного смачивания насадки. Это позволит получить надежное среднее значение, которое, однако, соответствует более низким температурам, чем рабочая температура.
Динамической (рабочей) УС колонны является количество жидкости, которое дополнительно удерживается во время ректификации в аппарате над кубом. Динамическую УС определяют следующим образом. Во время разгонки быстро удаляют колбонагре-ватель, по возможности также отсоединяют и колбу **, а всю стекающую жидкость собирают в измерительный сосуд устройства, показанного на рис. 95; целесообразно при этом отводить жидкость через холодильник.
Для косвенного определения общей удерживающей способности (УС) как насадочных, так и тарельчатых колонн в куб с находящимся в нем эталонным веществом, например к-гептаном, добавляют небольшое количество высококипящего вещества, например стеариновой кислоты или а-бромнафталина (пд = = 1,6580). В ходе разгонки легколетучий компонент смеси накапливается в колонне и соответственно увеличивается доля высококипящего вещества в расчете на оставшееся в кубе количество эталонного вещества. Во время разгонки через определенные промежутки времени из куба отбирают пробы и определяют концентрацию добавленного высококипящего вещества. Если добавляет стеариновую кислоту, то ее содержание определяют либо титрованием, либо взвешиванием перед и после упаривания пробы. Содержание
* Следует дополнительно учитывать утечку паров за пределы конденсатора (например, в ловушку или в производственное помещение), которая за 1 ч может достигнуть ощутимых размеров. Утечка вещества из системы происходит иногда в результате недостаточно полной конденсации паров (особенно легколетучих веществ). — Прим. ред.
** При работе с ядовитыми или легковоспламеняющимися веществами этого делать не рекомендуется по положениям техники безопасности работ в лабораториях. — Прим. ред.
150
Рис.
Зависимость общей удерживающей способности колонны от нагрузки для различных насадочных тел (при 730 мм рт. ст.):
/ — перфорированные полуцилиндры; 2 — седла из проволочной сетки; 3 — кольца Рашнга. Эталонное вещество — н-гептаи.
а-броинафталина можно определить рефрактометрическим методом. Общую УС колонны вычисляют по формуле:
УС=Ув[1-(х1/х2)] (131)
где УС — общая удерживающая способность; Ев — объем загруженной смеси; Xj и х2— концентрация добавленного вещества в куб перед разгонкой и во время перегонки соответственно.
Для получения сравнимых результатов статическую УС, а также динамическую и общую УС целесообразно указывать в пересчете на одну теоретическую ступень разделения или реальную тарелку. О зависимости общей УС от нагрузки опубликовано мало сведений. Коллинз и Ланц [200] приводят результаты, полученные для ситчатой колонны Ольдершоу диаметром 28 мм с 30 реальными тарелками (см. рис. 92). В зависимости от нагрузки общая УС изменяется между 43 и 60 мл, так что на одну реальную тарелку в результате перерасчета приходится в среднем 1,4—2,0 мл, а одной теоретической ступени соответствует в среднем 2,5—3,5 мл. В насадочных колоннах, по собственным данным автора, УС в расчете на одну теоретическую ступень разделения имеет величину того же порядка, что и для тарельчатых колонн, как это видно из рис. 96, иллюстрирующего зависимость общего удерживаемого количества w-гептана при 97 °C от нагрузки при остаточном давлении 730 мм рт. ст. [203]. Ректификацию проводили в колонне диаметром 19 мм, рабочая высота колонны составляла 812 мм.
Цуидервег [158] при расчете числа теоретических ступеней разделения п для периодической ректификации учитывает влияние УС на п и размер промежуточной фракции. Им была исследована зависимость «четкости» разделения от удельной общей УС, флегмового числа и числа теоретических ступеней для смесей с относительной летучестью от 1,07 до 2,42. С помощью введенного Цуидервегом так называемого полюсного расстояния можно определить оптимальное флегмовое число. Способ соответствует расчету по методу Мак-Кэба и Тиле при конечной концентрации хв = 5% (мол.).
4.10.6. РАБОЧЕЕ ДАВЛЕНИЕ
Под рабочим давлением понимают давление, установившееся при перегонке в головке аппарата. Его обычно измеряют в газовом (или воздушном) пространстве непосредственно за зоной конденсации. Рабочее давление следовало бы измерять в паровой фазе
151
Рис. 97.
Зависимость числа теоретических ступеней разделения, приходящихся иа 1 м высоты иасадки, луд от перепада давления по данным Дэвида.
Эталонная смесь бензол—этилеихлорид, концентрация бензола в кубовой жидкости х& =И4% (мол.).
перед зоной конденсации, т. е. на том же самом участке, где измеряют температуру. Но это сложно, поэтому трубки конденсатора подбирают с достаточно большим диаметром, чтобы
между давлением в паровом пространстве (перед зоной конденсации) и давлением в газовом пространстве (после зоны конденсации) не мог
вознйкнуть перепад. Возможность создания перепада давления особенно реальна при работе в условиях высокого вакуума; в этом
случае рекомендуется проводить сравнительные измерения перед и после зоны конденсации. Давление, установившееся в кубе, равно сумме давления в газовом пространстве (перед зоной конденсации) и перепада давления (гидравлическое сопротивление потока) по высоте колонны. Так как при работе под вакуумом увеличившийся объем паров сильно влияет на процесс разделения, то часто при ректификации применяют более крупные насадочные тела, чем это следует из соотношения диаметр колонны/диаметр насадочного тела = 10, или, по крайней мере, в верхнюю часть колонны загружают более крупные насадочные тела. В головке колонны устанавливается самое низкое давление, поэтому в верхней части колонны из-за более высокой скорости потока паров легко может произойти затопление насадки.
Вопрос о том, насколько благоприятно влияет применение вакуума при перегонке на число теоретических ступеней разделения, еще не выяснен окончательно. Вебер [204] установил, что разрежение оказывает лишь слабое влияние на число теоретических ступеней; при этом пропускная способность колонны снижается вследствие большего объема паров, а относительная летучесть под вакуумом обычно увеличивается (см. разд. 4.6.2); Майлз с сотр. [192] установил, что для различных насадочных тел ВЭТС имеет минимум при остаточном давлении порядка 200 мм рт. ст. Страк и Кинней [203] выяснили, что значения ВЭТС в интервале 50—100 мм рт. ст. проходят через слабо выраженный минимум, и при 100 мм рт. ст. разделяющая способность такова же, как и при атмосферном давлении (см. также разд. 4.8 и 4.10.4).
На основе обширного экспериментального материала Дэвид [191 ] однозначно показал, что число теоретических ступеней разделения тем меньше, чем ниже рабочее давление ректификации. Эти результаты находятся в полном согласии с данными Цуидер-вега [205], а также частично соответствуют опытным данным
152
Майлза [192]. Если построить графическую зависимость числа теоретических ступеней разделения, приходящегося на 1 м высоты насадки, пуц от перепада давления в колонне, то можно наглядно проследить влияние вакуума- на разделение. На рис. 97 подобные кривые построены по данным Дэвида [191] для смеси бензол— этиленхлорид. К аналогичным выводам пришел Киршбаум с сотр. [206], показавший, что в результате влияния относительной летучести изменение числа теоретических ступеней разделения, приходящегося на 1 м насадки, при переходе к вакууму не всегда сопровождается параллельным изменением степени обогащения, достигаемой в ректификационной колонне.
При исследовании влияния давления на массообмен в насадочной колонне с помощью меченых атомов 14С, 35S и 36С1 Зельвенский с сотр. [209] обнаружил, что снижение давления вызывает уменьшение разделяющей способности колонны.
Шапиро [207] вывел уравнение для расчета состава кубовой жидкости и жидкости на n-ой тарелке в зависимости от давления при ректификации идеальных бинарных систем с бесконечным флегмовым числом.
4.10.7. НАГРУЗКА И СКОРОСТЬ ПОТОКА ПАРОВ
Под нагрузкой понимают количество вещества, прошедшее в единицу времени через колонну и получаемое в виде конденсата в головке колонны. Нагрузка складывается из количеств флегмы и отбираемого дистиллята. В промышленности принято выражать нагрузку колонны через скорость потока паров, отнесенную к свободному сечению колонны. В лабораторной практике количество паров, проходящих через поперечное сечение колонны в единицу времени, рассчитывают только в тех случаях, когда нужно провести сравнение с полупромышленными или промышленными установками. Принято также измерять нагрузку в мл/(см2-ч).
Разделяющая способность колонны зависит от нагрузки (рис. 98) и тем сильнее, чем больше флегмовое число. Поэтому при испытаниях эффективности колонны необходимо точно указывать нагрузку; по предложению Вебера для получения сравнимых результатов испытания следует, как правило, проводить при нагрузке, равной 2/3 максимальной нагрузки. Под максимальной нагрузкой понимают нагрузку, несколько меньшую той нагрузки, при которой орошающая жидкость под действием поднимающихся паров удерживается во взвешенном состоянии и не может стекать вниз. При этом колонна захлебывается и не может работать в режиме противоточного массообмена.
Важная особенность насадочных колонн заключается в том, что эффективность насадочных тел небольшого диаметра (2—4 мм) сильнее зависит от нагрузки, чем эффективность насадочных тел большего диаметра (5—10 мм) (см. рис. 98). По некоторым данным [214], как было уже установлено выше, оптимальной нагрузкой
153
Рис. 98.
Зависимость числа теоретических ступеней, приходящихся на 1 м высоты, Луд от нагрузки по данным Штаге ([39 ] к гл. 1) для различных насадок: 1,2 — спирали 2х2х 0 0,2 мм и 4X4 х 0 0,4 мм, сталь V2A; 3,4 — коль-' ца Рашнга 6,5х 0 6 мм и 4,5х 0 4,5 мм, стекло; 5 — шарики 0 3 мм, стекло; 6, 7 — шарики 0 7,5 мм, 0 4 мм, матированное стекло; 8, 12 — кольца Рашнга 8X0,8 мм и 5,5х 0 5,5 мм, фарфор; 9 — кольца Рашнга 10Х 10 мм, глнна; 10 — спираль 0 3 мм, длина 1—5 мм, стекло; И — токарная стружка 0 1— 0 3 мм, ширина —2 мм; 13 — 17 — спирали соответственно 0 2 мм, 0 3 мм, 0 4 мм, 0 6 мм, 0 8 мм, сталь V2A; 18 — 20 — спирали с разомкнутыми витками соответственно 3x3x0,4 мм, 4х4х X 0 0,45 мм, ЗхЗХ 0 0,45 мм, сталь V2A; 21 — 23 — спнралн с сомкнутыми витками соответственно 2X2 х 0 2 мм, 3X3x0,45 мм, 4Х4х X 0 0,4 мм, сталь V2A.
Эталонная смесь: бензол — 1,2-днхлорэтан для кривых 1 —12 (опыты Шульце—Штаге); н-гептан —метнлциклогексаи для кривых 13—17 (опыты Кол-линга); н-гептан —метилцнклогексан для кривых 18—23 (опыты Брауэра).
является не самая низкая, а несколько меньшая, чем нагрузка, вызывающая явление захлебывания (см. рис. 83).
В лабораторных колоннах максимальная эффективность наблюдается не при верхней предельной нагрузке, как это имеет место по данным Дэвида для полупромышленных колонн, а при значительно меньших нагрузках, хотя и превышающих минимально допустимую нагрузку.
При сравнении лабораторных колонн с полупромышленными или промышленными колоннами необходимо иметь в виду, что скорости потока паров в лабораторных колоннах в среднем ниже,
154
т. е. находятся в интервале 0,02—0,5 м/с, в то время как в промышленных установках эта скорость достигает 0,1—2,0 м/с и выше [184]. В насадочных колоннах очень важно поддерживать скорость потока паров постоянной; в тарельчатых же колоннах колебания скорости потока паров меньше сказываются на эффективности процесса разделения.
От нагрузки зависят динамическая и общая УС, перепад давления и предельная скорость потока паров, которая в свою очередь определяется формой и размерами насадочных тел или размерами и конструкцией реальной тарелки, а также свойствами разделяемых смесей. В разд. 4.11 об этом сказано подробнее. О соотношении нагрузок в насадочных колоннах исчерпывающую информацию предоставили Штаге и Бозе ([39] к гл. 1).
4.10.8. ПРОВЕДЕНИЕ ИСПЫТАНИЙ
Подготовка колонн к работе. При испытании колонн на эффективность очень важна аккуратность в работе. Всю аппаратуру следует тщательно промыть и просушить. Ни в коем случае в колонне не должно оставаться даже следов воды. По этой причине перед испытанием рекомендуется оставлять включенным на ночь обогрев кожуха колонны. Насадочные тела перед загрузкой также следует тщательно очистить. При этом рекомендуется их промыть сначала в четыреххлористом углероде и трихлорэтилене, затем в горячем бензоле и, наконец, снова в трихлорэтилене. При заполнении колонны насадкой нужно следить за тем, чтобы не касаться руками насадочных тел. При работе с насадочными колоннами большое внимание уделяется способу укладки насадочных тел. Лучше всего опускать одновременно по 3—4 насадочных тела при постоянном постукивании деревянной палочкой по корпусу колонны. С помощью приспособления, показанного на рис. 87, достигается быстрая и неупорядоченная укладка мелких насадочных тел. После завершения очередного испытания насадочные тела выгружают из колонны, промывают, просушивают, снова загружают, после этого можно приступить к новому испытанию. Таким путем проверяют влияние способа укладки насадки на разделяющую способность колонны.
Очистку тарельчатых колонн проводить труднее, так как на тарелках может оставаться некоторое количество жидкости. В этом случае колонну пропаривают чистым легколетучим растворителем, а затем просушивают теплым воздухом, предварительно надев на нее обогреваемый кожух.
Эталонную смесь загружают в количестве, соответствующем восьмикратной УС колонны, которую определяют заранее, как это описано в разд. 4.10.5. Перегонную колбу подбирают такого размера, чтобы исходная смесь в начале испытания заполняла бы колбу на 2/3 объема. Содержание легколетучего компонента в эталонной смеси должно составлять 30—40% (мол.). Пока еще не
155
5
Рис. 99.
Пробоотборник по нормалям <Дестинорм» для работы при атмосферном давлении и под вакуумом.
Рис. 100.
Головка колонны по нормалям <Дестинорм» с устройством для капельного пробоотбора без застойных зон:
1 — паровая камера; 2 — термометр; 3 — капилляр; 4 — воронка; 5 — кран для сообщения с атмосферой; 6 — кран для отбора пробы; 7 — патрубок; 8 — приемник жидкости; 9 — мерный сосуд для флегмы.
установлено однозначно, в какой мере состав кубовой жидкости влияет на степень разделения [208].
Большое внимание уделяют приготовлению эталонной смеси. Нельзя без проверки применять выпускаемые промышленностью реактивы квалификации «чистый для анализа» или «чистый». Часто для контроля чистоты недостаточно определения одного только показателя преломления. Точный анализ возможен с помощью газовой хроматографии и инфракрасной спектроскопии [195]. Дополнительная очистка эталонного вещества не требуется в том случае, если экспериментально определенные физико-химические константы совпадают с теоретическими значениями и температура кипения вещества, измеренная термометром с ценой деления 1/10 °C, имеет отклонение, не превышающее 0,1 °C с учетом влияния колебаний атмосферного давления. Большинство веществ нуждается в химической очистке от сопутствующих примесей [210—212] и в последующей четкой ректификации при высоком флегмовом числе. При использовании недостаточно очищенных веществ возможно смещение калибровочной кривой По — содержание % (масс.), а также концентрирование сопутствующих примесей в головке колонны или кубе при испытаниях. Это может привести к искажению результатов измерения разделяющей способности колонн.
При построении калибровочной прямой п2° — содержание, % (масс.) берут тщательно очищенные вещества, приготавливают из них растворы, содержащие от 5 до 95% легколетучего компо-156
нента, и определяют их показатели преломления. Найденные значения наносят на диаграмму — содержание, % (масс.). Иногда с помощью уравнения Фенске (108в) для v = оо вычисляют необходимое число теоретических ступеней для ряда обогащений и строят график зависимости числа теоретических ступеней разделения от показателя преломления (см. рис. 71). По этому графику после отбора пробы и определения п® можно сразу же найти число теоретических ступеней разделения, соответствующее эффективности колонны.
При отборе проб объемом 0,1—0,2 мл из головки колонны и из куба нужно следить за тем, чтобы проба по возможности не соприкасалась со смазкой шлифа. До сих пор еще недооценивают возможность искажения значений измеряемых величин вследствие загрязнения пробы смазкой для кранов. Загрязнения пробы можно избежать, если для отбора проб из куба при атмосферном давлении и под вакуумом пользоваться устройством, показанным на рис. 99.
Применяемая при испытании приставка или головка также должна гарантировать при минимальном «мертвом» объеме возможность отбора жидкости по каплям без ее соприкосновения со смазкой крана. Аналогичное пробоотборное устройство показано на рис. 100. До установления стационарного режима работы колонны маленькая воронка 4 повернута вниз. Для отбора пробы воронку 4 поворачивают вверх и осторожно подсасывают жидкость путем присоединения патрубка 7 к вакуумной линии при медленном открывании зажима. После отбора 0,1—0,2 мл жидкости, которая капает в приемник 8 через большое отверстие в пробке крана 6, этот кран, а также зажим на вакуумной линии закрывают, а кран 5 осторожно и ненадолго открывают; благодаря этому находящаяся в воронке 4 жидкость вытесняется обратно в колонну. Приемник 8 сообщается с' атмосферой через патрубок 7. При работе в условиях атмосферного давления в патрубок 7 подают воздух для полного удаления жидкости из трубки для отбора проб, расположенной в нижней части приемника 8. Другие возможные способы пробоотбора обсуждаются в разд. 7.5.3 (см. также [39] к гл. 1).
Условием, обеспечивающим точность результатов испытаний, является также адиабатичность режима работы аппаратуры. Поэтому во избежание тепловых потерь необходимо тщательно изолировать асбестовым шнуром и стекловатой как куб, так и остальные узлы, вплоть до конденсатора (см. разд. 7.7). Так как при температуре даже ниже 80 °C всегда приходится принимать в расчет «дикую флегму», то при испытаниях нужно обязательно компенсировать потери тепла в колонне с помощью обогревающего кожуха. При этом регулирование обогрева’осуществляют автоматически по температуре внутри колонны (см. разд. 8.2.2). Обогревающие кожухи с продольным разъемом (см. разд. 7.7.3) имеют то
157
преимущество, что их можно монтировать после сборки стеклянной аппаратуры, а во время разгонки их можно в любой момент снять (при каких-либо неполадках). Участок между кубом и обогревающим кожухом, между кожухом и головкой колонны, а также участок, примыкающий к конденсатору, изолируют сначала слоем асбестового шнура толщиной 20 мм, на который накладывают обмотку из стеклянной шерсти, а поверх нее ленту из стеклоткани. Толщина изоляции должна составлять около 60 мм. Для большей надежности целесообразно применять ленточный электрообогрев.
Особенное внимание нужно обращать на правильный выбор точки для измерения температуры паров в приставке или в головке колонны, при котором было бы исключено смачивание шарика термометра переохлажденной жидкой флегмой. Кроме того, при перегонке под вакуумом должно быть гарантировано отсутствие перепада давления на участке между точками измерения температуры и давления, который мог бы исказить результаты измерения. В головке колонны по нормалям «Дестинорм» эти требования учтены. Температуру измеряют в паровой камере 1 (см. рис. 100), которая достаточно хорошо изолирована; применяют термометр 2 со стандартным шлифом, подвесной термометр или же встроенную термопару. Смачивание термометра исключено благодаря тому, что флегма в колонну возвращается через капилляр 3 и мерный сосуд для флегмы 9, расположенные ниже термометра. Остаточное давление измеряют непосредственно за охлаждаемой ловушкой с большим свободным сечением, поэтому между термометром 2 и манометром отсутствует какое-либо значительное сужение проходного сечения.
Выбор условий испытания эффективности колонн. Разделяющая способность колонны в условиях испытания в первую очередь зависит от нагрузки, которую поэтому следует во время опытов поддерживать строго постоянно. Скорость испарения целесообразно регулировать по перепаду давления в колонне, применяя контактный манометр (см. разд. 8.4.2); мощность обогрева контролируют по амперметру. Перед установлением заданной нагрузки режим работы насадочной колонны доводят до захлебывания с целью улучшения смачиваемости насадки. Для этого увеличивают мощность кипятильника, наблюдая за показаниями контактного термометра, до тех пор, пока в нижней части колонны не начнется процесс захлебывания, который затем распространяется по всей колонне, вплоть до головки. Захлебывание колонны поддерживают в течение примерно 15 мин и затем уменьшают мощность кипятильника, чтобы флегма снова свободно стекала вниз. Этот прием повторяют несколько раз, а затем с помощью контактного манометра устанавливают уровень необходимой нагрузки (см. также [39] к гл. 1).
Менее эффективно «холодное» захлебывание, когда колонну наполняют разгоняемой смесью и после сливания жидкости начинают ректификацию.
158
Рис. 101 иллюстрирует сильное влияние, какое оказывает «горячее» захлебывание на эффективность разделения. Испытания были проведены в колонне Подбельняка диаметром 25 мм с насадкой Хелигрид (см. рис. 274) при нагрузке 1000 мл/ч. Обнаружено, что стационарный режим массообмена в колонне устанавливается только через 8—10 ч, а захлебывание приводит к повышению числа теоретических ступеней разделения на 50% [213].
Предварительное захлебывание особенно благоприятно сказывается на разделяющей способности колонны при использовании насадки из проволочных спиралей. Эффективность же работы насадки, состоящей, например, из колец Рашига размером 4,5 X 6,0 мм, не определяется степенью их предварительного смачивания. Сведения о захлебывании под вакуумом приведены в разд. 5.4.1. Ленц [214] определил нагрузки, соответствующие захлебыванию, и оптимальные нагрузки для колонны с насадкой из стеклянных колец Рашига (5,5х4,0 мм) и из проволочных колец (сталь V 4А, 2 мм) при ректификации эталонных смесей На О—Da О (27%) и н-гептан—метилциклогексан. Диаметр колонны составлял 37,5 мм. Были исследованы числа теоретических ступеней разделения, приходящихся на 1 м рабочей высоты колонны, и перепад давления в колонне при различных скоростях потока паров. Нагрузка при оптимальном режиме для колец Рашига совпадает с нагрузкой при захлебывании колонны, а для насадки из проволочных колец она соответствует 81—88% максимальной нагрузки. Если насадочная колонна работает в так называемом эмульгаци-онном режиме, то интенсивность массообмена может повыситься в 6 раз с одновременным увеличением пропускной способности [214а].
После установления стационарного режима колонны при бесконечном флегмовом числе (примерно через 2 ч) из головки отбирают пробу объемом 0,1—0,2 мл; эту пробу для анализа не используют. Еще через 1 ч из головки и из куба отбирают пробы 0,1—0,2 мл и анализируют их. При этом следует руководствоваться теми же положениями, что и при отборе проб при исследовании фазового равновесия (см. разд. 4.6.3). Продолжают через каждый час отбирать и анализировать пробы, пока не будут достигнуты постоянные составы жидкости, взятой из головки и из куба.
Пробы должны быть по возможности малого объема, чтобы избежать возмущений, препятствующих установлению стационарного режима работы колонны. При этом характерно, что в корот-
Рис. 101.
Влияние предварительного захлебывания на эффективность разделения в колонне (по данным Брауэра с сотр.).
Пунктиром показана зависимость при работе колонны без предварительного захлебывания.
159
ких колоннах стационарный режим восстанавливается быстрее, чем в длинных колоннах с большим числом теоретических ступеней разделения.
При испытании длинных колонн необходимо иногда увеличивать интервал между отборами проб (более 1 ч). Время установления стационарного режима при использовании обычных эталонных смесей лежит в интервале 10—20 ч. Испытания рекомендуется проводить как при уменьшении, так и при увеличении нагрузок [208].
Определение разделяющей способности колонны в рабочих условиях проводят по методике, описанной в разд. 4.10.4, при этом устанавливают число эквивалентных теоретических ступеней разделения.
Расчет числа теоретических ступеней разделения. Если уже получено 3—5 постоянных значений концентраций дистиллята и кубовой жидкости, то можно испытание закончить и приступить к расчету числа теоретических ступеней разделения. Для повышения надежности данных испытание повторяют на другой день при тех же условиях.
При аналитическом определении п полученные значения хЕ (дистиллят) и хв (куб) подставляют в уравнение Фенске (1086), справедливое при v = оо. При графическом определении п применяют метод Мак-Кэба и Тиле (см. разд. 4.7.1). Если испытание проведено в рабочих условиях, т. е. при конечном флегмовом числе, то определяют число эквивалентных теоретических ступеней по методике, изложенной в разд. 4.10.4.
Число теоретических ступеней, соответствующее разделяющей способности колонны в рабочих условиях, можно определить также по уравнению, предложенному Оболонцевым и Фростом [202]:
lg xF-axE = “экв ’g (1-х£)-а(1-х£) (132)
Где a — доля отобранного дистиллята от загрузки куба, моль; хЕ и хр—содержание легколетучего компонента соответственно в дистилляте и в исходной смеси, % (мол.).
Оболонцев и Фрост предлагают следующую методику определения числа теоретических ступеней разделения колонны. После достижения стационарного режима колонны отбирают дистиллят при заданном конечном флегмовом числе до тех пор, пока количество отогнанного дистиллята приблизительно не станет равным массе (или числу молей) легколетучего компонента в первоначально загруженной кубовой жидкости состава хР. Таким путем получают числа эквивалентных теоретических ступеней разделения пэкв/и (см. разд. 4.10.4). При испытании по методу'Оболонцева и Фроста сначала работают в течение 3 ч при v — оо и выбранной нагрузке, затем переходят к режиму с конечным флегмовым числом. Так как пользоваться предложенным ими уравнением трудно, была разработана соответствующая диаграмма [84, 202].
160
4.10.9. НЕКОТОРЫЕ ДАННЫЕ ПО РАЗДЕЛЯЮЩЕЙ СПОСОБНОСТИ НАСАДОЧНЫХ
И ТАРЕЛЬЧАТЫХ КОЛОНН
По опубликованным данным практически невозможно сравнить между собой ВЭТС для различных насадочных колонн или коэффициенты полезного действия для тарельчатых колонн. Это объясняется тем, что испытания проводили с различными эталонными смесями в разных условиях и только в редких случаях приводили аппаратурно-технологические параметры, указанные в разд. 4.10 в качестве безусловно необходимых. Дополнительные трудности возникают из-за того, что чистота применяемых эталонных смесей не всегда была гарантирована, а растворение смазки для кранов в отбираемых пробах часто приводит к искажению результатов. По-видимому, в настоящее время необходимо перейти к стандартным методам испытания, чтобы таким образом обеспечить получение сравнимых данных. В свете последних научных достижений становятся необходимыми новые исследования эффективности важнейших насадочных и наиболее распространенных тарельчатых колонн, учитывающие эти достижения и основанные на использовании таких современных точных методов анализа, как инфракрасная спектроскопия, газовая хроматография и масс-спектрометрия.
В табл. 28 дана приближенная оценка эффективности ректификационных колонн; в ней приведены результаты испытания Таблица 28Б.
Результаты испытаний тарельчатых колонн [215]
Автор конструкции колонны или тип колонии
Кезом
Гроль — Олдершоу Зигварт
Бруун
Грайнер и Фридрихе
Штаге (тарелка с брызго-отбойниками)
Штаге (колпачковая тарелка с открытыми сливными патрубками) Штаге (колпачковая тарелка с брызгоотбойни-ками)
Шмюклер и Фриц Бранд
Колонны со стандартны-'s ми шлифами
Расстояние между тарелками, мм Диаметр колонны, мм
23—25 »#33
25 25
65 50
20 или 40 5
40 45
60
50
35—150 30—120
Общая УС тарелки, мл К. п. д. тарелки, % *
1,0 50
около 1—2 80—90
около 7,5 100—110
0,7 или 1,4 70—100
около 2— 2,5 64—66
70—90
80—95
3,75 75—95
80—90 40—90 40—95
* Указанные значения нельзя безоговорочно использовать для сравнения, они Дают только приближенное представление о коэффициенте полезного действия тарелки (см. разд. 7.3.3.)
6 Э. Крель
161
Таблица 28А.
Результаты испытаний эффективности насадочных колонн
Параметры насадки
Насадочные тела Материал Диаметр, мм Высота, мм Толщина стенки, мм Поверхность насадки в 1 л; м2
Кольца Рашнга Стекло гладкое 2,0 2,0 0,5 2,140
То же 2,0 2,0 0,6 1,680
» 3,0 3,0 0,7 1,320
» 4,0 4,0 0,6 1,109
» 4,5 4,5 0,5 1,382
» 5,0 5,0
» 6,0 6,0
» 6,5 6,5 0,5 1,109
» 10,0 10,0
Фарфор 5,5 5,5 1,1
То же 8,0 8,0 1,1
Глина 10,0 10,0 1,7
Проволочная сетка 3,0 3,0
То же 5,0
» 6,0
Кольца «Прим» Металл 2,2 2,5
Спирали Виль- Стекло 3,0 1,5—3,0 0,4
сона » 5,0 5,0 0,65
» 6,33
Разрезные » 2,1
стеклянные 3,1
спирали » 3,1
» 6,33
Спирали Сталь V2A 1,6 1,6 0,2
То же 2,0 2,0 0,2 4,547
4,0 4,0 0,4 1,489
Шарики Стекло гладкое То же 3,0 4,0 0,900
Стекло матированное 4,0 0,601
Стекло гладкое 6,0
Стекло матированное 7,5 0,450
Стекло гладкое 8,0
Седла Фарфор 4,0 1,500
То же 6,0 1,150
» 8,0
» 10,0 0,720
Условия определения числа теоретических ступеней разделения: а см. [216]; б — шим нагрузкам, большие ВЭТС относятся к большим нагрузкам; г) эталонная смесь к различным давлениям н к средней плотности орошения.
162
Параметры колонны ВЭТС по данным различных исследователей Литература
Диаметр, мм Поперечное сечение, см2 к « со со ст н о о АО О _ со 3 2 UfflS Шульце-Штаге [216 ]а Зигварт [2171° Торманн [213 Iе Майлз с сотр. [1921г Различные измерения
5—7
6—9
24 4,5 600 5,82—9,52
35 9,6 900 И,1 7—12
20 3,1 1250 15,5 8—14
24 4,5 600 5,00—14,3
50 19,6 950 18,2
24 4,5 600 3,69—7,5
24 4,5 600 7,07—11,30
24 4,5 600 7,23—12,80
25 4,9 1000 1,4—2,8 Разд. 7,8
20 3,1 600 4,35
50 19,6 1000 1,4—3,5
3—5
24 4,5 600 8,00—13,35
< 7—12
25 4,9 1067 7—9,2
10 0,8 800 4,8
13 1,3 1260 7,4 4—6
20 3,1 1250 9,5
25 4,9 1067 3,6—4,2
40 12,5 1000 1,4—2,0 Собствен-
ные дан-
ные
24 4,5 600 1,25—3,32 То же
24 4,5 600 1,82—5,00 2—5
! 24 4,5 600 6,00—6,06 5-6
6—7
f 24 4,5 600 5,82—7,22
!, 24 4,5 600 3,31—7,50
ho 7,0 450 4,7—6,3 Собствен-
30 7,0 450 6,0—9,0 ные дан-
|30 7,0 450 7,5—11,4 ные
зо 7,0 450 9,0—12,8
[остальные параметры не указаны; в) меньшие ВЭТС относятся преимущественно к меиь-|я-гептаи — метилциклогексаи и «-додекан — циклогексилцнклопеитаи; ВЭТС относятся
6*
163
насадочных и некоторых наиболее часто применяемых тарельчатых колонн. Различные конструкции насадочных и тарельчатых колонн рассмотрены подробно в разд. 7.8 и 7.3.3.
4.11. ГИДРАВЛИЧЕСКОЕ СОПРОТИВЛЕНИЕ, ПРЕДЕЛЬНАЯ СКОРОСТЬ ПАРОВ
И РАСЧЕТ ОСНОВНЫХ РАЗМЕРОВ КОЛОННЫ
Для ректификации при атмосферном давлении в лаборатории обычно применяют колонны диаметром 10—30 мм, а для ректификации под вакуумом — колонны диаметром 20—50 мм. При очень высоких нагрузках колонны диаметром 50 мм можно применять также для разгонки при атмосферном давлении. Если необходимо работать в режиме, близком к захлебыванию, чтобы обеспечить максимальную разделяющую способность, то следует предварительно точно рассчитать диаметр колонны, при котором можно приблизиться к желаемой нагрузке.
Напомним, что под нагрузкой колонны подразумевают количество паров вещества, проходящее в единицу времени через колонну и конденсирующееся в головке колонны с образованием флегмы и дистиллята. Поскольку насыпная насадка и насадки других видов оказывают сопротивление как поднимающимся парам, так и стекающей вниз жидкости, то нагрузку нельзя увеличивать беспредельно. С увеличением скорости испарения, т. е. нагрузки, растет разность давлений в головке и в нижней части колонны, которую называют перепадом давления в колонне (или гидравлическим сопротивлением колонны). Гидравлическое сопротивление колонны зависит от типа и размеров колонны и ее насадки, рабочего давления ректификации, физико-химических свойств смеси, а также от нагрузки или скорости паров. Перепад давления в колоннах с концентрическим зазором можно вычислить по формуле (188). Данные по гидравлическому сопротивлению колонн с вращающейся насадкой приведены в табл. 30 и 31.
Тарельчатые колонны имеют то преимущество, что их нагрузку можно снижать до очень низких значений, при этом их разделяющая способность даже увеличивается. Насадочные же колонны имеют определенную минимальную нагрузку, при которой еще обеспечивается противоточный массообмен. В промышленности во многих случаях колонны регулируют по перепаду давления во избежание захлебывания и для поддержания оптимальной нагрузки. Этот способ успешно применяют также для регулирования лабораторных установок (см. разд. 8.4.2).
Скорость паров (отнесенную к свободному сечению колонны), при которой в колонне начинает накапливаться жидкость, называют нижней предельной скоростью. Верхней предельной скоростью паров называют такую их скорость, при которой накопление жидкости в колонне достигает такой степени, что в тарельча-164
тых колоннах слой жидкости перебрасывается на вышележащую тарелку, а в насадочных колоннах в нижней части образуется пенящийся слой жидкости, который постепенно поднимается в верхнюю часть колонны.
После фундаментальных работ Маха [219], Барта [220] и Лева [221 ] появилось много статей, в которых были рассмотрены закономерности, связанные с гидравлическим сопротивлением насадочных колонн. При тщательном экспериментальном исследовании взаимосвязи между перепадом давления и разделяющей способностью насадочных колонн Брауэр [208] определил сопротивление насыпной насадки 13-ти различных видов (см. табл. 29) в зависимости от нагрузки по жидкости. Испытания проводили
Таблица 29.
Параметры насадочных тел, использованные Брауэром [208] при измерении гидравлического сопротивления колонн
Тиг насадочных тел Материал Размеры, мм Соотношение диаметров колонны и насадочного тела Свободный объем насадки м*/м8 Удельная поверхность насадки, м*/м8 Длина проволоки или ленты, из которой изготовлено насадочное тело, мм
1. Спирали с разомкнутыми витками V2A (сталь) 3X3X0,45 1 : 12,3 0,777 1802 43
2. Спирали с разомкнутыми витками То же 4X4X0,5 1 : 9,25 0,822 1480 51.
3. Спирали с разомкнутыми витками » 3X3X0,40 1 : 12,3 0,771 .1815 44
4. Спирали с сомкнутыми витками » 2X2X0,3 1 : 18,5 0,661 2680 32
5. Спирали с сомкнутыми витками » 3X3X0,45 1 : 12,3 0,704 1717 60
6. Спирали с сомкнутыми витками V4A 4X4X0,4 1 : 9,25 0,760 1543 100
•7. Кольца с перемычкой (свернутые из ленты) V2A 2X2X0,25 1 : 18,5 0,716 1965 10,5
8. Кольца с перемычкой То же 4X4X0,3 1 : 9,25 0,824 1176 18,7
9. Кольца с перемычкой » 6X6X0,4 1 : 6,16 0,811 946 24,1
10. 11. Кольца Рашига Эллипсы Фарфор Алюминий 5X5 3,7X7,4 1 : 7,4 0,475 0,306 977
12. 13. Шарики Кольца Рашига Стекло Фарфор 2 16X16 1 : 18,5 1 : 2,3 0,339 1786
105
Рис. 102.
Зависимость удельного гидравлического сопротивления насыпной насадки различных типов (см. табл. 29) от плотности орошения (по данным Брауэра).
Прямыми а—а, b—b, с—с отмечены границы гидродинамических режимов [см. уравнение (133)].
в колонне диаметром 37 мм с рабочей высотой 1 м при атмосферном давлении со смесью н-гептан — метилциклогексан. Результаты весьма тщательных измерений, представленные на рис. 102, позволяют сделать следующие выводы:
1) Все кривые похожи одна на другую. Точки излома, ограничивающие различные режимы, лежат на линии изломов а—а, b—b и с—с. 2) Во всех интервалах нагрузок, ограниченных
точками излома, ход кривых, отражающих изменение гидравлического сопротивления, можно описать с помощью уравнения вида:
^Plh—cfn (133)
где: п = 1,0 для области ниже а—а (ламинарный поток паров); п = 1,78 для области между а—а и Ь—Ь (турбулентный поток); п = 2,7 для области между b—b и с—с (турбулентный поток); п = 3,3 для области выше с—с (затопление, пено-образование);
Коэффициент с является функцией свойств паров и жидкости, а также параметров насадки.
Рис. 103 иллюстрирует взаимосвязь между гидравлическим сопротивлением насадки и ее разделяющей способностью. Линейная зависимость числа теоретических ступеней, приходящихся на 1 м рабочей высоты колонны, пуд для насадки из спиралей (см. табл. 29) от гидравлического сопротивления при турбулентном движении паров показывает, что во всем интервале нагрузок для турбулентного режима между переносом количества движения, тепла и вещества в процессе ректификации существует аналогия. Механизм массообмена при этом остается неизменным. Выше линии изломов с—с (см. рис. 103) пар в виде пузырей барботирует через накопившийся слой жидкости, причем процесс массообмена протекает уже по другому закону [203, 206, 208].
Каст [222] учел истинный путь, который проходит двухфазный поток и вместо высоты слоя насадки ввел понятие эффек
166
тивной длины (х7Ук; коэффициент у. является физическим параметром, характеризующим искривленность траектории потока. Каст установил, что его опытные результаты в области перехода от ламинарного потока паров к турбулентному в колоннах с насадочными телами размером до 50 мм, а также данные других авторов находятся в хорошем согласии с полученным им уравнением для расчета гидравлического сопротивления колонны. Тойч [223] вывел полуэмпирическое уравнение для упрощенного расчета перепада давления в колонне (в безразмерной форме); соответствующие исследования были ограничены насадочными телами размером не менее 25 мм. Указана область применимости этого уравнения. Путем обработки опытных данных Гелбе [224], изучавшим работу насадки из спиралей с разомкнутыми витками размером 4x4x0,5 мм в интервале рабочих давлений 10—740 мм рт. ст., Глазер [225] использовал в расчетах безразмерный параметр е:
<«>
где р — плотность паров, кг/м3; g — ускорение свободного падения, м2/с; Нк — рабочая высота колонны, м; ВЕП — высота единицы переноса, м; Др — перепад давления, кгс/м2; Хк — объем жидкости в колонне, м3.
Зависимость параметра е ог важнейших переменных также можно представить в безразмерной форме. Параметр е изменяется в зависимости от нагрузки; эта зависимость позволяет четко проследить взаимосвязь между переносом количества движения и вещества, а также учесть влияние давления.
На основе собственных и литературных данных Райхельт, а также Райхельт и Бласс провели анализ гидродинамических закономерностей, характерных для насадки из колец Рашига и шаров. В результате были получены уравнения для расчета гид-
Рис. 103.
Взаимосвязь между перепадом давления и разделяющей способностью насадки различных типов (см. табл. 29) (по данным Брауэра).
Рис. 104.
Зависимость гидравлического сопротивления насадки из фарфоровых седел размером 4—8 мм от скорости воздуха при плотности орошения водой 40 мл/(см2-с).
167
1,0 1,1 1,2 1,3 1,4 1,5 1,6 w, м/с
Рис. 105.
Зависимость гидравлического сопротивления иасадки из спиралей Вильсона размером 3 мм от скорости воздуха при расходах орошающей жидкости от 200 — 1800 мл/ч.
равлического сопротивления в однофазном потоке, проходящем через насадку из шаров и цилиндров.
Хекманн и Крель [226] исследовали гидравлическое сопротивление фарфоровых седел размером 4, 6 и 8 мм с использованием системы воздух—вода. Рабочая высота колонны составляла 1 м, а диаметр — 22 мм. Опытные данные, представленные на рис. 104, можно использовать только в качестве приближенной оценки перепада давления. Рис. 105 показывает, как сказывается увеличение расхода жидкости на гидравлическом сопротивлении насадки из спиралей Вильсона размером 3 мм. Очень выгодны (малый перепад давления) также кольца из проволочной сетки, как это видно из рис. 106 и 107. На них представлены результаты опытов, проведенных с использованием различных смесей.
Основываясь на опыте, приобретенном при стендовых испытаниях аппаратуры, Нойманн [227] ввел понятие «эквивалентной» скорости воздуха для оценки нагрузки по пару или жидкости.
Рид и Фенске [228] вывели уравнение для расчета перепада давления, которое Брауэр [208] привел к виду
Д<- ° X- wG ' <'®>
где р — абсолютное давление, кгс/м2; Др — разность давлений, кгс/м2; М — молекулярная масса; е0 = (VK — VH)/VK — свободный объем сухой насадки;
168
Ик — объем колонны, соответствующий рабочей высоте, м3; Vh — объем насадки, м3; Vp — объем удержанной жидкости, м3; Р — универсальная газовая постоянная, кгс-м/(кгс-°С); Т — абсолютная температура, К; Нк — высота слоя насадки (рабочая высота колонны), м; g — ускорение свободного падения, м2/с; т]—динамическая вязкость паров,кгс-с/м2; р — плотность паров, кгс-с2/м4; О — удельная поверхность насадки, м2/м3; D — удельный расход паров, м3/(м2 • с).
Сравнение результатов расчета по формуле (135) с экспериментальными данными дает очень хорошее согласие при оптимальном способе укладки насадочных тел.
Кривые на рис. 108 [229], а также данные, приведенные в табл. 30 [200], свидетельствуют о том, что физико-химические свойства перегоняемого вещества также существенно влияют на перепад давления. Данные табл. 30 относятся к ситчатой колонне Олдершоу. Следует указать на то, что в этом эксперименте применяли специально подобранные вещества, которые при температуре кипения имели неодинаковые значения плотности и поверхностного натяжения.
Умхольц и Винкль [230] определили зависимость перепада давления от нагрузки в ситчатых колоннах диаметром 25 мм,
О 2 6 6 8 10 12 16 16 18
Нагрузка, л/ч
Рис. 107.
Зависимость перепада давления, приходящегося иа одну теоретическую ступень разделения, в слое колец из проволочной сетки от нагрузки при остаточных давлениях от 10 до 735 мм рт. ст.
Диаметр колонны 5 см, рабочая высота 1 м, данные Тюркауфа.
Рис. 108.
Зависимость удельного гидравлического сопротивления колонны от скорости паров различных веществ (по данным Штаге):
/ — этанол; 2 — хлороформ; 3 — бензол; 4 — н-гептан; 5 — динзопро-пиловый эфир; 6 — дравннол; 7 — толуол; 8 — этнлацетат.
169
Таблица 30.
Гидравлическое сопротивление реальной тарелки Др ситчатой колонны Олдершоу при ректификации различных веществ
Диаметр колонны 28 мм, 30 реальных тарелок
н-Гептан (^6и°п = = 98,424 °C, рЭД = 0,68368) Мети лци к л ог екс а и (I760 = 101,10 °C, кип р2° = 0,76944) Ж Бензол (№$ = кип = 80,09 °C, = 0,87895) Муравьиная кислота (/760 _ 100 75 »с кип рЭД = 1,2206)
нагрузка, мл/ч Др, мм рт. ст. нагрузка, мл/ч Др, мм рт. ст. нагрузка, мл/ч Др, мм рт. ст. нагрузка, мл/ч Др, мм рт. ст.
1000 1000 1000 0,76 200 0,88
1500 0,50 1500 0,62 1500 0,82 400 0,95
2000 0,53 2000 0,63 2000 0,84 600 1,02
2500 0,55 2500 0,65 2500 0,87 800 1,09
3000 0,57 3000 0,67 3000 0,91 1000 1,18
3500 0,60 3500 0,71 3500 1,00 1125 1,23
4000 0,64 4000 0,77 4000 1,11
4500 0,70 4500 0,85 4280 1,17
5000 0,77 5000 0,93
5230 0,80 5230 0,97
Поверхностное натяжение использованных веществ изменяется в интервале 20— 40 дин/см (на границе с воздухом) при 20 °C.
в которых с шагом 25 мм установлены тарелки с долей свободного сечения 0,162, имеющие различное число отверстий неодинаковых диаметров. Из табл. 31 [192] видно, каким образом влияет рабочее давление ректификации на гидравлическое сопротивление и предельные нагрузки в колонне. В частности, согласно данным, приведенным в табл. 31, насадочным телам в форме шариков соответствует относительно узкий интервал рабочих нагрузок и большое гидравлическое сопротивление, что связано с малой долей свободного объема (0,26) насадки этого типа. Напротив, для насадок «Хели-грид» (см. разд. 7.3.4) имеется очень широкий интервал рабочих нагрузок и большая верхняя предельная нагрузка (см. также [39] к гл. 1).
Для расчета необходимого диаметра колонны следует знать верхнюю предельную скорость паров. На основании чисто теоретических рассуждений Шумахер [231 ] рассчитал константы скорости kn (м/с) для различных типов насадки по следующей формуле:
kn = P«K'n/rff,g или ®П = У kndnS/ Pfl (136)
где Рц — приведенная плотность паров; р^ = Рг/Рж" плотность паров (г/л)/плот-ность жидкости (г/л) (при рабочем давлении), te>n — предельная скорость паров; dH—диаметр насадочного тела, м; g — ускорение свободного падения.
170
При использовании этой формулы предполагают постоянство вязкости паров и жидкости и учитывают влияние на предельную скорость паров типа и размеров насадочных тел, плотности паров и жидкости, но игнорируют влияние вязкости паров и жидкости и поверхностного натяжения жидкости.
Для константы скорости kn установлены следующие значения в зависимости от вида насадки: 0,01 для колец Рашига, 0,021 для колец «Прим», 0,019 для седел, 0,004 для кусковой насадки, 0,0126 для спиралей (по данным автора).
Таблица 31.
Перепад давления и предельные нагрузки для различных насадочных тел при остаточном давлении 20—760 мм рт. ст. в колонне диаметром 25 мм с рабочей высотой 1,07 м
Тип насадочного тела Размер насадочного тела, мм Остаточное давление, мм рт. ст. Перепад давления, мм рт. ст. Предельная нагрузка, г/ч
мин. макс. мни. макс.
«Хели-грид» — 50 5 28 1200 1650
100 5 28 650 1600
150 6 27 550 1100
760 1 27 500 5500
Шарики (неплотная упа- 3,6 20 6 26 450 700
ковка) 50 5 32 500 900
100 6 35 700 1150
150 5 32 1000 1400
760 3 27 125 2500
Шарики (плотная упаков- 3,6 20 3,5 31 380 720
ка) 50 3,5 30 620 900
100 4 30 800 1170
150 4 35 1000 1300
Седла 6,3 20 2 8 450 800
50 1 8 600 1100
150 2 8 900 2200
760 2 800 2600 4800
Стеклянные спирали с тре- 6,3 20 1 5 450 900
мя витками 50 1 5 800 1400
150 1 5 1000 2200
760 1 — 1200 1600
Одинарные стеклянные 6,3 20 2 14 450 900
витки 50 3 14 970 1200
150 3 18 1450 2000
760 2 — 2400 —
171
Рис. 109.
Номограмма Шумахера для определения предельной скорости паров для насадок.
— диаметр насадочного тела.
Для быстрого расчета искомой предельной скорости паров была разработана номограмма (рис. 109), применение которой поясним на следующем примере.
Пусть нужно определить предельную скорость паров при ректификации органической кислоты нормального строения С? при 20 мм рт. ст. (/|®п = = 125,0 °C). Насадочные тела — седла размером 4 мм. Определяем необходимые величины:
kn =0,019
20
рп = -ЧС|а • 130'0,01605 = 0,105 [по формуле (32)] оУо
0^= —= (при 20 мм рт. ст.) = 0,0001165 = 0,1165-10-8
« pF 900 (г/л) v г '
На номограмме рис. 109 необходимые построения для определения предельной скорости показаны пунктирной линией. Через точку пересечения вертикальной линии, соответствующей константе скорости 0,0190, и горизонтальной линии, соответствующей диаметру d = 4 мм, проводят линию, параллельную 172
наклонным линиям сетки. Из точки пересечения этой параллельной линии с вертикальной линией, соответствующей значению = 0,12, проводят горизонталь до вертикальной шкалы предельных скоростей и считывают искомую величину 2,5 м/с.
Конечно, с такой предельной скоростью паров колонна работать не может, поэтому берут 50—70% от найденного значения (60% соответствуют скорости 1,5 м/с).
Усложним поставленную задачу, пусть теперь нужно определить необходимое поперечное сечение колонны для перегонки кислоты С7 нормального строения при 20 мм рт. ст. и нагрузке 400 г/ч. Для расчетов понадобится формула
Р-22 400 (273 -1- 0 760
щп-3600-273р
(137)
где f — поперечное сечение колонны, см2; D — расход паров в колонне, моль/ч; t — температура перегонки, °C; wn — выбранная скорость паров, см/с; р — рабочее давление процесса перегонки, мм рт. ст.
Выполнив простейшие арифметические операции в формуле (137), получим
DT 22 400-760
wnp ’ 3600-273
(138)
При нагрузке D = 400 г/ч = 3,4 моль/ч при = 150 см/с требуется колонна с поперечным сечением:
Отсюда рассчитываем радиус колонны
г = Vf/л = К7,9/3,14 = 1,6 см
(140)
Для ректификации при атмосферном давлении формула (138) приводится к виду
/ =0,022807/к>п (см2)
(141)
Для сравнения подсчитаем, какая предельная нагрузка может быть при перегонке н-гептана в колонне диаметром 30 мм при атмосферном давлении. Предельная скорость паров при работе с насадкой из седел размером 4 мм составит 0,38 м/с. Отсюда
D = /аУп/0,028871 = 7,9-38/(0,0288-371) = 35,5 г-моль/ч = 3550 г/ч = 5200 мл/ч
(142)
То, что такие высокие нагрузки вообще возможны в колонне диаметром 30 мм, доказывают опыты Коллинга [171 ]. Сравнительный расчет был проведен главным образом для того, чтобы показать, насколько существенно увеличивается скорость паров и, следовательно, уменьшается нагрузка при переходе к пониженному давлению.
Автору до сих пор не известны какие-либо специальные методы расчета предельной скорости паров для лабораторных тарельчатых колонн. Уравнения, справедливые для промышленных колонн, в данном случае не могут быть использованы, так как дают большие погрешности. Опыт показывает, что тарельчатые колонны могут быть нагружены на х/3 от нормальной нагрузки насадочных колонн того же диаметра. Это обусловлено тем, что, во-первых, слой жидкости на тарелках и, во-вторых, паровые патрубки со
173
сравнительно узким поперечным сечением приводят к увеличению гидравлического сопротивления.
Вопросы гидродинамики потоков в насадочных колоннах и в зернистых слоях подробно рассмотрел Барт [220], который, в частности, отметил, что насадочные тела с острыми кромками вызывают в два и три раза большее гидравлическое сопротивление по сравнению с насадочными телами округленной формы.
На основании анализа гидродинамических закономерностей однофазных потоков, движущихся в слоях насадки, Дэвид [191] наметил последовательность расчета числа теоретических ступеней разделения в насадочной колонне с кольцами Рашига, имеющими размеры от 8 до 50 мм. Дэвид исходил при этом из той предпосылки, что перепад давления, скорость паров и плотность паров вследствие их сильного влияния на разделяющую способность насадочной колонны должны быть учтены в любой расчетной методике. Исходя из известного уравнения для определения потери напора в трубопроводах, коэффициент трения можно представить в следующем виде:
g = (143)
дапРпЛ
где Др — перепад давления, мм вод. ст.; d„—диаметр насадочного тела, м; шп — скорость паров, отнесенная к свободному сечению колонны, м/с; рп — удельный вес паров, кгс/м3; h — высота слоя насадки, м.
Отношение числа теоретических ступеней разделения, приходящихся на 1 м насадки, пуд к коэффициенту трения £ было принято в качестве базовой величины, не зависящей от критерия Рейнольдса. Эта величина, имеет постоянное значение (0,13) для всех смесей, использованных Дэвидом. Он принимает, что эффективность разделения имеет максимальное значение при верхней предельной скорости паров. Однако в лабораторных колоннах эта скорость не намного превышает ту скорость паров, которая соответствует минимально допустимой нагрузке. Поэтому соотношение
попт = 0,13g (144)
имеет силу также для лабораторных колон, если эффективность разделения максимальна (попт отнесено к 1 м рабочей высоты колонны).
При обобщении экспериментальных данных было получено следующее эмпирическое уравнение для расчета рабочей высоты колонны Нк:
Нк = 1 +(« — 0,13g)(10+ </н)/120 (м) (145)
Уравнение (145) справедливо для флегмового числа v — оо, т. е. для соотношения расходов жидкости и паров равного 1. В результате обработки результатов 115 опытов с использованием системы вода—воздух Бек [244] предлагает новые уравнения для насадочных тел размером 8—80 мм и для колонн диаметром от 150 до 1200 мм;
174
4.12. ТЕПЛОВЫЕ РАСЧЕТЫ
Тепловые расчеты процесса лабораторной перегонки проводят редко, поскольку в данном случае затраты энергии по сравнению с полупромышленными или промышленными установками весьма незначительны. Обычно в лабораториях перегонку проводят при большем или меньшем избытке тепла, а фактическую потребность в электрической энергии регулируют с помощью дополнительных сопротивлений. В лабораторной практике газ до сих пор еще применяют при дистилляции по методу Энглера, при аналитических разгонках, как средство обогрева масляных, песочных бань и бань с металлическими теплоносителями. Применения открытого газового пламени для нагревания избегают при перегонке веществ с высоким давлением паров ввиду возможной опасности перегрева жидкости, растрескивания аппаратуры или взрыва. В настоящее время предпочтение отдают электрическому обогреву при помощи закрытых колбонагревателей или нагревательных устройств, в которых электрическая спираль защищена слоем изоляционного материала. Для достижения невысоких температур применяют инфракрасное излучение (в видимой и невидимой частях спектра), которое обладает всеми преимуществами радиационного обогрева [232]. Применение токов высокой частоты для нагревания в лабораторных условиях находится еще только в стадии проверки.
При периодической перегонке разделяемую смесь, загруженную в куб, сначала нагревают до температуры кипения. При дальнейшем подводе тепла (в соответствии с энтальпией испарения смеси) происходит испарение. Скорость испарения зависит от количества тепловой энергии, подводимой в единицу времени. При непрерывной ректификации часть тепла подводят к исходной смеси уже в теплообменнике для предварительного подогревания исходной смеси. Основная часть тепловой энергии расходуется для нагревания куба.
Обозначим общий расход тепла через Q; тогда уравнение общего теплового баланса для различных видов перегонки без учета тепла, необходимого для подогревания кубовой жидкости до температуры кипения, будет иметь следующий вид:
для дистилляции
Q = Qk = Qa Qn == Окон 4- Qn (146)
для периодической ректификации
Q = Qk = Од 4" Q<b 4" Qn — Qkoh 4" Qn (147)
для непрерывной ректификации
Q — Qk 4" Опит — Од 4" Оф 4~ Qn = Окон 4" Qn (148)
где Qk — количество тепла, подводимое в кубе (колбе); QnHT — количество тепла, подводимое при непрерывной ректификации в теплообменнике предварительного подогрева; 0д—энтальпия дистиллята (в парообразном состоянии); Оф — эн-
175
Рис. 110.
Схема теплового баланса при дистилляции (а), периодической (б) и непрерывной (в) ректификации.
тальпия флегмы (в парообразном состоянии); Qn—тепловые потери; QKOh— количество тепла, отводимое в конденсаторе при конденсации паров и охлаждении дистиллята.
Эти уравнения справедливы при условии, что дистиллят и флегма охлаждены до температуры, при которой смесь поступает в куб. Дополнительные пояснения к приведенным соотношениям даны на рис. ПО.
Чтобы дать представление о затратах тепла при лабораторной перегонке, приведем следующий пример. Пусть необходимо разделить смесь бензол—толуол [50% (масс.) ] при атмосферном давлении с флегмовым числом 2 при нагрузке колонны 0,5 кг/ч и загрузке 2 кг исходной смеси (20 °C). Физико-химические параметры компонентов этой смеси, необходимые для дальнейших расчетов, таковы:
Бензол
°С........................ 80,1
Теплоемкость ср при 50 °C, ккал/(кг-°С)................... 0,43
Энтальпия испарения ДЯу, ккал/кг........................ 93,8
(при 80,1 °C)
Толуол
110,6
0,43
86,5 (при 109,7 °C)
* 1 ккал = 4168,84 Дж, 1 Дж = 2,388- 10-4 ккал.
Принимаем, что испаряется только бензол, хотя в действительности с повышением температуры в кубе испаряется и некоторая часть толуола (вследствие увеличения концентрации толуола 176
при отгонке бензола). Обусловленная этим допущением незначительная погрешность несколько завышает расход тепла, так как энтальпия испарения для толуола меньше, чем для бензола. Тепловые потери оценить весьма трудно; они складываются из тепловых потерь в кубе, головке колонны и, главным образом, в самой колонне. Тепловые потери в кубе в значительной степени можно уменьшить применением подходящего теплоизолирующего материала (см. разд. 7.7). Методы устранения тепловых потерь в самой колонне изложены ниже. Затраты тепла на подра л > смеси до 80 °C составляют г*" 1
QK = в — Л) ср 0п (149) 1
QK=2(80— 20) 0,43 ккал = 51,6 ккал (150) I
+ Тепловые потери (10%) ККал I
56,8 ккал I
Затраты тепла на испарение дистиллята и флегмы составляют L
QK=(E + R)&HV -( Qn (151)1
Qk = (0,5 + 1,0)93,8 ккал/ч = 140,7 ккал/ч (152) 1
, -г 14,0 ккал/ч 1
+ Тепловые потери (10%) т——---’
r v ' 154,7 ккал/ч
где В — количество исходной смеси в кубе, кг; С — температура в гдццс! [Lj-гонки, °C; 4—температура исходной смеси. °C.
Исходя из эквивалентного соотношения между электрической и тепловой энергией
1 кВт = 860 ккал (153)
получим, что для проведения данной разгонки необходим электрический подогреватель с фактической мощностью
154,7/860 = 0,18 кВт = 180 Вт (154)
Расчет поверхности конденсатора обсуждается в разд. 7.4.
Формула (191) может быть использована для расчета греющей поверхности теплообменника для подогрева питающей жидкости при непрерывной ректификации (в этой формуле Q — количество тепла, подводимое к жидкости; F — поверхность теплообмена, Д/ — разность между температурой греющей среды и температурой нагреваемой жидкости). Средняя разность температур вычисляется по формуле (192).
В лабораториях обычно приходится работать со стеклянными приборами; в случае применения стекла в качестве конструкционного материала коэффициент теплопередачи можно принять приблизительно равным 2,5-10-2 ккал/(см2-ч-°C). При использовании обычных круглодонных колб для перегонки при нормальных скоростях испарения поверхность колбы вполне достаточна для подвода необходимого тепла.
Чтобы обеспечить возможно более полное исключение тепловых потерь, в качестве тепловой изоляции колонн используют
177
.Рис. 111.
Зависимость тепловых потерь в изолированной колонне от рабочей температуры.
1 — колонна с вакуумированной рубашкой; 2 — колонна с изоляцией из стекловолокна, толщина слоя 50 мм; 3, 4 — колонны, изолированные магнезией (85%), толщина слоя изоляции 25,4 мм (3) и 50,8 мм (4).
1,2 — данные Роуза [158]; 3, 4 — данные
Юнга [233].
в первую очередь материалы с очень низкой теплопроводностью (см. разд. 7.7), например магнезию, асбест, минеральную вату и стеклянную шерсть [1531. Однако это не всегда полностью достигается (табл. 32).
Юнге [233 ] провел измерения тепловых потерь в колонне с вакуумированной рубашкой и в колонне с тепловой изоляцией из стекловолокна толщиной 50 мм. Результаты этих исследований в зависимости от температуры внутри колонны приведены на рис. 111; на рисунке представлены также данные Роуза [153], взятые из табл. 32.
* Тепловыми потерями в ректификационных колоннах никак нельзя пренебрегать при балансовых опытах. «Дикая» флегма, вызванная тепловыми потерями в колонне, может быть исключена лишь в том случае, если дополнительно обогревать изоляционный кожух. Только адиабатически работающая колонна может давать наилучшие и воспроизводимые результаты.
Тепловые потери в теплоизолированной колонне можно рассчитать по следующей формуле:
'к-2лНкЫ Qn=z 2,3 lg (</H3/rfK)
где К — теплопроводность теплоизоляционного материала, ккал/(м-ч-град); Qn — тепловые потери, ккал/ч; Нк — высота колонны, м; At — разность между Таблица 32.
Тепловые потери в колонне со слоем изоляции из магнезии (85% оксида магнии)
Колонна диаметром 25,4 мм и высотой 30,5 см
(155)
Перегоняемое вещество Разность температур /760 — 25, кип °C Толщина изолирующего слоя, 25,4 мм Толщина изолирующего слоя 50,8 мм
тепловые потери 1 «дикая» флегма, мл/ч тепловые потери «дикая» флегма, мл/ч
кал/ч кал/см2 кал/ч кал/см2
Бензол 55 4 800 20 54 3450 14,3 42
Толуол 85 7 600 31,6 101 5400 22,4 73
п-Цимол 151 13 800 57,2 238 9800 40,7 170
178
температурой внутри колонны и температурой окружающей среды; с/из — наружный диаметр тепловой изоляции, м; dK — диаметр колонны, м.
Пример. Нужно рассчитать тепловые потери в колонне диаметром 25 мм и высотой 5 м. В качестве изоляции использована стеклянная шерсть в виде крошки [X = 0,055 ккал/(м-ч-°C)] толщиной 62,5 мм. Температура внутри колонны составляет 76,6°С, комнатная температура 18°С. По формуле (155) находим:
_ 0,055.6,28-5.58,6 .. , ,
Qn =-----2 31о-6---= 56,5 ккал/ч (156)
Таким образом, несмотря на изоляцию, имеют место тепловые потери (56,5 ккал/ч), которые следует компенсировать дополнительным обогревом во избежание накопления жидкости у стенок колонны.
4.13. ХАРАКТЕРИСТИКА РАЗДЕЛЯЕМЫХ
ВЕЩЕСТВ И ПОЛУЧЕНИЕ КРИВЫХ РАЗГОНКИ
При лабораторной перегонке наряду с температурой кипения определяют также такие характеристики дистиллята, как плотность. показатель преломления, температуры затвердевания и плавления, а иногда также молекулярную массу и иодное число (рис. 112). Точную характеристику продукта можно получить, измеряя несколько Показателей (особенно при аналитических разгонках). Фракционный анализ дистиллята только по температуре кипения в большинстве случаев приводит к ошибочному заключению [234].
В последнее время для идентификации фракций все шире привлекают инфракрасную спектроскопию, масс-спектроскопию, а также газовую хроматографию. Для анализа все чаще применяют проточные приборы, обеспечивающие непрерывность контроля.
Физико-химические константы разделяемых веществ обычно берут из справочников и руководств (см. в [235]), однако к этим данным следует относиться весьма критически. Часто опубликованные данные значительно расходятся между собой. Даже для таких веществ, как бензол и 1,2-дихлорэтан, указанные значения физико-химических констант расходятся между собой на величину, которой нельзя пренебречь. Обычно отдают предпочтение более поздним данным, относительно которых можно предположить, что они определены с максимально возможной точностью. Если на диаграмму разгонки нанести теоретические значения констант чистых веществ (см. рис. 84), то оценка полученных результатов значительно упростится.
К сожалению, в литературе не всегда можно найти все необходимые значения физических констант, и требуется либо пересчитать физические константы для других условий, либо рассчитать их заново.
Фундаментальный труд Рида и Шервуда [235] посвящен физическим свойствам газов и жидкостей и методам их расчета. Методы расчета термодинамических параметров газов и жидкостей весьма удачно систематизированы также и в книге Хехта с сотр. [236 ].
179
Показатели преломления в разных справочниках часто указываются для различных температур, поэтому не исключена возможность применения интерполяции. Для членов гомологического ряда веществ графическая зависимость показателя преломления от числа атомов углерода для различных температур, как показал Штаге [237], имеет вид равносторонней гиперболы. Используя эту закономерность, можно корректировать соответствующие диаграммы (рис. 113). Следует учитывать также, что отклонение температуры на 1 °C приводит к изменению показателя преломле; ния в среднем на 5 единиц в четвертом знаке после запятой, поэтому необходимо очень точно регулировать температуру в ячейке рефрактометра, по возможности с точностью до 0,1 °C. Очень удоб-
Рис. 112.
Диаграммы разгонки бензиновой фракции.
180
1&30
1,kW
е W
1,390
1,380
1,310 .
10 15 20 25 30
пс
Рис. ИЗ.
Зависимость показателя преломления карбоновых кислот нормального строения от числа атомов углерода для различных температур.
ные номограммы для расчета различных физических констант чистых веществ приведены в работах Харбанда [2381, а также Орличека и Пелля [239].
При проведении перегонки при атмосферном давлении необходимо постоянно следить за барометрическим давлением. Отклонения порядка 20 мм рт. ст. могут привести, например, к из
менению температуры кипения бензола на 1 °C. Даже если рабочий барометр точно откалиброван по прецизионному барометру, необходимо вводить дополнительную поправку, учитывающую температурную деформацию столбика ртути и шкалы. С помощью этой поправки результаты измерений приводят к 0 °C, так как согласно определению единица давления 1 мм рт. ст. соответствует температуре 0 °C. Номограмма Хойсслера (240] позволяет без дополнительных расчетов сразу определить скорректированное значение барометрического давления (рис. 114). Номограмма основана на зависимости:
6кор=6[1 — (а — ₽Н1 (мм рт. ст.) (157)
где 6кор — скорректированное барометрическое давление; Ь — отсчитанное барометрическое давление; а — коэффициент линейного расширения ртути (0,000182 на 1°С); Р — коэффициент линейного расширения шкалы (для латуни — 0,000019, для стекла — 0,000008); t — комнатная температура, °C.
В соответствии с правилами калибровки стеклянных термометров (см. разд. 8.2.1) термометр погружают в калибровочную жидкость до деления шкалы, соответствующего измеряемой температуре. Если столбик ртути не погружен полностью в жидкость или пар, температуру которых измеряют, то следует внести поправку на выступающий столбик ртути. Для измерения средней температуры выступающего участка ртутного столбика рядом : с рабочим термометром устанавливают вспомогательный термометр. Скорректированные показания рабочего термометра определяют по формуле:
| ^кор = 1 + ka {t — ^ср) (158)
[где t — показание рабочего термометра; k — кажущийся коэффициент объемного расширения ртути в стекле (для термометров из йенского стекла № 16 k = Р= 0,000157, а йенского стекла № 2954 k = 0,000163); а — длина выступающего ктолбика ртути, выраженная в градусах; <Ср — средняя температура выступаю-|цего столбика ртути, показываемая вспомогательным термометром.
181
Номограмма Коуля [2411 построена на основе зависимости (158) при k = 0,000156. Применительно к перегонке при атмосферном давлении номограмма дополнительно позволяет определить температуру кипения точно при 760 мм рт. ст. (обычно для удобства сравнения результаты пересчитывают относительно этого давления).
Корректировка температуры кипения базируется на зависимости:
= (Ь — 760) dt/dp (159)
где dtldp — производная температуры кипения от рабочего давления в мм рт. ст. (зависит от природы вещества). .
Комбинирование уравнений (158) и (159) приводит к соотношению, позволяющему пересчитывать температуру кипения для 760 мм рт. ст.
К =- [ka (t — t0) ] — (b — 760) dt/dp (160)
Номограмма, приведенная на рис. 115, облегчает приведенный выше расчет. Этой номограммой пользуются следующим образом.
Сначала вычисляют t — t0, затем полученную на шкале А точку соединяют с соответствующей точкой на шкале а. Затем точку пересечения на шкале Б соединяют со значением барометри-
Рис. 114.
Номограмма Хойсслера для корректировки показаний барометра.
182
Рис. 115.
Номограмма Коуля для корректировки показаний термометра.
ческого давления (в мм рт. ст.) на шкале В. Точка пересечения последней прямой со шкалой dtldp дает скорректированное значение t, соответствующее полному погружению в жидкость термометра и давлению 760 мм рт. ст.
Пример. Допустим, что при перегонке под атмосферным давлением температура в головке колонны соответствует точке кипения фенола и составляет t = = 179,8 °C. Средняя температура выступающего ртутного столбика оказалась равной /ср = 30°С (она измерена вспомогательным термометром, шарик которого подведен к середине непогруженного участка ртутного столбика). Длина выступающего столбика соответствует а = 40 °C, барометрическое давление 750 мм рт. ст.; для фенола dtldp = 0,045.
Находим t— tcp— 149,8 °C. Соединим точку 150 на шкале А с точкой 40 на наклонной шкале а, и на шкале Б получим точку пересечения 0,95. Это и есть поправка на столбик ртути. Эту точку соединяем с точкой 750 на шкале В и получаем окончательную поправку ~Н,4°С в точке пересечения линии 0,045 шкалы dtldp с прямой, соединяющей шкалы Б и В. Таким образом, скорректированная температура равна (179,8 °C + 1,4 °C) = 181,2 °C (см. рис. 115).
Аналогичную методику для уточнения температур кипения 1ри 760 мм рт. ст. предлагает Рекхард [57], который определил
183
Выход дистиллята, °/q
Рис. 116.
Ступенчатая диаграмма непрерывной разгонки сырой смеси жирных кислот.
6
Mff 8 10 1?. /4 1618 20
пс
Рис. 117.
Столбиковая диаграмма распределения постоянно кипящих фракций по числу атомов углерода для сырой смесн жирных кислот.
групповые константы с в уравнении (18) (разд. 4.4) для 400 органических веществ. Данный способ применим также и к многокомпонентным смесям.
Для записи значений измеряемых величин в лабораторных условиях очень удобно пользоваться печатными бланками (карты опытов). С помощью записей в этих бланках значительно упрощается построение диаграммы разгонки. Диаграмму разгонки можно строить либо по возрастанию выхода дистиллята (см. ниже рис. 118), либо по возрастанию температуры (см. рис. 84, 112). В то время как первый способ еще применяют в промышленности для анализа многокомпонентных смесей (нефть, бензин), все большее распространение получает второй способ. Согласно этому способу на оси ординат откладывают значения температуры кипения, а на оси абсцисс — выход дистиллята. Для повышения точности отсчета шкалу температур обычно строят в более крупном масштабе, чем шкалу выхода дистиллята. Целесообразно также (если при этом не снижается наглядность) наносить на диаграмму разгонки все измеренные величины, в том числе температуру куба, и кривую изменения рабочего давления. После того как на основании результатов измерений идентифицированы определенные фракции, на диаграмме для большей наглядности проводят утолщенные вертикальные линии, разграничивающие отдельные фракции. На рис. 84 приведен пример разделения головного погона жирных кислот в результате ректификации с большим числом тео
184
ретических ступеней при 20 мм рт. ст. Для бинарных смесей удобна также такая форма диаграммы, для построения которой на оси у откладывают значения концентрации дистиллята, а на оси х — выход дистиллята (см. рис. 90).
Для оценки результатов непрерывной ректификации, в частности смесей гомологов, большую пользу может оказать построение ступенчатой (идеализированной) диаграммы [242] (рис. 116). На ее основе можно построить столбиковую диаграмму фракционного состава смеси (рис. 117). Наглядные симметричные диаграммы Майер—Грольмана и Веселовского [243] также позволяют быстро проанализировать результаты разделения, полученные при разгонке по Энглеру или при аналитической ректификации многокомпонентных смесей, например моторных топлив, сланцевых масел, смол. По этому методу на ось у наносят значения температуры кипения, а на ось х (вправо и влево от оси у) — выход дистиллята в процентах от общего количества по фракциям, укладывающимся в температурные интервалы не уже 10 °C. На диаграмме получают площади (их обычно заштриховывают), напоминающие по форме репу или луковицу и дающие наглядное представление о результатах разгонки. Дополнительно слева на симметричной диаграмме приводят ряд чисел, соответствующих количествам дистиллята (в %), которые были получены с момента начала разгонки до определенной температуры; справа на диаграмме наносят числа, показывающие выход дистиллята (в %) для определенных температурных интервалов. На диаграмме разгонки (рис. 118, б) значение 180 °C соответствует верхнему температурному пределу бензиновой фракции, а 325 °C — верхнему температурному пределу фракции среднего масла.
Рис. 119-
Симметричная диаграмма разгонки обычного (а) и специального (б) дизель-юго топлива.
185
На рис. 118 дано сравнение обычной и симметричной диаграмм разгонки для сланцевого масла.
Основные преимущества симметричной диаграммы разгонки заключаются в том, что эти диаграммы позволяют быстро сравнивать количество кубового остатка (27%) с общим количеством дистиллята и по ним легко установить температуру, при которой начинается термическое разложение (по утолщению площади диаграммы при температуре около 320 °C). Кроме того, путем наложения диаграмм, нанесенных на кальку, можно быстро сравнить результаты различных разгонок.
Даже рядом изображенные симметричные диаграммы площадей, построенные для различных разгонок, дают быстрый и надежный способ сравнения полученных результатов. На рис. 119 сравниваются две симметричные диаграммы разгонки для обычного и специального дизельного топлива.
4.14. ПРИМЕРНАЯ ПОСЛЕДОВАТЕЛЬНОСТЬ
РАСЧЕТА ПРОЦЕССА РАЗДЕЛЕНИЯ
Ниже рассмотрена рабочая схема решения задач разделения, приведенная также в табл. 4.
4.14.1. ПЕРИОДИЧЕСКАЯ РЕКТИФИКАЦИЯ
ПРИ АТМОСФЕРНОМ ДАВЛЕНИИ
В лабораторию регулярно поступают относительно большие количества смеси (около 10 л в неделю) бензол — н-гептан, которую нужно разделить методом периодической ректификации. Дистиллят должен содержать 99% (мол.) бензола, а кубовый остаток не более 2% (мол.) бензола.
Характеристика компонентов разделяемой смеси:
Бензол н-Гептан
Содержание, % (мол.) . . 60 40
Молекулярная масса . . 78,11 100,20
у760 °C ^КИП’ . . 80,1 98,42
Плотность (при 20 °C), г/см3 . . . . . 0,87895 0,68368
Относительная летучесть 1,77
1. Предварительная оценка необходимого числа теоретических ступеней разделения. При атмосферном давлении разность температур кипения компонентов смеси равна 18,3 °C. При условии идеального поведения смеси необходимо либо 9 теоретических ступеней разделения (расчет по диаграмме на рис. 72), либо 16 теоретических ступеней [расчет по формуле (96) ]. Как показано ниже в п. 3, исходная смесь не является идеальной, поэтому необходимое число теоретических ступеней разделения должно быть увеличено.
186
Пример ясно показывает, что по разности температур кипения можно получить только грубо приближенную оценку для п.
2. Рабочее давление разгонки. Разделение целесообразно проводить при атмосферном давлении, поэтому отпадает необходимость выбора оптимального рабочего давления ректификации. Следует только учесть, что температура кипения должна быть скорректирована относительно давления 760 мм рт. ст. (см. рис. 114, 115); при этом необходимо внести поправку на выступающий ртутный столбик.
3. Расчет числа теоретических ступеней разделения и флегмового числа. Кривая равновесия системы бензол — н-гептан известна (см. табл. 26, разд. 4.10.3), она асимптотически приближается к диагонали [при увеличении концентрации до 100% (мол.) 1.
Возьмем за основу величины, указанные в разд. 4.10.3, и построим кривую равновесия [диаграмма размером 50x50 см с делениями в % (мол.)].
С помощью диаграммы равновесия пар — жидкость определим условия ректификации, как это описано в разд. 4.7.1. Рабочая линия процесса ректификации для укрепляющей части колонны [хв = 60% (мол.) и хЕ = 99% (мол.)] пересекает ось ординат в точке у = 32. Из (78) следует
/мнн = 99/32 — 1 = 2,09
Выберем рабочее флегмовое число v = 9, и по (75) получим
У =99/10 =9,9
Рабочая линия, проведенная через у = 9,9 и хЕ = 99,0 для рабочего флегмового числа v = 9, служит базой для графического изображения ступеней разделения между этой линией и кривой равновесия. Для начала разгонки находим число теоретических ступеней п = 25.
Концу разгонки соответствует дистиллят состава хЕ = 99% (мол.) и кубовая жидкость концентрацией 2% (мол.). При этих условиях рабочая линия пересекает ось ординат в точке у = 3, поэтому
^мнн = 99/3 — 1 = 32
Определяем, что этому рабочему числу соответствует 25 теоретических ступеней; берем за основу v = 50, откуда получаем у — 99/51 =1,94
Соединим точки у = 1,94 и хЕ = 99,0. Между новой рабочей линей и кривой равновесия строим ступенчатую линию. Как и сле-овало ожидать, получаем п = 25 теоретических ступеней.
Таким образом, получены следующие параметры ректификации: исло теоретических ступеней разделения п = 25; флегмовое :исло в начале ректификации v = 9; флегмовое число в конце ектификации v = 50.
187
4. Определение нагрузки, количества дистиллята и времени установления рабочего режима. Прежде всего пересчитаем концентрацию бензола из % (мол.) в % (об.) (см. разд. 3.4) по формуле (15):
78,11-06.0,879
78,11.0,6-0,879+ 100,2.0,4-0,684 “ /о ^°°о
Таким образом, около половины загрузки куба останется в кубовом остатке. Объем жидкости, соответствующий удерживающей способности (УС) колонны (около 100 мл), следует отобрать как промежуточную фракцию, и при следующей разгонке ее загружают в колбу вместе с исходной смесью.
Если требуется получать 200 мл/ч дистиллята, то при рабочем флегмовом числе v — 9, соответствующем началу разгонки, нагрузка колонны составит 2000 мл/ч.
Если нагрузку колонны постоянно поддерживать на уровне 2000 мл/ч, то в конце разгонки при флегмовом числе v = 50 можно отбирать только 2000/51 40 мл/ч дистиллята. Для отбора
дистиллята в конце разгонки со скоростью 200 мл/ч потребовалось бы увеличить нагрузку колонны до 200-51 = 10 200 мл/ч. На практике, это, конечно, неосуществимо, так как при повышении нагрузки в 5 раз колонна не обеспечит требуемой эффективности разделения, соответствующей 25 теоретическим ступеням. При постоянной же нагрузке по мере обеднения кубовой жидкости легколетучим компонентом флегмовое число нужно постепенно увеличивать с 9 до 50.
Параметры периодической разгонки, обеспечивающие заданное разделение, можно рассчитать по уравнению Биллета [135]. Метод основан на столь громоздких вычислениях, что для ознакомления с ним следует обратиться к оригиналу.
В заключение определим, какой порядок имеет время установления стационарного режима, т. е. время, в течение которого устанавливается состав дистиллята, равный 99% (мол.) (см. разд. 4.8.3). Для этого нужно знать общую УС колонны. Для приближенной оценки (см. разд. 4.10.5) можем принять, что в насадочных колоннах УС составляет 3 мл на одну теоретическую ступень разделения. Примем, что общая УС колонны равна 75 мл.
По формуле (123) вычисляем фактор интенсивности массообмена (R = 2000/3600 = 0,55 мл/с):
I =25-0,55/75 =0,18 с’1
Подставив найденное значение в формулу (124), получим приближенное значение пускового времени для начальных условий ректификации:
тп =26а/(0,18-2) =676/0,36 = 1877 с « 30 мин
Таким образом получаем (при условии достаточного подвода тепла в кубе): нагрузка колонны — 2000 мл/ч; скорость отбора дистиллята в начале разгонки при v = 9 — 200 мл/ч н пусковое время в начале разгонки — 30 мин.
188
5. Расчет размеров колонны. Для расчета размеров колонны, обеспечивающей нагрузку 2 л/ч, прежде всего необходимо знать скорость потока паров, предельную для выбираемой насадки. Пусть нужно использовать насадку из фарфоровых седел размером 4 мм. Ход расчета предельной скорости паров подробно описан в разд. 4.11. Приведенная плотность паров составляет:
РЛ=Рп/Рпс=2>7/87«,95 = 3,07-Ю-з
Величину рп рассчитывают j по формуле (32):
рп = (78,11 -760/353,35) 0,01605 = 2,7 кг/м3 * * 6
Пользуясь номограммой, приведенной на рис. 109, находим, что предельная скорость паров дапр для заданных условий при значении константы k = 0,019 (для седел) составит 0,49 м/с. Она может быть вычислена по формуле (136):
аупр = КО,019-0,004-9,81/0,00307 = 0,49 м/с
Исходя из того, что рабочая скорость паров составляет 60% от предельной скорости, находим, что дапр = 0,3 м/с (или 30 см/с). Далее по формуле (141) вычисляем необходимую площадь поперечного сечения колонны:
/=0,0228-22,5-353,35/30= 6,0 см2
Нагрузка колонны равна D = 2000 мл/ч = 1758 г/ч = 22,5 моль/ч.
Окончательно устанавливаем, что диаметр колонны равен 30 мм (/ = 7,06 см2), при этом удельная нагрузка составит 285 мл/(см2-ч). Для седел размером 4 мм при указанной удельной нагрузке можно принять, что ВЭТС = 8 см (см. табл. 28Д). Таким образом, колонна должна иметь следующие размеры:
Внутренний диаметр ..................... 30 мм
Рабочая высота ....................... 2000 мм
Общая высота (включая стандартные шлифы) ............................... 2150 мм
Методика тепловых расчетов приведена в разд. 4.12. Количество тепла, необходимое для нагревания смеси до точки кипения,
вычисляют по формуле (149), а для испарения дистиллята и флегмы — по формуле (151). Поверхность теплообмена в конденсаторе
рассчитывают по уравнению (191).
Отводимое из конденсатора тепло можно определить по формуле (155); пример из разд. 4.12 поясняет ее использование.
6. Испытание колонны на эффективность (см. разд. 4.10.8) нельзя провести с применением разделяемой смеси. Даже если содержание легколетучего компонента в кубе довести до 2%
(мол.), для обогащения дистиллята до 99% (мол.) при v = оо потребуется всего только 18 теоретических ступеней разделения. Испытание обычно проводят, используя смесь н-гептан — мети лциклогексан при нагрузке 2 л/ч. Если в этих условиях не бу-
189
дет достигнуто 25 теоретических ступеней разделения, то высоту колонны следует соответственно увеличить. Значение ВЭТС, равное 8 см для седел размером 4 мм, при удельной нагрузке 285 мл/(см2-ч), выбрано только ориентировочно.
Установка должна быть хорошо изолирована. Колонну снабжают заранее рассчитанным компенсационным обогревательным устройством (см. разд. 7.7.3). Применение автоматической головки колонны (см. разд. 7.5.3) позволяет проводить перегонку почти без наблюдения за установкой. В головке колонны целесообразно установить контактный термометр, связанный через реле с сигнальным звонком (см. разд. 8.2.2). Контактный термометр настраивают на температуру кипения бензола. Как только последует сигнал, начальное флегмовое число, равное 9, следует увеличить до 20. Точно так же флегмовое число увеличивают до 50.
Нагрузку целесообразно регулировать по гидравлическому сопротивлению с помощью контактного манометра (см. разд. 8.4.2). В зависимости от количества исходной смеси в качестве куба можно использовать подвесной сосуд из йенского стекла (см. разд. 7.6.1) или круглодонную колбу емкостью от 4 до 10 л, обогреваемую с помощью колбонагревателя (см. разд. 7.7.1).
О других возможностях автоматизации сообщается в гл. 8; о сборке установок см. также гл. 9.
4.14.2. НЕПРЕРЫВНАЯ РЕКТИФИКАЦИЯ
Способы расчета процесса непрерывной ректификации изложены в разд. 4.7.2 [пример — разделение смеси жирных кислот Св—С7 состава 81% (мол.) Св при 20 мм рт. ст., хЕ = 96%, хА ~ 0,5% ] и в гл. 9 (пример — разделение многокомпонентной смеси жирных кислот С*—С8 при 20 мм рт. ст.). В разд. 5.2.2 описана аппаратура и способы проведения непрерывной ректификации.
4.14.3. ВАКУУМНАЯ ПЕРЕГОНКА
Расчет вакуумной ректификации следует в том же порядке, что и атмосферной ректификации. Дополнительно в расчет условий процесса вакуумной перегонки входит: 1) выбор оптимального рабочего давления разгонки (разд. 4.6.2 и 4.10.6); 2) построение кривой равновесия идеальной смеси для заданного рабочего давления (разд. 4.6.1, табл. 10); 3) определение зависимости объема паров от давления (разд. 4.11); 4) расчет диаметра труб и герметичности аппаратуры (разд. 5.4.1); 5) измерение и регулирование давления (разд. 8.3).
4.15. ИСПОЛЬЗОВАНИЕ ЭВМ ПРИ РАСЧЕТЕ КОЛОНН
Применение электронных вычислительных устройств в практике лабораторной и пилотной ректификации несомненно эффективно, промышленная же ректификация с ее разнообразными проблемами регулирования сегодня уже не мыслима без использования вычислительной техники. Благодаря автоматизации технологических процессов в настоящее время появилась возможность управлять многостадийными процессами разделения. Важнейшие
190
критерии для сравнения различных управляющих машин при автоматизации лабораторных установок рассмотрены Блаттом и Фляйс-снером [259].
Различают цифровые вычислительные машины (ЦВМ) и аналоговые вычислительные машины (АВМ), а также их комбинацию в виде гибридной вычислительной системы [2481. ЦВМ выдает результаты в виде цифровых таблиц, в А^ВМ результаты выводятся в виде изменяющихся во времени напряжений, которые различными способами могут быть изображены в графической форме. Подробные сведения о методах математической обработки с помощью ЭВМ можно найти в специальной литературе [248, 249].
Практически все технические расчеты процесса перегонки можно проводить с помощью ЭВМ. Уже применение настольных вычислительных машин дает возможность определить давление паров, параметры фазового равновесия и оптимальные условия для получения максимального числа теоретических ступеней разделения, а также рассчитывать флегмовые числа. Применение ЭВМ типа IBM 7040, например при исследовании работы насадочных колонн в переходном режиме, обеспечивает выигрыш во времени благодаря быстрому определению состава вещества (в мольных долях) на основе данных по плотности и показателям преломления [162].
Разработано большое число программ для расчета термодинамических параметров систем, состоящих из нескольких фаз [109]. Для расчета фазового равновесия многокомпонентных смесей предложена обширная программа на языке Фортран [891.
Процессы дробной перегонки (дистилляция по Энглеру, аналитические разгонки по методу Американского общества по испытанию материалов, см. разд. 7.2), применяющиеся, главным образом, для контроля качества продукции, сопряжены, как показал Рот [250], с очень сложными термодинамическими закономерностями. Соответствующую математическую модель процесса можно рассчитать, используя, например, ЦВМ типа ZRA 1.
Приняв за основу метод Мак-Кэба и Тиле, Шубринг [2511 разработал программу расчета процесса разделения бинарных смесей, предназначенную для ЭВМ IBM 705. Допустив вполне практически приемлемые упрощения, в соответствии с которыми мольная энтальпия испарения не зависит от состава смеси, а энтальпия растворения равна нулю, с помощью этой программы можно рассчитать число теоретических ступеней разделения для двухкомпонентных смесей, как идеальных, так и неидеальных, в том числе азеотропных смесей. С помощью перфокарт в имеющуюся программу закладывают данные по равновесию, концентрации питающей жидкости, дистиллята и кубовой жидкости. Время решения одной задачи составляет от 15 с до 5 мин. Напечатанная таблица, полученная на ЭВМ, дает для каждой задачи последовательность возможных значений числа теоретических ступеней разделения в зависимости от флегмового числа или от
191
Рис. 120.
Упрощенная схема, поясняющая процедуру расчета числа теоретических ступеней разделения на ЭВМ IBM 705 (по данным Шубринга [251]).
отношения количества образующихся паров к количеству кубового продукта. Упрощенная блок-схема, представленная на рис. 120, поясняет ход расчета, а на рис. 121 приведена итоговая диаграмма, показывающая зависимость числа теоретических ступеней от флегмового числа для непрерывного разделения бинарной смеси. Применяя метод номограмм для отражения взаимосвязи между и vn, можно на одной и той же диаграмме представить зависящие друг от друга параметры укрепляющей и исчерпывающей частей колонны [2511.
Силей [252] применил ЭВМ для расчета оптимальных параметров лабораторной колонны Олдершоу диаметром 31,8 мм с 12 реальными тарелками в исчерпывающей части и 10 реальными тарелками в укрепляющей части. На ЭВМ Electric KDF 7 (Англия) были проанализированы 280 процессов разделения. В качестве эталонной смеси использовали смесь метилциклогексан— толуол. При этом за расчетное число теоретических ступеней разделения принимали то значение, которому соответствовала минимальная погрешность. Были изучены возможные погрешности, возникающие при измерении состава смеси, при определении положения и наклона рабочей линии, а также погрешность данных по равновесию.
Рис. 122 иллюстрирует изменение общей относительной погрешности для интервала флегмовых чисел 1,0—1,3 в исчерпыва-192
ющей части колонны. Форма кривых для укрепляющей части колонны аналогична.
Эти кривые располагаются на графике тем выше, чем больше флегмовое число; наименьшие значения п, при которых кривые определены, изменяются от 1,0 до 0,7.
Использование ЭВМ необходимо при точном расчете процесса разделения многокомпонентных смесей в ректификационных колоннах [264]. О возможности математического моделирования предварительной разгонки сырой нефти сообщают Киршбах, Френ-цель и Штранкмюллер [265]. Ляйбниц и Шулер [253] описывают процедуру расчета процесса разделения идеальных многокомпонентных смесей при использовании ЭВМ ZRA 1. Вопросы расчета фазового равновесия в неидеальных многокомпонентных растворах рассмотрены Нагелем с сотр. [254]. Аналоговые вычислительные машины широко применяют при технологических расчетах процессов массо- и теплопередачи [255]. В последние годы были усовершенствованы, главным образом, те методы, которые невозможно реализовать без помощи ЭВМ [6]. Имеются в виду прежде всего так называемые матричные методы расчета Ванга и Хенке [256], Саржента и Мертага [257], а также Стэйнт-хорпа с сотр. [258].
Рис. 121.
Итоговая диаграмма разделения (251].
Условия процесса: а = 1,5; Xg ~ 99,9% (масс.); х% = 65%, х& — 0,1%
Рис. 122.
Относительная суммарная погрешность определения числа теоретических ступеней в укрепляющей части колонны прн разных флегмовых числах Эталонная смесь метилциклогексан—толуол.
7 Э. Крель
193
При исследованиях гидравлического сопротивлёйий, а Также массо- и теплопередачи в неподвижных слоях насадки Брауэр и Мьюс [260] разработали программу на алгоритмическом языке Фортран, предназначенную для расчета перепада давления и высоты слоя с учетом структуры насыпной насадки.
Вагнер и Бласс [262 ] описывают метод расчета процесса ректификации в тарельчатой колонне, реализуемый на гибридной ЭВМ; в качестве примера для расчета выбрана система дихлорэтан—толуол. Метод позволяет рассчитать изменение во времени составов дистиллята и кубовой жидкости при разделении бинарных смесей по заданным рабочим параметрам процесса при известной кривой равновесия.
Теоретические и практические вопросы, связанные с исследованием динамических характеристик промышленных ректификационных колонн, рассмотрены Кёлером и Шобером [264].
5
Обычные методы перегонки
После проведения ориентировочных расчетов процесса разделения решается задача его технологического и аппаратурного оформления. При выборе рационального метода перегонки руководствуются такими показателями, как имеющееся количество исходного сырья, желаемая производительность колонны и качество дистиллята. При этом следует учитывать физико-химические свойства разделяемых веществ, а также аппаратурные и энергетические возможности.
5.1. КЛАССИФИКАЦИЯ МЕТОДОВ ПЕРЕГОНКИ ПО КОЛИЧЕСТВУ
ПЕРЕРАБАТЫВАЕМОГО ПРОДУКТА
Размеры дистилляционной и ректификационной колонны должны находиться в точном соответствии с количеством перерабатываемого вещества. При необходимости можно, конечно, большое количество сырья перерабатывать и в небольшой колонне, затрачивая на это много времени; в другом крайнем случае при проведении процесса в слишком большой установке увеличиваются потери продукта и кубовые отходы. В лабораториях часто требуется разделять смеси в количествах, измеряемых миллиграммами, и наоборот, необходимо перерабатывать от 5 до 10 кг/ч сырья с применением полупромышленных методов. В связи с проблемами масштабного перехода от малых аппаратов к большим все большее значение приобретают исследования процессов дистилляции и ректификации на пилотных установках.
5.1.1. МИКРО- И ПОЛУМИКРОПЕРЕГОНКА
В препаративной микротехнике обычно работают с количествами вещества менее 0,1 г. Разделение смеси при загрузке до 5 мл методами перегонки целесообразно отнести к области микротехники, а до 50 мл — к области полумикроперегонки. При этом следует принципиально различать дистилляцию (простую перегонку), фракционную перегонку и ректификацию. Штаге и Гем-мекер [1 ] указывают на эффективность разделения смесей в микроколичествах в совокупности методами перегонки и газовой хроматографии. Для более полного использования газовых хрома-
Т
195
Рис. 123.
Прибор Боденхаймера для микро дистилляции:
1 — конденсационная трубка; 2 — насадка из стеклянного порошка; 3 — капилляр с пробой жидкости; 4 — нагревательный блок с термометрами.
Рис. 124.
Горизонтальная колонка Янцеиа для микроректификации.
тографических методов часто возникает необходимость в предварительном разделении смесей путем перегонки. Авторы дают превосходный обзор по ректификационным колоннам различных типов, обсуждают конструктивные усовершенствования, а также рассматривают средства автоматизации процесса разделения. Штаге [1а] подробно описывает современные приборы для микроперегонки.
Для дистилляции веществ в количестве от нескольких миллиграмм до 1,0 г применяют дистилляционные капилляры, трубки с шарообразными расширениями и маленькие колбы. Колонны с очень низкой удерживающей способностью по жидкости, например полые колонны, колонны из концентрических трубок (щелевые трубчатые колонны) и колонны с вращающейся лентой применяют для разделения 1—25 мл вещества; при объеме исходной смеси от 25 до 50 мл можно также использовать насадочные и тарельчатые колонны.
С помощью прибора, описанного Боденхаймером [2], можно проводить фракционную перегонку нескольких миллиграмм жидкой смеси (рис. 123) и одновременно измерять температуру кипения смеси с точностью 1—2 °C. Трубка 1, один конец которой сужен до диаметра 1 мм, помещена в нагревательный блок 4. Температуру измеряют двумя стеклянными термометрами. Исследуемую пробу жидкости помещают в запаянный конец капилляра 3, остальной объем капилляра 3 заполняют стеклянным порошком 2. Выступающая из нагревательного блока расширенная часть трубки 1 имеет шлифованную изнутри поверхность молочно-белого цвета.
Температуру нагревательного блока равномерно увеличивают со скоростью 4—6 °С/мин. В момент приближения к точке кипения на стенках конденсационной трубки наблюдается запотевание, и при достижении точки кипения стекло в этом месте стано-196
вится прозрачным. По длине смоченного (прозрачного) участка можно оценить количество дистиллята, если перед этим провести калибровку с эталонными веществами. Чтобы выделить более мелкие фракции, нагревают то место трубки, где сконденсировалась первая фракция, при этом ее пары конденсируются в трубке 1 на большем расстоянии от нагревательного блока, для чего конец трубки охлаждают влажной фильтровальной бумагой.
Горизонтальный ректификатор Янцена (рис. 124) позволяет проводить ректификацию образцов массой от 1 до 100 мг. Смесь загружают в капиллярную трубку длиной 1200 мм и диаметром 5,5—5,7 мм [3]. В нагревательном цилиндрическом кожухе создают Перепад температур в 30 °C и с помощью тока водорода обеспечивают перенос вещества в направлении понижения температуры. При этом легколетучие компоненты перемещаются дальше, чем труднолетучие. Прибор предназначен специально для разделения высококипящих веществ. Рис. 125 иллюстрирует результаты разделения смеси додекановой и тетрадекановой кислот. Фишер [За 1 описывает новый усовершенствованный автоматический прибор такого типа.
Для разделения смеси в количествах от 0,05 до 0,5 мл обычно применяют трубку с шарообразным расширением (например, как на рис. 126). Эту узкую (диаметром 5—7 мм) тонкостенную трубку заполняют следующим образом. Шарик нагревают, и после охлаждения разделяемая жидкость благодаря образовавшемуся разрежению втягивается в трубку; жидкость можно загружать также с помощью капиллярной пипетки. Техника перегонки в такой трубке, особенно способом обогрева, подробно описана Бертеле и Хумелем [36].
56
52
48
44
40
10 20 30 40 50 60 70
Длина трубни, см
Рис. 125.
Результат разделения смеси додекановой и тетрадекаиовой кислот в го* ризоитальной колонке Янцеиа [3 ].
Рис. 126.
Прибор Бабкока для микродистилляции под вакуумом из трубки с расширениями.
197
Рис. 127.
Прибор для микроперегонки 00100.
Прибор имеет перегонную микроколбу емкостью 5 мл и приемные пробирки емкостью 6 мл (6 шт.) или 2 мл (9 шт.).
Изготовитель: фирма Юхайм, Золингген.
Рис. 128.
Прибор Креля для мнкроперегонки (от 1 до 10 мл смеси) при атмосферном давлении и под вакуумом:
/ — капельницы; 2 — проволочные сетки.
Изготовитель: народное предприятие «Комбинат технического стекла», Ильмен ау.
В микромасштабе можно проводить даже вакуумную перегонку. На рис. 126 показан прибор Бабкока [4], с помощью которого получают три фракции 1, 2 и 3. Чтобы собрать некоторое количество дистиллята, не прерывая процесса перегонки, в микроприборах применяют поворотные вакуумные приемники (рис. 127).
Любое количество фракций дистиллята можно отобрать при микроперегонке в приборе Креля (рис. 128). Этот прибор применяют для перегонки как при атмосферном давлении, так и под вакуумом. Его можно видоизменять в зависимости от количества исходной смеси. Так, при 1—2 мл вещества целесообразно работать с полой трубкой. При необходимости можно вставить сверху в качестве брызгоотбойников одну или две конические проволочные сетки, которые несколько улучшают разделение. Если исходное количество вещества составляет 3—5 мл, то на стеклянной перфорированной перегородке размещают стеклянные кольца Рашига или спирали размером 4x4 мм. При 5—10 мл вещества можно применять спирали размером 2x2 мм, насыпанные на коническую проволочную сетку.
Сначала ректификацию проводят при закрытом кране тонкой регулировки (о = оо) до тех пор, пока в системе не установится постоянная температура. Затем приступают к работе при любой 198
рабочей нагрузке и любом флегмовом числе, применяя капельницы 1 и кран для регулирования этих параметров. Благодаря таким приемам можно получать точные и воспроизводимые результаты анализов малых проб вещества по температурам кипения.
В отличие от полых трубок (см. разд. 7.3.1) колонна Вигре обладает большей орошаемой поверхностью за счет большого числа заостренных глубоких вмятин, кроме того в средней части она имеет ввод для питающей смеси. Шрадер и Рицер [5 ] использовали такую колонну в качестве дефлегматора в приборе для микроперегонки (рис. 129). Он содержит плоскодонную колбу 1, которая обеспечивает постоянную поверхность испарения, и приемник 3 со стеклянными трубками 2 емкостью 0,1 мл. Дистиллят стекает к трубкам 2 по стеклянной нити 4. Разумеется, прибор можно применять только для разделения высококипящих веществ, так как в нем не предусмотрен водяной холодильник. Более вместительный прибор Кленка [6] также имеет только воздушное охлаждение (рис. 130). Колонка длиной 8,5 или 13 см со спиральной металлической лентой длиной 120 мм и вакуумированной рубашкой обеспечивает хорошее разделение при очень малом количестве смеси.
Как насадочные, так и тарельчатые колонны вследствие их значительной удерживающей способности по жидкости можно применять только для проведения полумикроперегонки (см. разд. 4.10.5). Причем следует стремиться к тому, чтобы при
Рис. 129.
Прибор Шрадера—Рицера для микроперегонки с колонкой Вигре: 1 — плоскодонная колба; 2 — стеклянные трубки; 3 — приемник; 4 — стеклянная нить.
Рис. 130.
Прибор Кленка со спиральной вставкой для микроректификации более 4 мл вещества.
Рис. 131.
Прибор Гульда с сотр. для молекулярной микродистилляции по методу «холодного пальца» :
1 — «холодный палец»; 2 — Кольцевой выступ; 3 — выемка; 4 — капилляры; 5 — приемник.
199
Таблица 33.
Параметры колонок для аналитической полумикроректификации
Колонна Внутренний диаметр, мм Длина, см га £ л о, га ч « 2 > Объем фракции, мл 1 Нагрузка, мл/ч Скорость отбора дистиллята, мл/ч Флегмовое число ВЭТС, см УС колонны, мл
Колонна с вращающейся лентой 5 90 100—150 0,5 10—30 0,5—1,5 40 1,1 1,9
Колонна «Гипер-кол» 8 60—90 20—100 0,5— 1,0 20—30 0,5—1,5 40 0,7 48
Колонна с узкой кольцевой щелью 8 60 10—50 0,5 20—30 0,2—0,5 100 0,9 3,2
заданном исходном количестве вещества размеры колонны были бы по мере возможности минимальными. В качестве насадочных тел при полумикроперегонке можно применять спирали размером от 1,6 до 2 мм (см. разд. 7.3.2 и 7.8).
Для тонкого разделения веществ в очень малых количествах более эффективны щелевые трубчатые колонны (см. разд. 7.3.1) и колонны с вращающейся лентой (см. разд. 7.3.5). Колонны обеих разновидностей также широко применяют Для аналитических разгонок и вакуумной микроректификации высококипящих веществ [7].
Винтерс и Динерштайн [8 ] описывают устройство, состоящее из восьми полумикроприборов для аналитической разгонки; устройство имеет центральную измерительную ячейку.
Технические характеристики некоторых приборов для аналитической полумикроперегонки приведены в табл. 33 [8].
Шпигельберг [9] сообщает о разделении эфирных масел в колонне с вращающейся лентой при 4—13 мм рт. ст. Примеры разделения высокомолекулярных органических соединений в щелевой трубчатой колонне приведены Янценом и Витгертом [3], а также Фишером [7].
Молекулярную дистилляцию (см. разд. 5.4.4) в микромасштабе можно проводить либо по методу «холодного пальца», либо по принципу падающей пленки, т. е. в тонком слое. На рис. 131 [10] показан прибор, основанный на методе «холодного пальца». Разделяемую смесь загружают в выемку 3; пары конденсируются на «холодном пальце» 1. Дистиллят скапливается у кольцевого выступа 2, откуда по каплям стекает во вращающийся приемник 5 с капиллярами 4.
Для перегонки веществ в количествах менее 4 г Брегер [11] предложил оригинальный прибор, с наружной поверхностью
200
испарения и внутренней поверхностью охлаждения (рис. 132). Этот небольшой прибор с длиной рабочего участка 185 мм имеет ту особенность, что он может вращаться вокруг горизонтальной оси (проходящей через его середину). Благодаря этому исходную смесь, а также и дистиллят можно многократно (при различной мощности обогрева) испарять из падающей пленки. Перед началом перегонки смесь дегазируют. Прибор Пашке с сотр. [12] предназначен для микроперегонки приблизительно 0,5 г вещества (рис. 133). Исходным веществом пропитывают цилиндрик 1 из стеклянной ваты, который подвешивают на крючке. Прибор вакуумируют, затем включают систему обогрева кожуха 2. Количество отогнанного вещества определяют по шкале 3.
Опыт показывает, что нагревание малых колб при микроперегонке сопряжено с трудностями. В колонне часто наступает захлебывание. Поэтому диаметр колонны должен составлять не менее 6—8 мм. Весьма удобен для нагревания малых колб микроколбо-нагреватель, в котором используется инфракрасное излучение в невидимой части спектра. Применение инфракрасного излучения позволяет без особых затруднений проводить вакуумную ди-
Рис, 132.
Прибор Брегера для молекулярной микродистнлляции меиее 4 г вещества (принцип «падающей пленки»):
1 — участок измерения температуры; 2 — нагревательный кожух; 3 — колба для исходной смеси; 4 — приемник для дистиллята; 5 — конденсатор; 6 ~ обогреваемая поверхность.
Рис. 133.
Прибор Пашке с сотр. для молекулярной микродистилляции 0,5 г вещества: 1 — цилиндрик из стеклянной ваты; 2 — нагревательный кожух; 3 — шкала.
201
стилляцию, так как в этом случае кипение жидкости происходит более равномерно и без «бросков».
Пфайл [13] дает обстоятельный обзор препаративной микротехники, в котором также описаны приборы для микроперегонки. В этой связи следует указать на микровесы Айгенбергера [14], с помощью которых можно сравнительно быстро определить плотность вещества при очень малых количествах пробы. Более простая установка сконструирована Клазеном [15].
5.1.2. АНАЛИТИЧЕСКАЯ РЕКТИФИКАЦИЯ
Цель аналитической ректификации состоит в том, чтобы из двух или многокомпонентной смеси извлечь отдельные компоненты с возможно более высокой степенью чистоты. Степень чистоты устанавливают путем определения физических констант вещества, например коэффициента преломления, плотности, точки затвердевания или плавления, а также молекулярной массы. Так как обычно не известно, какие компоненты и в каких количествах содержатся в разделяемой смеси, то анализ с применением ректификации следует проводить периодическим способом. Для аналитической ректификации применяют колонны с достаточным числом теоретических ступеней разделения (в разд. 4.7—4.12); головка колонны должна обеспечивать точное регулирование нагрузки и флегмового числа. Чтобы получить точное представление о количественном соотношении разделяемых компонентов необходимо, чтобы промежуточная фракция была как можно меньше. Промежуточной фракцией является количество дистиллята, которое отбирают между фракциями сравнительно чистых (или весьма чистых) компонентов. По мере отбора промежуточной фракции в ней постепенно уменьшается содержание легколетучего компонента (см. рис. 56). Количество загрузки выбирают исходя из содержания того компонента, который необходимо выделить и который находится в исходной смеси в минимальном количестве. Далее необходимо стремиться к тому, чтобы отношение
Общая УС , т = _-----------------------------
Доля выделяемого компонента в загрузке
было по возможности минимальным, что позволит уменьшить количество промежуточной фракции. О благоприятном влиянии УС колонны при этом не может быть и речи, поскольку при аналитической ректификации работают с высоким флегмовым числом (см. разд. 4.7.1). Поэтому для аналитических разгонок следует выбирать колонны малого диаметра, так как отрицательное влияние общей УС колонны растет одновременно с увеличением числа теоретических тарелок.
По данным Штаге [16], наименьшая из выделяемых фракций должна иметь объем по меньшей мере в десять раз превышающий общую УС колонны, применяемой для разделения. Это означает, 202
например, что при общей УС, равной 120 мл, объем загрузки должен составлять 12000 мл при выделении в чистом виде компонента, содержание которого в исходной смеси составляет 10%. Это относится обычно только к разделению близкокипящих компонентов, когда требуется очень большое флегмовое число, а степень чистоты выделяемых компонентов приближается к 100%.
Наряду с насадочными колоннами для проведения аналитических работ особенно пригодны высокоэффективные щелевые трубчатые колонны, колонны с вращающейся лентой, а также колонны с регулярной насадкой, обладающие очень низкой общей УС (см. разд. 7.3.1, 7.3.4 и 7.3.5).
При разделении многокомпонентных смесей, когда исходная смесь имеется в достаточном количестве, сырую смесь предварительно разделяют на отдельные фракции путем расширительной дистилляции (см. разд. 5.4.2) или же методом обычной непрерывной ректификации (см. разд. 5.2.2). При этом к эффективности разделения не предъявляют высоких требований, а объем отдельных фракций определяют заранее путем предварительной пробной разгонки (см. разд. 7.2) сырья. Фракции, полученные при грубом разделении, затем дополнительно разделяют на исходные компоненты путем аналитической ректификации, используя то благоприятное обстоятельство, что соотношение т при этом значительно уменьшается.
В качестве примеров практического применения метода аналитической ректификации многокомпонентных смесей следует указать на работы Кюнхансса с сотр. [17] по разделению легкого масла бурого угля, работы Проффта и Бухманна [18] по получению хинолиновых оснований, а также ректификационное разделение смеси синтетических аминов жирного ряда, проведенное Фюрстом и Шветликом [19]. Приборы «Дестинорм» применяют, в частности, при разделении смесей аминовых кислот минерального происхождения путем ректификации сложных метиловых эфиров соответствующих ацетиламиновых кислот [20] и при разделении буроугольных смол для определения их состава [21 ]. Гутвассер и Мюллер [22] разработали специальный пленочный испаритель для фракционной перегонки масляных жирных кислот при остаточном давлении 0,1 мм рт. ст. Мэир с сотр. [23] показал, что рациональным сочетанием обычной перегонки, азеотропной ректификации, адсорбционных методов и метода зонной плавки можно добиться четкого разделения керосина на отдельные компоненты.
Нередко бывает так, что имеющейся исходной смеси недостаточно для выделения из нее в чистом виде компонентов, содержащихся в незначительном количестве. В этих случаях нужно применять колонны, имеющие особенно низкую УС по жидкости, например роторные колонны, и в особенности колонны с вращающейся лентой (см. разд. 7.3.5). При использовании насадочных или тарельчатых колонн за счет повышения флегмового числа
203
при появлении промежуточной фракции добиваются того, чтобы в данных условиях разделения размер этой фракции был по возможности наименьшим. Ситчатые и колпачковые колонны (см. разд. 7.3.3) редко применяют для аналитической ректификации. Однако по сравнению с насадочными колоннами они имеют то преимущество, что для них можно определить градиент концентраций в верхней части колонны путем отбора и анализа проб с верхних тарелок. Например, если концентрация легколетучего компонента на третьей тарелке сверху начинает снижаться, то можно сразу же увеличить флегмовое число. Таким путем добиваются малого размера промежуточных фракций.
При аналитической ректификации используется также следующий способ разделения: сначала концентрируют компоненты в сравнительно большой колонне, затем ректификацией во второй меньшей колонне (с малой УС) получают чистые вещества. Для очень трудных вариантов тонкой аналитической ректификации, когда для разделения требуется более 100 теоретических ступеней разделения, целесообразно применять щелевые трубчатые или многотрубчатые колонны (см. разд. 7.3.1), а также колонны с насадкой из проволочных спиралей или из проволочных сеток (см. разд. 7.3.4). В щелевых трубчатых колоннах при работе в условиях вакуума достигают значений ВЭТС ниже 5 мм [7].
При отсутствии высокоэффективной насадки (ВЭТС ль 2 см) нередко из-за ограниченной высоты помещения возникают трудности с получением необходимого числа теоретических ступеней разделения. В помещении высотой 3,5 м при рациональном конструктивном оформлении установки можно разместить колонну с рабочей высотой около 2,5 м.
В этом случае наилучшие результаты можно получить, если работать с двумя последовательно включенными колоннами, как это показано на рис. 134. Такой прием хорошо себя оправдал, в частности при аналитической ректификации оснований буроугольных смол [241. Дистиллят первой колонны непрерывно подают в нижнюю часть второй колонны; вторая колонна может иметь меньшие размеры. Нагрузки и флегмовые числа для обеих колонн также могут различаться. Необходимо только следить за тем, чтобы количество жидкости в кубе сохранялось постоянным. Этот комбинированный способ работы имеет то преимущество, что вторая ректификация (если она необходима) происходит одновременно с первой. В то время как содержание легколетучего компонента в кубовой жидкости первой колонны значительно уменьшается [примерно с 50 до 1 % (мол.)], концентрацию кубовой жидкости второй колонны поддерживают постоянной в возможно более узких границах [около 90—95% (мол.)], постепенно повышая флегмовое число в первой колонне. Таким образом, во второй колонне поддерживается почти стационарный режим непрерывной ректификации, преимущество которого можно использовать (см. разд. 5.2.2). В кубе второй колонны накаплива-204
ются высококипящие компоненты, так что этим способом можно получить очень маленькую промежуточную фракцию. После прекращения подачи питающей жидкости в первую колонну кубовую жидкость второй колонны можно переработать периодической ректификацией или проанализировать содержание в ней легколетучего компонента.
Для аналитической ректификации следует обязательно применять такую приставку или головку, которая обладает возможно меньшим «мертвым» объемом. Под «мертвым» объемом понимают
Рис. 134.
Схема установки из двух последовательно соединенных колони по нормалям «Дестинорм» .
205
методу деления потока паров или
Рис. 135.
Диаграмма разгонки смеси растворителей: 1 ~ дистилляция по Зиглеру; 2 — пробная ректификация; 3 — четкая ректификация.
количество жидкости, которое задерживается в приставке или в головке колонны и не принимает участия в противоточном массообмене. Автоматические головки колонн, работающие по жидкости (см. разд. 7.5.3), имеют
то важное преимущество, что они практически не создают «мертвые» объемы. Следует также иметь в виду, что возможно загрязнение дистиллята за счет растворения в нем смазки крана. Это обстоятельство обычно не принимается во внимание, хотя может привести к существенным ошибкам. В связи с этим разработаны устройства, обеспечивающие отбор дистиллята без его контакта со смазкой для кранов (см. разд. 4.6.3). В последнее время в автоматических головках колонны (см. разд. 7.5.3) применяют магнитные клапаны, наилучшим образом отвечающие этим требованиям. При аналитической ректификации большое внимание уделяется достижению наиболее высокой степени чистоты выделяемого продукта. В связи с этим следует рассмотреть вопрос о том, что лучше: непрерывно отбирать дистиллят как определенную долю от всего образующегося конденсата или работать с периодическим отбором дистиллята, когда конденсат поочередно либо полностью направляют в колонну как флегму, либо полностью отбирают в качестве дистиллята. В последнем случае флегмовое число определяется отношением времени подаа^^щегм^г^лительности отбора дистиллята. При работе с
средняя концентрация легколетучего компонента в дистиллят^ полученном
^ДДДДДЦ^^^Д/иднак^оыл^усИМш^^^^^^^^^^^» более полное разделений
смеси достигается, когда дистиллят отбирают
а не через каждые 30 с [25 ]. Если нужно поддерживат^^^геевы-' сокие значенг^Фл^^овоп^шсла, как например при ректификации изотопов |ИИИИМЩДДУго по соображениям технического порядка работают (см. разд.
5.1.4). —....... ... ‘ ----
Очевидно, анализы смесей по температурам их кипения можно также выполнять и при разделении их в простых дистилляционных приборах, если разность температур кипения компонентов относительно велика или если нужно определить только температурные пределы кипения. Соответствующая аппаратура описана в разд. 7.2. В этом случае очень трудно получить правильную кривую
206
разгонки, как это иллюстрирует рис. 135 на примере анализа смеси растворителей. Как видно из рисунка, начало кипения смеси по приведенным кривым не совпадает. Это объясняется тем, что при дистилляции по Энглеру (см. разд. 7.2) начало кипения принято регистрировать в момент отрыва первой капли от трубки конденсатора. При проведении же пробной ректификации (рис. 135) с той же первоначальной загрузкой куба сначала работают при бесконечном флегмовом числе до установления стационарного режима. В этом случае значительно точнее улавливается начальный момент кипения, которому в действительности соответствует 36 °C, как это было установлено для данной смеси методом четкой ректификации. При дистилляции по Энглеру кипение начинается при 59 °C, а при пробной ректификации — около 45 °C.
Значительная затрата времени на аналитическую четкую ректификацию (например, 120 ч для разделения нефтяной фракции с интервалом кипения от —30 до +260 °C) послужила стимулом для моделирования процесса ректификации с использованием специальной газовой хроматографической аппаратуры [261. При этом получаются опытные значения концентраций, которые сравнимы с результатами разделения в ректификационной колонне с числом теоретических ступеней разделения 100. Указанным способом можно анализировать как сырые нефти, так и нефтяные фракции соединений с числом атомов в углеродной цепи от 1 до 40. «Прибор для одновременной аэрографии и ректификации» с помощью небольшого встроенного компьютера позволяет получать кривые «температура кипения — концентрация [% (масс.)]». Площади под этими кривыми непрерывно интегрируются и подсчитанные значения через каждые 10 с регистрируются самописцем. На анализ указанной выше нефтяной фракции (от —30 до +260 °C) требуется всего лишь около 1 ч [27].
5.1.3. ПРЕПАРАТИВНАЯ И НАРАБОТОЧНАЯ ПЕРЕГОНКА
После идентификации всех компонентов смеси методом аналитической ректификации обычно ставится очередная задача — получить эти компоненты в достаточно больших количествах. Если разность температур кипения велика и нет необходимости в тонком разделении, то для наработки продукта вполне можно обойтись простыми дистилляционными приборами (рис. 136, а), собранными из стандартных деталей. На рис. 136, б показана модификация прибора с колонной. Эти приборы, разумеется, работают с неизмеряемой «дикой» флегмой. Для улучшения воспроизводимости результатов используют приставку или головку колонны, обеспечивающую точную регулировку нагрузки и флегмового числа (см. разд. 7.5). Приемник дистиллята Аншюца и Тиле (см. рис. 136), предназначенный для работ под вакуумом, можно при-
207
страница zuo
При работе с низкими флегмовыми числами средняя концентрация легколетучего компонента в дистилляте, полученном с периодическим отбором, всегда меньше, чем при непрерывном отборе. Однако было установлено, что при очень большом флегмовом числе (порядка 100) более полное разделение смеси достигается, когда дисстилят отбирают через каждый 1 час, а не через каждые 30 с. [25]. Если нужно подержать более высокие значения флегмового числа, как например при ректификаии изотопов (V= 1000-10000) то по соображениям технического порядка работают с периодическим отбором дистилята (см. разд. 5.1.4).
Рис. 136.
Приборы из стандартных деталей для дне тилляции (а), для ректификации (б).
менять также и при ректификации под атмосферным давлением. Кроме того, при высоких нагрузках можно также проводить простую перегонку с дефлегмацией (см. разд. 5.2.3) и так называемую расширительную перегонку (см. разд. 5.4.2).
“ б Лабораторные установки обыч-
но снабжены стандартными шлифами, поэтому диаметр колонны обычно не превышает размера, при котором можно еще применять шлиф NS45. Полупромышленные колонны из стекла, снабженные сферическими, коническими или плоскими шлифами, имеют наибольший номинальный диаметр около 150 мм. Для пилотных установок колонны изготавливают из стекла, керамического материала или металла, причем при выборе материала следует учитывать вероятность коррозии. Номинальные диаметры пилотных колонн лежат в интервале от 100 до 400 мм.
Штаге [281 дает обзор установок, применяемых для препаративного разделения в лабораториях и на опытных производствах. Хампель [29], занимавшийся проблемой получения ультрачистых растворителей, пришел к выводу, что наряду с экстракцией перегонка по-прежнему является самым эффективным методом очистки растворителей (свыше 99,9%). Высокой степени очистки достигают при ректификации спиртов, простых эфиров, бензола, пиридина и ряда углеводородов. Например, содержание примесей в четыреххлористом углероде может быть доведено до 10“4% и менее.
5.1.3.1. Полупромышленные установки
Размеры полупромышленных аппаратов должны соответствовать ожидаемой нагрузке н необходимой разделяющей способности (см. разд. 4.11). Стандартные детали и узлы для полупромышленных установок обычно изготавливают со шлифами NS10, NS14,5, NS29 и NS45. Непрерывно действующие установки, рассчитанные на более высокие нагрузки, описаны в разд. 5.2.2, 5.4.2 и 5.4.5. Для ректификации при нагрузках 20 л/ч была, в частности, разработана полупромышленная установка согласно нормалям «Де-стинорм», в комплект которой входят колонны диаметром 50, 70 и 90 мм. Она пригодна для работы как при атмосферном давлении, так и под вакуумом (остаточное давление до 1 мм рт. ст).
208
На рис. 137 дана схема полупромышленной установки непрерывного действия, собранная из стандартных деталей. При сборке полупромышленной установки часто копируют схемы промышленных установок.
Детали колонн снабжены коническими шлифами, причем для установки термометров и патрубков для жидкостей используют конические шлифы NS 14,5; в опасных участках предусматривают более подвижные сферические шлифы размером 35 мм (см. разд. 3.1).
Для обеспечения тонкого регулирования количества дистиллята головку колонны (см. рис. 137) снабжают многоходовым краном; флегмовое число можно контролировать с помощью градуированных мерников флегмы и дистиллята. Охлаждающий змеевик, вмонтированный в паровую трубу, обеспечивает возможность работы с дефлегмированием паров. Количество полученной при этом флегмы можно рассчитать по изменению температуры охлаждающей воды или же измерить с помощью градуированной бюретки. Аналогичную головку применяют в полупромышленных установках также при автоматическом регулировании флегмового числа по принципу деления потока жидкости (см. рис. 312).
При непрерывной ректификации питающую жидкость предварительно подогревают во вспомогательном теплообменнике, выполненном в виде U-образной трубки (рис. 138). При частично закрытом кране 1 уровень жидкости в теплообменнике поднимается до соединительной трубки 2; в этот момент начинается рециркуляция жидкости. В ходе рециркуляции смесь может нагреваться до 250 °C.
На рис. 137 показана только принципиальная схема установки. Степень автоматизации установки определяется ее насыщенностью контрольно-измерительной и регулирующей аппаратурой (см. гл. 8).
Для периодической разгонки в качестве куба обычно применяют круглодонные колбы емкостью лишь до 10 л; для разделения сырья в больших количествах используют подвесные кубы (см. рис. 316) или металлические кубы с плоскими или сферическими шлифами, на которые сверху устанавливают остальные стеклянные приборы. Целесообразно использовать куб из стали V2A емкостью 25 л, снабженный греющей баней и электронагревателями мощностью по 3 кВт с трехступенчатым регулированием (см. разд. 7.7.2). В связи с этим следует отметить, что одно из преимуществ непрерывной ректификации как раз и состоит в том, что даже при нагрузках до 20 л/ч можно использовать кубы емкостью всего 2—5 л (см. разд. 7.6.1).
Тарельчатая колонна Креля (рис. 139) диаметром 90 мм, предназначенная для использования в полупромышленной установке, выполненной по нормалям «Дестинорм», имеет встроенные устройства для перетока жидкости, как и в промышленных колоннах. Благодаря этим устройствам протекаемый в такой колонне
209
^Рис. 137.
Схема непрерывно действующей полупромышленной установки по нормалям «Дестинорм» при нагрузке до 20 л/ч. Изготовитель: народное предприятие «Комбинат технического ” *
стекла», Ильменау.
Рис. 138.
Подогреватель питающей жидкости для колонн непрерывной ректификации:
1 — край; 2 — соединительная трубка. Изготовитель: народное предприятие «Комбинат технического стекла», Ильменау.
Рис. 139.
Тарельчатая колонна Креля с внутренними перетоками (диаметр 50, 70 и 90 мм).
Изготовитель: народное предприятие «Завод технического стекла Штюцербах».
процесс ректификации более соответствует промышленному. Краны для отбора проб и термометры могут быть установлены на любой тарелке, поэтому в ходе ректификации можно следить за изменением концентрации и температуры по высоте колонны (прежде всего в ее верхней части).
Колонны с колпачковыми тарелками «Лабодест» конструкции Штаге (см. разд. 7.3.3) также имеют промежуточные перетоки, они изготавливаются с номинальными диаметрами до 100 мм. Зависимость коэффициента полезного действия тарелки от ее конструкции рассмотрена в разд. 7.3.3.
5.1.3.2. Пилотные установки
Для изготовления пилотных ректификационных установок * в качестве конструкционного материала обычно применяют стекло. Этот материал имеет то существенное преимущество, что при проведении ректификации можно визуально контролировать гидродинамические процессы, протекающие в колонне и других частях установки. К тому же заводы технического стекла в каталогах предлагают большой выбор стандартных деталей с плоскими шлифами и с трубками, присоединительные концы которых снабжены кольцевыми выступами со сферическими или плоскими торцами [165]; благодаря этим стандартным деталям значительно облегчается сборка установок для перегонки в промышленных условиях. Обычно применяют боросиликатное стекло, отличающееся химической стойкостью, а также стойкостью к воздействиям повышенных и быстро изменяющихся температур (см. разд. 7.1). Ниже приведены дополнительные сведения [301 относительно технических изделий из стекла «Разотерм» (тип стекла 320 по стандарту TGL 7209), выпускаемых народным предприятием «Йенским заводом стекла Шотт».
Толщина стенок в 5—8 мм обеспечивает достаточную механическую прочность стеклянных приборов. Кроме того, низкое значение коэффициента линейного расширения стекла «Разотерм» (а = 3,3 10 6) обуславливает стойкость к изменениям температуры.
Сборку пилотных установок проводят чаще всего по агрегатному принципу, применяя стандартные элементы, например трубы с наружными кольцевыми выступами на концах, фасонные части труб, теплообменники, круглодонные колбы, цилиндрические сосуды, колпачковые колонны с номинальными диаметрами 200, 400 и 500 мм (рис. 140), царги для колонн, запорные органы, дозировочные насосы, а также контрольно-измерительные и регулирующие приборы. Уже выпускаются стандартные и типовые установки для обработки воды путем дистилляции и ионного обмена, вакуумные испарители циркуляционного типа производитель-
* См. примечание на с. 11.
211
Рис. 140.
Ректификационная установка из стекла «Разотерм» с колпачковой колонной диаметром 400 Мм.
Изготовитель: народное предприятие «Йенский завод стекла Шотт», Иеиа.
Рис. 141.
Вакуумный циркуляционный испаритель производительностью 100 л/ч (по воде).
Изготовитель: народное предприятие «Йенский завод стекла Шотт», Иена.
ностью от 5 до 100 л/ч по дистилляту (рис. 141), установки для регенерации растворителей. Кроме того, выпускаются и специальные типовые узлы, например автоматические делители флегмы и клапаны с автоматическим приводом. Применение стандартных узлов из разных материалов дает возможность соединять стеклянные колонны с металлическими или фарфоровыми кубами в том случае, когда для проведения периодической ректификации требуются более вместительные кубы, чем изготавливаемые из стекла (до 200 л) (см. разд. 7.7.2).
Применение стандартных прямых, фасонных и переходных участков труб, а также запорных органов обеспечивает бездефектный монтаж замкнутых трубопроводных систем [165]. Диаметры выпускаемых труб от номинального размера 15 мм ступенчато увеличиваются до 500 мм. Прямые участки труб, которые в настоящее время изготавливаются длиной до 3 м, а также фасонные части соединяют с помощью накидных фланцев, при этом сферические 212
или плоские торцы труб, снабженных на концах кольцевыми выступами, допускают угловое смещение до 3°. Предусмотрены также различные соединения специального назначения (винтовые соединения для труб с заплечиками, фланцевые соединения для гладких труб).
Показанные на рис. 140 и 141 ректификационные установки (габаритная высота до 14,5 м) снабжены парогенератором с нагревателями. Испарительная способность одного нагревателя (с плоской спиралью), имеющего номинальный размер 200 или 300 мм, составляет 20 л/ч воды при давлении насыщенного пара 2 ати. Достоинством этих установок является возможность увеличения охлаждающей и греющей поверхности при необходимости. При расчете теплообменника коэффициент теплопередачи k можно принять равным 400 ккал/(м2-ч-°С) при нагревании жидкостей паром (через стенку). При конденсации паров в холодильнике принимают k =300 ккал/(м2 ч °С), а при охлаждении жидкостей k = 150 ккал/(м2 ч °C).
Стеклянные детали, трубопроводы и пилотные установки фирмы Квикфит герметичны при работе под вакуумом. Концы труб снабжены конусными переходами; при номинальных диаметрах труб от 15 до 225 мм — сферические шлифы. Толщины стенок лежат в интервале 2—10 мм. Конструкционные элементы фирмы Квикфит могут быть дополнительно защищены покрытием из полиэфиров, стойким при температуре до 150 °C [1651.
Проявляется тенденция к созданию стандартной аппаратуры для перегонки, в том числе циркуляционных испарителей и сборных конструкций из стеклянных приставок и кубов, выполненных из других материалов. Эта тенденция распространяется также и на автоматизированные пилотные установки непрерывного действия с электромагнитными делителями флегмы, регуляторами уровня и расхода (рис. 142). Применением различных вентилей из стекла можно значительно упростить обслуживание подобных установок. Такие вентили подробно описаны в разд. 7.2.1.
Для автоматизации пилотных установок из стекла дополнительно требуется регулирование систем нагревания кубовой и питающей жидкости (см. разд. 7.7.2); кроме того, необходимы приборы для регулирования флегмового числа (см. разд. 8.4.1 и рис. 142) и скорости испарения (см. разд. 8.4.2). Для измерения и дозирования расхода газа и жидкости в пилотных установках были разработаны специальные типовые приборы из стекла (см. разд. 8.6).
Для создания устройств, предназначенных для дозирования агрессивных жидкостей (температура до 100 °C, кинематическая вязкость жидкостей до 30 сСт и плотность до 2 г/см3), особенно рекомендуется комбинирование стекла и политетрафторэтилена. На «йенском заводе стекла Шотт» разработан ряд бессальниковых дозировочных насосов (рис. 143), используемых в комплекте со стандартным приводом РАЕ 32/50 (насосный завод «Зальцведель»),
213
Рис. 142.
Рис. 143.
Головка дозировочного насоса из стекла с тефлоновым сильфоном.
Предназначена для установки на специальном приводе РАЕ 32/50 (стандарт TGL 17-747907). Изготовитель:’ народное предприятие «Иенскнй завод стекла Шотт», Иена.
Рис. 144.
Схема газового проходного вентиля с условным проходом 15 мм.
Изготовитель: народное предприятие «Иенскнй завод стекла», Иена.
Установка для непрерывной вакуумной ректификации (высота 7,2 м, ширина 1,9 м) с колонной диаметром 150 мм:
1,7 — холодильники (0,2 м2); 2 — кипятильник (0,5 м2); 3 — теплообмен* ник для питающей жидкости (0,14 м2); 4 — дефлегматор (1,4 м3); 5 — магнит; 6 — регулятор флегмы.
Изготовитель: фирма «Стеклянная техника», Висбаден, Шнрштайи.
Номинальные расходы лежат в пределах 63 и 650 л/ч при максимальном напоре от 3,5 до 2,5 кгс/см2. Повышенной коррозионной стойкостью обладает также газовый проходный вентиль с условным заводом (рис. 144). Корпус вентиля выполнен из стекла, мембрана и основание мембраны — из политетрафторэтилена. Вентиль закрывают поворотом шпинделя на 230°.
214
Для разделения агрессивных веществ, разрушающих стекло, предназначены перегонные аппараты, изготовленные из фарфора (рис. 145). Наименьший куб имеет емкость 50 л; номинальный диаметр колонны не менее 50 мм.
Фирма Подбильняк (Чикаго) поставляет автоматизированные пилотные ректификационные установки серии «ректификатор» четырех типов, предназначенные для разделения в периодическом или непрерывном режимах (рис. 146). Установки фирмы Подбильняк имеют следующую характеристику:
емкость куба ................
диаметр колонны .............
рабочая высота колонны . . . рабочее давление ............
рабочая температура . , . .
45—450 л
76—304 мм
1,5—3,0 м
от 3 мм рт. ст. до 136 ат
от —40 до +350 °C
В колоннах преимущественно применяется насадка «Хели-пэк» или «Окта-пэк» (см. разд. 7.8). Предусмотрен обогрев электрический, паровой или с применением теплоносителя «Даутерм».
Рис. 145.
Полупромышленная периодическая ректификационная установка из фарфора Изготовитель: народное предприятие «Керамический завод Хешо-Кала».
Рис. 146.
Автоматизированная пилотная ректификационная установка Подбильияка Изготовитель: фирма Подбильияка, Департамент обрабатывающей промышленности, Франклин парк, Иллинойс, США.
215
Карпентер и Хельвиг [31 ] описывают типовые детали и узлы, применяемые в пилотных установках. Жордан в книге [32], в которой рассмотрены полупромышленные опытные химические установки, в первую очередь обсуждает проблемы переноса опытных результатов на промышленные установки.
Часто приходится решать вопрос о том, что можно ли вместо проведения исследований на пилотных установках ограничиться применением чисто расчетных методов, основанных на масштабном переходе от малых аппаратов к большим. Однако для ректификационного разделения веществ еще нет методов для достаточно точного математического описания процесса с учетом всех решающих факторов. Поэтому опытно-промышленные испытания по-прежнему остаются важнейшим источником сведений, необходимых для масштабного моделирования [33]. В первую очередь это относится к насадочным колоннам, для которых гидродинамические характеристики газового и жидкостного потоков играют особую роль (см. разд. 4.2). Кроме того, для оценки стоимости ректификационных колонн с целью уменьшения капиталовложений необходимо знать зависимость разделяющей способности и перепада давления от нагрузки. Эту зависимость для большинства колонн до сих пор нужно устанавливать экспериментально. Чтобы можно было сравнивать различные колонны, для их испытаний следует подбирать одинаковые смеси и испытания проводить в одинаковых условиях (см. разд. 4.10 и 4.11).
5.1.3.3. Получение дистиллированной воды
Типичным примером наработочной перегонки в лабораторных условиях является получение дистиллированной воды, осуществляемое чаще всего по непрерывному способу.
Аппараты для дистилляции воды обычно работают при постоянном уровне жидкости в перегонном кубе. Это достигается благодаря тому, что предварительно нагретая в конденсаторе вода поступает в куб при непрерывном отводе ее избытка, что и обеспечивает регулирование уровня. Благодаря применению кварцевого стекла гарантируется высокая степень чистоты дистиллята (см. рис. 147).
Большое число предложенных конструкций дистилляторов указывает на то, что до сих пор еще не найден наиболее экономичный способ их работы. Поэтому для более экономичного использования тепла переходят на внутренний электрообогрев и предусматривают достаточный слой тепловой изоляции. Производительность лабораторных аппаратов лежит в интервале 0,5— 2,1 л/ч. Производительность от 40 до 50 л/ч обеспечивают широко применяемые дистилляционные аппараты, изготовленные из меди, никелированные (до зеркального блеска) или оцинкованные внутри. Они прочны и при хорошей теплоизоляции имеют высокий коэффициент полезного действия.
216
Шоттом и др. разработан автоматический аппарат для дистилляции воды с электрическим обогревом, выполненный из дюралюминия 50 (рис. 148). Отдельные модели этого прибора дают от 10 до 70 л/ч дистиллированной воды с электропроводностью менее 0,5 мкСм при условии, что аппарат подпитывают полностью обессоленной водой. Народное предприятие «Йенский завод стекла Шотт» выпускает дистилляционные установки с электрическим и паровым обогревом производительностью от 8 до 110 л/ч. Бидистилляторы с электрическим обогревом имеют производительность 8 и 24 л/ч. Для них специально разработан погружной нагреватель THQ 6 (номинальная мощность 6 кВт).
Представляет интерес аппарат с эффективным использованием тепла, работающего по принципу «per descensum» (нисходящего потока) (см. рис. 36). Такой аппарат имеет к. п. д. 95%. Аналогично работает прибор Кюлльманна [35], в котором получают дистиллят без какой-либо примеси газа; с помощью дополнительной приставки можно одновременно получать одно- и двукратно дистиллированную воду (рис. 149).
Бидистиллятор 1600 Зельнера [36] очень экономичен и дает от 1,3 до 1,6 л/ч продукта (рис. 150). Тепловая энергия к нему
Рис. 147.
Непрерывный дистиллятор «Пюратор» из кварцевого стекла для получения однократно дистиллированной воды (2 л/ч):
1 — перегоииый куб; 2 — внутренний нагреватель; 3 — дистилляционная приставка; 4 — теплообменник; 5 — трубка для измерения уровня; 6 — головка с колпаком и капельницей.
Изготовитель: народное предприятие «Кварцевое стекло», Берлин.
Рис. 148.
Автоматический дистиллятор воды нз дюралюминия 50 производительностью 10 — 70 л/ч.
А — охлаждающая вода; Б — газ для пневмоавтоматики; В — отработанная вода; Г — газовая защита; Д — исходная вода.
Изготовитель: фирма Шотт, Майнц,
217
Рис. 149.
Аппарат Кюлльманна для одновременного получения одно* и двукратно дистиллированной воды.
Изготовитель: фирма Кройцер—Крайлер, Мюнхен.
Рис. 150.
Дистиллятор воды 1600 Зельнера Изготовитель: фирма Шотт, Майнц.
подводится только на первой испарительной ступени, на которой из водопроводной воды получается монодистиллят. Вторичное выпаривание монодистиллята на второй испарительной ступени проводят при 30—40 °C под вакуумом, создаваемым водоструйным насосом. При этом используется теплота конденсации первичных паров, благодаря чему экономится электроэнергия. Водопроводная вода, необходимая для работы водоструйного насоса, служит первоначально в качестве охлаждающей среды. Часть воды после прохождения конденсатора и насоса используется для питания первой испарительной ступени аппарата. При том же потреблении энергии, что и для простой дистилляции, и при минимальном расходе водопроводной воды получают бидистиллят высокой степени чистоты.
Весьма полезна предварительная очистка питательной воды в ионообменном фильтре, как например в аппарате для дистилляции воды, выполненном из инертного по отношению к воде кварца фирмы «Хереус-Кварцшмельце» (Ханау). В последнее время применяют горизонтальные цилиндрические перегонные кубы. Разбрызгивание кипящей воды (брызгоунос) и образование 218
аэрозоля в этих аппаратах сведено к минимуму благодаря тому, что паровые пузыри движутся по кратчайшей траектории, а зеркало жидкости достаточно велико [36а].
5.1.4. РАЗДЕЛЕНИЕ ИЗОТОПОВ
Химические элементы, встречающиеся в природе, большей частью являются смесями изотопов. Их можно назвать многоизотопными элементами, в то время как изолированные изотопы представляют собой собственно чистые элементы. Изотопы элемента занимают одно и то же место в периодической системе элементов, поэтому они обладают одинаковыми зарядами, но разными массами атомных ядер. Чистые элементы 12С и 13С различаются, например, только тем, что в первом случае атомное ядро содержит шесть, а во втором случае — семь нейтронов. Атомные массы, которыми оперируют в химии, представляют собой усредненные значения атомных масс отдельных изотопов.
Изотопы были впервые открыты в радиоактивных веществах. Большая часть нерадиоактивных элементов также содержит определенное число стабильных изотопов. В табл. 34 приведены некоторые стабильные изотопы, которые могут быть выделены, главным образом, путем ректификации [37].
В органической химии для метки молекул используют прежде всего тяжелые изотопы элементов С, Н, О и N. Вплоть до углерода отсутствуют подходящие радиоактивные изотопы с достаточно большим периодом полураспада, поэтому стабильные изотопы 2Н, 13С, 18О и 15N имеют особенно большое значение для химических исследований, а также в биологии, медицине и геологии.
Элементы с нечетным зарядом ядра обычно состоят лишь из одного или двух изотопов с нечетными массовыми числами (за исключением Н, Li, В и N). Элемент Hg имеет семь стабильных изотопов (массовые числа 196—204) с относительной разностью в атомных массах порядка 0,5%. Все процессы разделения, направленные на обогащение и получение изотопов, основаны на использовании различия атомных масс, обусловливающем их неодинаковые свойства, поэтому относительная разность атомных масс является показателем степени трудности разделения. Точно также относительная естественная концентрация изотопов в исходном сырье имеет важное значение для процесса разделения. Из элементов, указанных в табл. 34, соотношение естественных концентраций изотопов особенно благоприятно в смесях 10В— nB, 20Ne—22Ne и 35С1—37С1.
Для обогащения или полного разделения изотопов применяют методы диффузии, термодиффузии, электролиза и обменные реакции. Обогащение можно также проводить с помощью методов осаждения и центрифугирования. Ректификационные методы разделения применяют для получения изотопов 4Не, D, 10В, 13С, 15N, 18О, 22Ne, 37С1 и 36Аг. Обстоятельный обзор методов получе-
219
Таблица 34.
Стабильные изотопы, относительная разность их атомных масс и их относительная естественная концентрация
Элемент Атомный номер Массовое число Относительная разность атомных масс, % Относительная естественная концентрация, %
Н (D) 1 1 100 99,9844
2 0,0156
Не 2 3 33,3 1,3-10~4
4 99,9999
В 5 10 10 18,83
11 81,17
С 6 12 8,35 98,9
13 1,1
N 7 14 7,15 99,62
15 0,365
О 8 16 «О—17О 6,25 99,757
17 1?О—18О 5,9 0,039
18 «О—18О 12,5 0,204
Ne 10 20 2’Ne—21Ne 5,0 90,51
21 21Ne—22Ne 4,75 0,28
22 20Ne—22Ne 10 9,21
S 16 32 32S_33S 313 95,06
33 33s—3«s 3,03 0,74
34 3«s—”S 5,9 4,18
36 0,016
Cl 17 35 5,7 75,4
37 24,6
Ar 18 36 ”Ar—38Ar 5,55 0,307
38 звДг—«Ar 5,25 0,060
40 з’Аг—«Ar 11,11 99,633
ния и применения стабильных изотопов дают Бродский [38] и Ветцель [39], они рассматривают специальные проблемы ректификации изотопов. Все большее значение приобретает комбинация химического изотопного обмена и ректификации, получившая название «обменной ректификации» [40]. Анализ стабильных изотопов проводят преимущественно методом масс-спектрометрии [39]. Разработаны также другие методы анализа изотопов, которые описали Мерсеа [41 ], а также Мюллер и Мауэрсбергер [42].
При обогащении стабильных изотопов методом ректификации в качестве сырья используют, главным образом, газы; лишь дейтерий и 18О получают из воды. Соотношения давлений паров для подобных смесей изотопов указаны в табл. 35. Разделение всех смесей, за исключением соединения бора ВС13, требует, разумеется, значительных затрат на охлаждение. Кроме того, для достижения обычной степени разделения смесей изотопов за исключением изотопов гелия и водорода требуется более 500 теоретический ступеней разделения. Кун с сотр. [43], применив большое число теоретических ступеней разделения, определил относительную летучесть для соединений изотопов с температурами кипения 80 °C.
Таблица 35.
Соотношения давлений паров для смесей изотопов
Смесь Температура, К Р°/Р° а л т Литература
h2-d2 18,65 3,6 [48]
3Не—«Не 2,19 5,4 [48]
иВС13—10ВС13 260 1,003 [52, 53]
UBF3—10BF3 170 1,01 [53]
12С16О—13CleO 68,2 1,010 [48]
«(у «О—12С18О 68,2 1,007 [48]
12С2Н4—12Ci3CH4 120 1,0014 [54]
169,3 1,0019
12С2Н8—12С13СНв 130 1,0022 [54]
165 1,0006
12СН4—13СН4 97,55 1,0099 [54]
111,8 1,0070
14N2—14N15N 63,3 1,006 [48]
««N2—««N15N 71,5 1,005 [55]
UNi’O—15N16O 117 1,03 [55]
UNi’O—14№8O 120 1,04 [50]
leQi’O—18O18O 63,14 1,01286 [49]
74,05 1,00912
20Ne—22Ne 24,6 1,043 [48]
ЗвДг_4°Аг 83,8 1,006 [48]
128Хе_136Хе 161,4 1,000 [48]
Р® — давление паров соединения, содержащего легкий изотоп, р® — давление па-ров соединения, содержащего тяжелый изотоп.
221
5.1.4.1. Разделение изотопов низкотемпературной ректификацией
Методы низкотемпературной ректификации (см. разд. 5.3.1) обычно применяют для разделения смесей изотопов Н—D, 10В— ПВ, 12С—13С, а также изотопов инертных газов — гелия, неона и аргона.
Благодаря большой разности атомных масс и больших различий в давлениях насыщенных паров изотопов получение дейтерия из газовой смеси Н2—D2 путем низкотемпературной ректификации теоретически представляется сравнительно простым. Селлерс и Аугуд [441 подробно и систематически изучили проблемы низкотемпературной ректификации систем HD—Н2, 16О18О—1вО1вО, а также О2—N2. Разделение проводили в колпачковой колонне высотой 27 м. Позднее исследования низкотемпературной ректификации смеси Н—D были проведены Тиммерхаусом с сотр. [45]. Для разделения использовали пилотную установку с колонной диаметром 150 мм, содержащую ситчатые тарелки, и определили ряд параметров для данной системы, важных с точки зрения разделения. Небольшую лабораторную колонну для ректификации смеси Н2—HD описал Вайссер [46].
В настоящее время для промышленного производства тяжелой воды применяют крупномасштабные установки [47]. Значительные трудности аппаратурного характера возникают при разделении газовых изотопных смесей. Поэтому лабораторное получение изотопов при температуре кипения жидкого азота и жидкого воздуха пока еще слишком дорого. Однако если ректификационную установку присоединить к промышленной установке для получения кислорода из жидкого воздуха, то концентрирование изотопов 36Аг, 18О и 16N может оказаться очень экономичным [48, 49]. По-видимому, очень выгодна низкотемпературная ректификация NO при одновременном получении 16N и 18О [501, а также ректификация СО при концентрировании 13С [511.
Методом низкотемпературной ректификации в колонне с 130 теоретическими ступенями разделения Клузиус и Мейер [48] ежесуточно обогащали 15 л аргона до концентрации 0,6% 36Аг (вместо 0,307% в природном аргоне). Для этого применяли насадочную колонну высотой 3 м, изготовленную из латунной трубки с внутренним диаметром 12 мм. Насадка состояла из проволочных спиралей размером 2 Х2 мм, выполненных из нержавеющей стали. На рис. 151 показана схема специально для этой цели изготовленного перегонного куба емкостью 250 мл и конденсатора, охлаждаемого жидким азотом. Бевилогуа с сотр. [164] сообщает о получении изотопов 22Ne и 20Ne, а также о концентрировании 21Ne ректификацией при 28 К:
В полупромышленной колонне диаметром 12 мм и высотой 12 м, заполненной металлическими кольцами диаметром 1,2 мм (рис. 152), Мюленпфордт получал в год 4,5 кг BF3 с 83% 10В.
222
Колонна на верхнем конце имеет- сборник для разделяемого вещества, а на нижнем конце — участок для отбора обогащенного вещества, что обусловлено более низкой летучестью 10BF3. Необходимым условием высокой разделяющей способности является равномерная подача жидкости BF3 в верхний конец колонны. Такая подача обеспечивалась специально разработанным дозировочным приспособлением.
а 6
Рис. 151.
Перегонный куб (а) и конденсатор (б) установки низкотемпературной ректификации для обогащения88 Аг:
/ — платиновый впай; 2 — нагреватель; 3 — перегонная колба для жидкого аргона; 4 —- место отбора; 5 — сильфон из томпака; 5 — насадка; 7 — латунная трубка; 8 — место спая стекла и металла; 9 — сосуд из пнрекса; 10 — верхний и нижний патрубки для отбора; 11 — медная сетка; 12 — резиновая трубка; 13 — двойная стенка из латуни; 14 — высоковакуумирован* ная полость; 15 — сифон для жидкости; 15 — патрубок для отвода паров азота;
17 — трубка из мельхиора; 18 — термометр для измерения температуры жидкого азота; 19 — сосуд с жидким азотом; 20 — свинцовая дробь; 21 — медная пластина.
Рис. 152.
Установка низкотемпературной ректификации для концентрирования 10BF3 конструкции Мюленпфордта:
I.— испаритель с электрообогревом; II — колонна для очистки сырья от SiF4 и HF; III — колонна с насадкой; IV — двойной конденсатор для паров BF3; V — резервуар для исходной жидкости с электрообогревом; VI — вспомогательный конденсатор; VII — конденсатор для паров этилена; VIII - буферный резиновый баллон; IX — резервуар для жидкого воздуха; 1, 2 — патрубки для присоединения к диффузионному насосу; 3,4 — линия для присоединения манометра; 5 — буферная емкость, заполненная водородом; 6 — линия для подачи этилена и водорода; 7 — линия подачи исходного BF3; 8 — линия для отвода обедненной фракции BFS; 9 — линия для отвода обогащенной фракции 10BF3; 10 — линия для отвода загрязнений.
223
5.1.4.2. Получение D и 180 путем ректификации воды
В данном разделе рассмотрен метод получения D и 18О путем ректификации природной воды при атмосферном и пониженном давлениях.
Водород существует в виде трех изотопов: ГН — водород, 2Н — дейтерий и радиоактивный 3Н — тритий. Тритий встречается в природе в ничтожных количествах.
Кислород существует в виде трех стабильных изотопов: 16О, 17О и 18О. Теоретически возможны следующие модификации воды (без учета трития 3Н):
|2Н1вО2Н| 1Н1«О2Н
2Н17О2Н Щ1’02Н
|*Н18О1Н| 2Н1«О2Н Щ18О2Н
Наибольший интерес представляют тяжелая вода D216O(2H2ieO) и вода 1Н218О, обогащенная изотопом кислорода.
, Физико-химические свойства D2O и Н2О следующие [37, 56 ]:
D2O Н2О д
Плотность pf°, г/см3 . . . 1,1059 0,9982 0,1077
Температура плавления, °C 3,82 0,00 3,82
Температура кипения, °C 101,42 100,0 1,42
Температура, при которой
жидкость имеет максималь-
ную плотность, °C .... 11,6 4,0 7,6
Динамическая вязкость, сП 1,260 1,009 0,251
Поверхностное натяжение,
дин/см 67,80 72,75 4,95
Показатель преломления
«20 nD 1,32844 1,33300 0,00456
Как видно из приведенных данных, разность температур кипения D2O и Н2О составляет 1,42 °C, а разности значений плотностей и температур, соответствующих максимумам плотности, очень малы.
Исходным сырьем для получения тяжелой воды является природная вода, содержащая 0,0146 % (ат.) D (1 : 6850). В дождевой воде концентрация D несколько выше, а именно 0,0200% (ат.) (1 : 5000) [56]. Поэтому природную воду, подвергаемую ректификации, можно рассматривать как смесь низкокипящего компонента Н2О (100 °C) и высококипящего компонента D2O (101,42 °C).
224
Поскольку в природной воде наряду с Н2О и D2O содержатся молекулы HDO, относительная летучесть компонентов данной смеси а' определяется выражением:
ХНРО + 2хРгО \ / 2уНгО ~Ь -УНРО
2хНгО + ХНРО J \ ^HDo+^D.O
(161)
В соответствии с работой Юри [57 ] а' можно выразить также через отношение давлений паров чистых компонентов:
а = ]/ Рщо/РРгО (162)
Это уравнение выведено на основе предположения, что давление паров HDO является средним геометрическим значением давлений паров Н2О и D2O, а константа равновесия реакции
Н2О 4-D2O^2HDO
(163)
в паровой фазе равна 4,0. Рис. 153 иллюстрирует зависимость а' от температуры, полученную Киршбаумом [56].
Аналитическим путем установлено, что число теоретических ступеней разделения смесей Н2О и D2O минимально при возможно меньшем рабочем давлении и, следовательно, при низких температурах. Однако при проведении ректификации в таких условиях трудно конденсировать пары дистиллята, и производительность колонны значительно ограничена из-за большой нагрузки по пару. При использовании тарельчатых и насадочных колонн имеют место такие потери напора и допустимая скорость
потока паров, которые существенно увеличивают длительность процесса ректификации и делают данный метод разделения экономически невыгодным. Вследствие этого при проведении подобной ректификации обычно идут на компромисс, работая при давлении 50—125 мм рт. ст. Значения а' при температурах 40—60 °C,
р,мм ргп.сгп.
соответствующие этому интервалу давления, лежат в пределах 1,06— 1,05.
При ректификации природной воды графический метод Мак-Кэба и Тиле неприменим для расчета числа теоретических ступеней разделения, так как с одной стороны кривая равновесия очень полога, а с другой — рабочие концентрации D2O очень близки к предельным значениям, а именно: х0 = 0,000146 D2O
Рис. 153.
Зависимость а' = [/Л/'н2оА,Е>гО от температуры.
8 Э. Крель
225
(ат. доли) — исходная концентрация; хк0;, = 0,998 D2O — конечная концентрация; а' = 1,059 (при 40 °C и 55,3 мм рт. ст.).
Для данного примера уравнение Фенске дает в первом приближении следующее значение минимального числа теоретических ступеней разделения при работе с бесконечным флегмовым числом:
«мин = Г 1g^°-!(1~Хо] / 1g а' 1 - 1 = 308 (164)
Необходимое число теоретических ступеней разделения при оптимальной скорости отбора кубового продукта составляет —700 [58]. Следует иметь в виду, что в данном случае нужным компонентом является высококипящее вещество D2O, которое при периодической ректификации накапливается в кубе, а при непрерывной перегонке выводится из него. В качестве дистиллята Получается вода с концентрацией D2O менее 0,0146% (ат.).
Согласно работе Куна [59] эффективность элементарного акта разделения подобной идеальной системы изотопов, имеющая место при однократном частичном испарении жидкой смеси, определяется соотношением:
При данной температуре величина 6 характеризует бинарную смесь, поскольку она не зависит от значения исходной концентрации х0 (в интервале 0,99—10*3 мол. долей). Величину 6 называют параметром разделения. Зная давления паров чистых компонентов и предположив, что тепловой эффект смешения этих компонентов равен нулю, 6 можно рассчитать по уравнению:
Р°/Р° = *в (166)
где р® и — давление паров чистого низкокипягцего и высококипящего компонента соответственно.
Если относительная разность давлений насыщенных паров компонентов мала, то значение 6 можно определить по следующим формулам:
МРи-рЖ (167)
6==а — 1 (168)
Для смесей с тепловым эффектом смешения равным нулю, к которым относится большинство смесей изотопов, 6 можно приближенно рассчитать по уравнению:
6= 1о,7 ДТ /Тр (169)
где ДТ — разность температур кипения двух компонентов при атмосферном давлении; Тр — температура кипения низкокипящего компонента при рабочем давлении ректификации.
226
В табл. 36 приведены значения параметра разделения 6 для бинарных смесей, полученные в предположении, что оба компонента подчиняются правилу Трутона и зависимости их теплот парообразования от температуры примерно одинаковы.
Эффективность ректификационной колонны с определенным числом теоретических ступеней разделения п, возникающую при совмещении большого числа элементарных актов разделения, определяют по формуле:
,1,~Г|1<>н) =епа (170)
х0/(1—х0)
При этом необходимое число теоретических ступеней разделения, требующееся для конкретного процесса ректификации, рассчитывают по уравнению:
(‘/о
Это уравнение соответствует известному уравнению Фенске (1 086) при v = оо.
Минимальное флегмовое число для заданного процесса разделения можно определить в первом приближении по формуле: ^мин = Z/T = (%кои — Хо)/[6хо (1 — х0)] (172)
где Z — количество жидкости, циркулирующей в кубе (соответствует флегме)! т — количество отбираемого кубового продукта (соответствует отбираемому дистилляту).
При отборе кубовой жидкости, обогащенной высококипящим изотопом, максимально возможный относительный выход кубового продукта оценивают по уравнению:
(т/2)макс = Х()6 (1 — Хо)/(%кон — ^о) (173)
Максимальному выходу кубового продукта, определяемому выражением (173), соответствует бесконечное число теоретических ступеней разделения. Поэтому действительное количество отбираемой фракции должно быть несколько меньшим.
Таблица 36.
Параметр разделения 6 и относительная летучесть а для смесей изотопных соединений
Разделяемая смесь Рабочая температура, °C Разность температур кипения» °C Параметр разделения д Относительная летучесть а
H2O-D2O 70 1,6 0,05 1,05
Н216О—Н218О 70 0,165 0,005 1,005
12СС14—13СС14 35 0,036 0,00125 1,00125
C3SC14—Сз7С14 35 —0,003 0,00010 1,0001
8*
227
t/Z
Xo , 7о(мОЛ.)
Рис. 154.
Зависимость числа теоретических ступеней разделения от относительного выхода кубового продукта т/Z, полученная Куном (значение х^ и *кон в мольных долях):
1 — х0 = 0,002, ткон = 0,015, б =- 0,0045; 2 — = 0,002, *Кон = 0,020,
б = 0/0045; 3 — х0 = 0,002, xRoH = 0,020, б = 0,0065.
Рис. 155.
Зависимость необходимого числа теоретических ступеней разделения от концентрации D2O в исходной смеси при v = «.
Давление: 760 мм рт. ст. (100 °C) и 100 мм рт. ст. (50 °C); *кон — = 99,8% (мол.).
Необходимое число теоретических ступеней разделения п, требующееся для конкретного процесса ректификации при конечном флегмовом числе [или при относительном выходе кубового продукта, меньшем чем (т/Х)макс ], можно определить по диаграмме, полученной Куном (рис. 154).
При стационарном отборе кубовой фракции величину п рассчитывают по формуле:
<174>
На рис. 154 приведены в качестве примера зависимости необходимого числа теоретических ступеней разделения от относительного выхода кубового продукта, полученные для процесса обогащения воды стабильным изотопом 18О.
Из рассмотрения кривой 3 видно, что для повышения концентрации изотопа 18О с 0,2% (мол.) до 2% при а = 1,0065 необходимы следующие условия:
t/Z=10~6, «=388; т/Z = Ю'1, «=409; t/Z=2-10-«, «=437; t/Z=5.10-\ «=567.
Другие методы расчета числа теоретических ступеней разделения и удобные номограммы приведены в работах Хубера [61] и Хильтбруннера с сотр. [62].
Кун с сотр. [60] при получении D2O сначала обогащал природную воду путем электролиза до содержания D2O 1,0% (мол.) 228
и только потом применял ректификацию. В промышленности используют также и обратный путь. На рис. 155 показаны зависимости необходимого числа теоретических ступеней разделения от концентрации D2O в исходной смеси, полученные при бесконечном флегмовом числе и хкон = 99,8% (мол.) для температур 50 °C (100 мм рт. ст.) и 100 °C (760 мы рт. ст.). Материальный баланс двухступенчатого аппарата Куна наглядно показан на рис. 156, из которого видно, что относительный выход кубового продукта составляет на первой ступени 1 : 1270, а на второй-—1 : 78.
Сложности, возникающие в лабораторных условиях при такой скорости отбора кубовой жидкости, затрудняют аппаратурное оформление установки. Поясним детали процесса получения D2O на примере ректификационной установки Куна [59] (рис. 157). Исходная смесь (природная вода), находящаяся в сосуде 1, испаряется и через обогреваемые снаружи капилляры 2 равномерно поступает в трубчатые колонны, заполненные насадкой из проволочной сетки. Расход паров контролируют по показаниям дифференциального манометра 3, подсоединенного к емкостям 4 и 5. Нижние части трубчатых колонн в зоне куба 10 подогревают водя-
Рис. 156.
Схема материального баланса двухступенчатой установки Куна для получения D2O.
Высота разделяющей части 530 см (ступень 1) и 680 см (ступень 2), ВЭТС = = 1,8 см (ступень 1) и 1,7 см (ступень 2), давление в головке колонны 120 мм рт. ст. (ступень 1) н 60 мм рт. ст. (ступень 2).
Рис. 157.
Принципиальная схема ректификационной установки Куна с трубчатыми колоннами для получения D2O:
1 — сосуд для исходной смеси; 2 — обогреваемые капилляры; 3 — дифференциальный манометр; 4, 5 — емкости; 6 — конденсатор; 7 — буферная емкость; 8 — сборник конденсата; 9 — барометрическая труба; 10 — куб; 11 — приемная камера; 12 — клапан; 13 — приемник кубового продукта; 14 — испаритель.
229
Зависимость относительной летучести для смеси Hgieo—Hgl8o от температуры По данным: О — Достровского; Ливлнна и Времена; Мак-Вильямса, Пратта, Делла и Джойса; Д — Вэхла и Урея; □ — Ризен-фельда и Чаига; X — Жаворонкова, Уварова и Севрюговой.
Рис. 159.
"Схема металлической ректификационной установки для обогащения воды изотопом Н21вО.
1 —• основной трубопровод для подачи воды; 2 — трубопровод-для подачи дистиллированной воды; 3,4 — основные конденсаторы; 5 — переточная труба; 6 — линия для исходной смеси; 7 — дефлегматор; 8 — магнитный клапан; 9 — линия к вакуумному насосу; 10 — маностат; 11 — дифференциальный манометр; 12 — манометр кипятильника; 13 — ректификационная колонна; 14 — расходомер; 15 — контрольная головка; 16 — штуцер для отвода продукта; 17 — медная труба (диаметр 25,4 мм) между кипятильником и колонной; 18 — кипятильник; 19 — сборник; 20 — штуцер для выхода сточной воды.
ним паром, поступающим из испарителя 14. Расход греющего пара выбирают таким образом, чтобы обеспечивалось испарение всей жидкости, находящейся в этих частях колонн. Через капилляры, размещенные внизу трубчатых колонн, в камеру 11 отбирают пары путем создания в ней некоторого разряжения. Скорость отбора должна соответствовать выбранному относительному выходу кубового продукта. Посредством клапана 12 осуществляют контроль за расходом отбираемых паров, которые затем конденсируют в расположенном ниже холодильнике. Конденсат стекает в приемник кубового продукта 13. Пары, поднимающиеся по колоннам, выводят через штуцер, присоединенный к емкости 5, и полностью конденсируют в конденсаторе 6. Конденсат, поступивший в сборник 8, насосом подают в сосуд 4, откуда часть его отводят по барометрической трубе 9 в точном соотношении с количеством исходной воды, подаваемой в колонны через капилляры 2. Для работы под вакуумом вакуумный насос присоединяют к сосуду 7 и к штуцеру, размещенному над приемником кубового продукта 13.
Производительность данной установки при разделении природной воды достигает 1,5 кг/ч. Ректификацию проводят в две ступени. На первой ступени воду обогащают от 1 до 50% (мол.) D, 230
а на второй — от 50 до 99,8% D. Нагрузку поддерживают примерно^на 10% ниже нагрузки, соответствующей пределу захлебывания. При заполнении трубчатых колонн кольцами Рашига из проволочной сетки ВЭТС составляет 1,7—1,8 см. Применение насадочных колонн обусловлено высоким коэффициентом поверхностного натяжения воды, что затрудняет равномерное распределение жидкости по стенкам полых трубчатых колонн. Обеспечение равномерного распределения жидкости по насадке является основной проблемой при получении тяжелой воды (см. разд. 4.2).
Соотношение содержаний изотопов кислорода в естественных условиях примерно составляет 1вО : 17О : 18О = 2500 : 1 : 5. При ректификации природной воды кубовая жидкость обогащается изотопом 18О. Как видно из рис. 158, относительная летучесть компонентов смеси Н21вО—Н218О примерно на порядок ниже, чем для смеси Н2О—D2O. При 100°C (760 мм рт. ст.) а составляет около 1,004, а при 50 °C (ПО мм рт. ст.)- 1,007. Данный график
взят из работы Достровского и Равива {53 ]. Значительный разброс опытных данных частично объясняется трудностями экспериментального определения коэффициента а. Сравнительно высокая концентрация изотопа 18О в природной воде, равная 0,204% (ат.), упрощает условия проведения ректификации.
При работе на двухступенчатой ректификационной установке Кун достигал обогащения воды до 90% (ат.) 18О. Достровский с сотр. [53], применяя комбинированную установку, получал повышение концентрации 18О до —95% и 17О до —2,0%. Предварительное обогащение от 0,2% до 1,6% 18О проводили в 10 параллельно включенных колоннах (диаметр каждой колонны 100 мм). При ректификации на многоступенчатом каскаде из колонн (диаметр колонн от 30 до 100 мм) с расходом исходной смеси 800 мл/ч при относительном выходе кубового продукта 1,37-Ю"2 конечная концентрация 18О достигала до 99,8%. Наибольшее обогащение 17О получалось в средней части каскадной установки, в которой концентрация 17О примерно составляла 10%. В указанных процессах особенно хорошо себя зарекомендовала насадка Диксона в виде колец Рашига из фосфористо-бронзовой сетки 100 меш. с S-образными перемычками [63].
На рис. 159 показана схема непрерывно работающей одноступенчатой ректификационной установки, использованной автором для обогащения природной воды изотопом 18О до концентрации 5,8%. Испытания, проведенные на этой установке при 300 мм рт. ст., позволили с большой точностью определить значение а при данном давлении и температуре 76 °C, оказавшееся равным 1,0068 [64]. Уваров с сотр. [54, 65], работая на непрерывно действующей при атмосферном давлении ректификационной колонне диаметром 52 мм, заполненной на высоту 9,5 м насадкой из треугольных спиралей размером 2,0X1,6 мм, добился обогащения 18О от 3 до 24,5 %. ВЭТС составляла около 1 см. Более вы-
Рис. 160.
Металлическая ректификационная установка для непрерывного получения Н218О.
сокое обогащение тяжелой воды изотопом кислорода путем ректификации получил Сташевский [70].
На рис. 160 показана установка непрерывного действия, снабженная медной колонной диаметром 76,2 мм и высотой 9,14 м. Насадка выполнена в виде колец Рашига из проволочной сетки диаметром 1,6 мм. Установка автоматизирована и снабжена специальными аппаратами для электролиза кубовой жидкости, обогащенной 18О. Выделяющийся в виде газа изотоп 18О вступает в реакцию с водородом, также образующимся при электролизе. В качестве конечных продуктов получают таким образом Н218О и D216O.
Если при обычной ректификации время установления стационарного режима лежит в интервале от нескольких минут до 24 ч, то при ректифика-
ции изотопов этот период может длиться несколько недель и даже месяцев. Формулы для определения данного периода времени, представленные в ранних работах, не являются удовлетворительными. Кун с сотр. [59] предложил
Таблица 37.
Результаты простого и двойного концентрирования
Вещество ”эфф Параметры
верхняя часть колонны (13С)
*b,/*b2 (Рл~Рт>+т + ~ + °C
Хлороформ 610 0,62 —8-Ю-4 +0,02
Четыреххлористый углерод 80 0,90 —1,3-10-* +0,033
Метанол 140 0,97 —2 • 10~4 +0,0055
Бензол 80 0,98 —2,5-10~4 +0,006
и рт — давление паров вещества, содержащего легкий и тяжелый изотоп соответст изотопа в кубе и в головке колонны соответственно; /л и tT — температура кипения вещест
232
для расчета времени установления стационарного режима установок с многотрубчатыми и насадочными колоннами (времени пуска) следующую формулу:
>в| <I7S’
где Н — удерживающая способность колонны по жидкости; Z — расход исходной смеси.
Другие сведения по вопросу о получении D и 18О можно найти в работах Джексона и Пигфорда [67], Розена [68] и Бродского [38]. В книге Лондона и Кохена [69] рассмотрена общая теория разделения изотопов. В ней содержатся также сведения об оптимизации условий разделения с использованием каскада ректификационных колонн, которая играет важную роль в промышленной ректификации, в том числе при разделении изотопов [71 ].
5.1.4.3. Получение других изотопов
В настоящем разделе приведены дополнительные примеры использования ректификации для концентрирования изотопов. Относительная разность молекулярных масс ДМ и естественная концентрация наиболее благоприятны у изотопов С и С1:
12С 98,9% „ зьС1 75 4о/
Д/И=8,35% Д/И = 5,7%
1»С 1,1% 37С1 24,6% 0
Кун [59], проводя ректификацию в многотрубчатой колонне, обладающей эффективностью около 250 теоретических ступеней разделения, добился увеличения концентрации 13С на 7%. Концентрацию определяли с помощью масс-спектрометра. При этом был установлен интересный непредсказуемый факт, что тяжелый изотоп углерода 13С накапливается в низкокипящей фракции, а изотоп 37С1 — в высококипящей фракции. Подобный эффект
изотопов при ректификации
процесса
куб (”С1) куб (•’ О)
*B/*BS (Рл-Рт)/Рт 'л~ + °С *b/*b2 (рл-рт)/рт t„-t, °C л т
1,06 1,003 + Ю“Б +3-10-4 —0,003 —0,001 1,50 3-10-" —0,07
венио, при температуре ректификации (34,6 °C); и xg2 — концентрация выделяемого ва, содержащего легкий и тяжелый изотоп соответственно.
233
возникает и при разделении смеси изотопов бора 10В—ПВ. Баерчи и Кун [72] показали, что причиной более высокой летучести тяжелого изотопа является вклад тепловых колебаний его молекул в дисперсионное взаимодействие.
Было установлено, что при ректификации СС14 происходит двойное разделение изотопов. Относительная летучесть смеси 12С—13С примерно составляет 1,00125, что соответствует разнице температур кипения около 0,036 °C (см. табл. 36). Эффект накопления 13С в верхней части колонны наблюдался также при ректификации хлороформа, бензола и метанола. При ректификации хлороформа, кроме того, происходит концентрирование 37С1 в кубе, а при ректификации метанола в кубе накапливается 18О (табл. 37) [43].
Траузер с сотр. исследовал возможность использования молекулярной дистилляции и дистилляции на «коротком пути»* для концентрирования изотопа лития. Он разработал одно-и многоступенчатую установку и определил, что относительная летучесть разделяемой смеси изменяется на первой ступени в пределах от 1,052 до 1,064 при изменении температуры в интервале от 535 до 627 °C. Подобным образом можно разделять и изотопы ртути.
5.2. КЛАССИФИКАЦИЯ МЕТОДОВ
ПЕРЕГОНКИ ПО СПОСОБУ
ПРОВЕДЕНИЯ ПРОЦЕССА
В лаборатории процессы дистилляции и ректификации проводят как при периодическом режиме работы колонны, так и при непрерывном режиме. Необходимость повышения пропускной способности установок приводит к применению полунепрерывных и непрерывных методов работы. В специальных случаях можно применять метод парциальной конденсации, который при правильном использовании обладает определенными преимуществами по сравнению с другими методами перегонки.
5.2.1. ПЕРИОДИЧЕСКАЯ И ПОЛУНЕПРЕРЫВНАЯ перегонка
Под периодической перегонкой понимают процесс разделения некоторого количества смеси, однократно введенного в куб. Различают простую перегонку и фракционную разгонку. При простой перегонке весь дистиллят собирают в одном приемнике. В результате этого процесса исходная смесь разделяется на две части: дистиллят и кубовый остаток. Дистиллят, полученный при фракционной разгонке, состоит из отдельных порций (фракций). Основное отличие периодического процесса от непрерыв-
* Определение этого термина см. в разд. 5.4.4. — Прим. ред. 234
Рис. 161.
Установка с колонной я подогревателем исходной смеси для полунепрерывной вакуумной ректификации.
ного заключается в том, что в периодическом процессе в результате отбора дистиллята непрерывно изменяется состав и количество кубовой жидкости.
Эффективность использования тепла и производительность обычной лабораторной установки можно повысить, применяя полунепрерывный метод работы. От периодического метода он отличается тем, что в куб по мере отбора дистиллята добавляют исходную смесь. Кубовая жидкость обогащается высококипящей фракцией. Исходную смесь подают непосредственно в куб или несколько выше него. При этом данную смесь предварительно подогревают до температуры, близкой к температуре кипения (см. рис. 138). Размер выбранного куба должен соответствовать общему количеству разделяемой смеси и заданному объему высококипящей фракции.
Полунепрерывный метод перегонки применяют в тех случаях, когда нужно отделить низкокипящую фракцию или сконцентрировать смесь, содержащую растворенные твердые вещества. В последнем случае непрерывной подачей исходной смеси предотвращают загустевание или даже затвердевание кубовой жидкости. По истечении определенного промежутка времени подачу исходной смеси прекращают и содержимое куба разделяют периодической перегонкой или выводят в качестве кубового остатка. Непрерывный отбор кубовой жидкости невозможен, поскольку с исходной смесью в куб поступает низкокипящая фракция, которая предварительно должна быть отделена. На рис. 161 показана ректификационная установка полунепрерывного действия с колонной и подогревателем исходной смеси в виде сосуда с обогревающим кожухом.
5.2.2. НЕПРЕРЫВНАЯ РЕКТИФИКАЦИЯ
В отличие от периодической и полунепрерывной ректификации при непрерывном методе работы исходную смесь непрерывно подают в колонну через штуцер, расположенный между укрепляющей и исчерпывающей ее частями (рис. 162). После пуска установки все условия проведения процесса ректификации остаются постоянными. Исходную смесь, предварительно подогретую до температуры, равной температуре жидкости в колонне в месте ввода исходной смеси, разделяют до заданного соотношения на дистиллят и кубовый продукт, которые имеют постоянный состав.
235
Рис. 162.
Вакуумная ректификационная установка непрерывного действия с устройством для подачи исходной смеси (по нормалям «Дестииорм»):
1 — устройство для подачи исходной смеси; 2 — пульт управления; 3 — подогреватель исходной смеси; 4, 8 — мерные бюретки; 5 — термостатированный сосуд для исходной смеси; 6 — головка колонны с мерной бюреткой; 7 — штуцеры для присоединения к вакуумной линии; 9 — укрепляющая часть колоииы; 10 — тройиик для ввода исходной смеси; 11 — исчерпывающая часть колонны; 12 — контактный манометр; 13 — измеритель расхода жидкости, поступающей в куб; 14 — пробоотборник кубовой жидкости; 15 — куб; 16 — нагреватель куба.
Удерживающая способность колонны по жидкости также стабилизируется при постоянных разности температур и градиенте концентраций (см. разд. 4.7.2).
Основное преимущество непрерывной ректификации состоит в том [28], что разделяемая смесь находится в мягких температурных условиях. Кроме того, при непрерывной работе часто удается достигнуть производительности лабораторной установки, такой же, как и для полупромышленных установок периодического действия. Непрерывно работающие лабораторные установки производительностью 10—20 кг/сут можно использовать для получения различных продуктов, например термически нестойких фармацевти-
ческих препаратов, для отгонки растворителей и т. д. Пропускная способность лабораторных установок составляет 0,5— 5 л/ч. Сильно агрессивные вещества, вызывающие коррозию металлической аппаратуры, обычно разделяют в стеклянных установках непрерывного действия. На основе опытных данных, полученных с использованием таких установок, с достаточной степенью надежности можно разрабатывать полупромышленные и промышленные установки из фарфора, технического стекла или металла.
Другое преимущество непрерывной ректификации состоит в том, что при стационарном режиме работы колонны получают дистиллят и кубовый продукт постоянного состава. Кроме того, затраты тепла оказываются существенно ниже, чем в периодическом процессе. При работе на лабораторных ректификационных установках обычно легко обеспечивают постоянство состава исходной
236
смеси, являющееся необходимым условием для непрерывного процесса. Точный состав разделяемой смеси можно предварительно установить путем аналитической ректификации или посредством пробной перегонки (см. разд. 7.2).
5.2.2.1. Области применения непрерывной ректификации
По основным областям применения непрерывную лабораторную ректификацию можно разбить на следующие группы.
1) Опытная ректификация с применением промышленных методов. Эта ректификация используется при разработке процессов разделения в полупромышленном или промышленном масштабе, для сравнительной ректификации, моделирующей промышленный процесс, и для демонстрационных исследований в учебных целях.
2) Предварительное разделение на фракции больших количеств веществ (около 50—100 л). Этой ректификацией пользуются для отделения низкокипящей фракции от высококипящего остатка и для выделения среднекипящей фракции.
3) Наработочная ректификация при производительности установки до 20 кг/сут (см. разд. 5.1.3). С помощью этой ректификации разделяют термически нестойкие смеси, например эфирные масла, ароматические вещества, выделяют чистые вещества из смесей, очищают сырье и растворители, получают фармацевтические препараты.
В разработке промышленных ректификационных установок отчетливо видна тенденция к переходу от периодических методов разделения к непрерывным. Это заставляет и исследовательские лаборатории заниматься данной проблемой.
Методы разделения, реализуемые в крупных установках, необходимо предварительно разрабатывать в лаборатории при одинаковом аппаратурном оформлении процесса. Экспериментальным путем можно быстрее решить поставленную задачу, чем посредством расчетов, особенно при исследовании разделения многокомпонентных смесей. Очевидно также, что лабораторные исследования — это наиболее экономичный путь исследований, так как проведение опытов с использованием промышленных установок требует значительно больших затрат материалов, энергии и времени. Вследствие введения принципа сборки лабораторной установки из отдельных стандартных деталей появилась возможность с помощью лабораторной аппаратуры в значительной мере воспроизвести промышленную установку и благодаря этому смоделировать (конечно, в уменьшенном масштабе) процесс разделения. Таким образом, на основе лабораторных исследований можно проектировать полупромышленные и промышленные установки.
До настоящего времени после разработки метода разделения в лаборатории ставилась задача найти путь перехода от лаборатор
237
ного способа разделения к промышленной установке. Появление сборных комбинированных установок в значительной степени исключило такой путь *. Исследования по ректификации, описанные в разд. 5.2.2.4, проведены с учетом особенностей промышленных установок. Подобные лабораторные установки позволяют также исследовать процессы перегонки, уже внедренные в промышленность, с целью изучения причин нарушений в работе больших установок путем сравнительной и модельной ректификации [73]. Можно также уточнить конструктивные (геометрические) параметры промышленных ректификационных колонн, предназначенных для точного фракционирования изотопов, посредством опытной ректификации в промышленном масштабе. С помощью специальных расчетных программ можно найти распределение концентраций по высоте испытуемой ректификационной колонны, находящееся в хорошем соответствии с опытными данными. На основании этого можно определить оптимальные конструктивные (геометрические) параметры промышленных колонн (см. разд. 4.15; [73а]).
Для разделения бинарных смесей с большой разностью температур кипения преимущества непрерывного метода работы особенно очевидны. На сравнительно простых установках можно проводить ректификацию непрерывно в течение продолжительного времени, достигая сравнительно высокой производительности в условиях работы как при атмосферном давлении, так и под вакуумом. В установке, состоящей из двух ректификационных колонн, можно непрерывно разделять тройную смесь (см. разд. 4.9). Известно, что для разделения смеси, состоящей из п компонентов, необходимо п— 1 колонн. Однако, смеси с числом компонентов больше трех вследствие значительных аппаратурных затрат следует разделять не за один проход, а прежде выделить две или три фракции из нескольких компонентов, после чего эти фракции периодически разделить на чистые вещества.
Если разделяемая смесь имеет большой высококипящий остаток, то сначала следует отделить его в пленочной ректификационной колонне (см. разд. 5.4.3), поскольку высокомолекулярная фракция исходной смеси может легко разлагаться или полимеризоваться при ректификации в обычных условиях. Такое предварительное разделение в мягких температурных условиях дает воз-
* Следует, однако, иметь в виду, что эффективность ректификационной колонны существенно зависит от ее геометрических размеров и лабораторные данные не могут быть в полной мере использованы для разработки промышленной установки. Моделирование ректификационной аппаратуры является предметом специальных исследований. Необходимо также учитывать, что процесс разделения, проводимый в установках из металла, часто отличается от процесса, проте-каемого в стеклянной аппаратуре, особенно при разделении термически нестойких веществ. Вследствие этого необходимо одновременно проводить исследования термической стойкости компонентов исходной смеси в присутствии материала, из которого предполагается изготавливать промышленную установку. — Прим, ред.
238
Исходная смесь С4—С30
4 остаток > С20
4 непрерывная пленочная ректификация
4
Ci—С2о ' 4
непрерывное самопроизвольное испарение
4 4
фракция Cj—С4 С4—С20
4 Г“
периодическая ректификация непрерывная ректификация
I
4 4
непрерывная ректификация непрерывная ректификация
периодическая периодическая периодическая периодическая
ректификация ректификация ректификация ректификация
Рис. 163.
Схема разделения многокомпонентной смеси Ct—С20
можность значительно повысить выход высококипящих компонен-тов. Низкокипящую фракцию можно отделить от основной части методом расширительной перегонки (см. разд. 5.4.2), позволяющей вести процесс с высокой скоростью (до 3 л/ч). С помощью этих двух вышеописанных методов можно выделять основную часть смеси, свободную от высококипящего остатка, а затем фракционировать ее на обычной ректификационной установке непрерывного действия. Схема разделения многокомпонентной смеси Сг—С20 наглядно поясняет подобный процесс (рис. 163).
5.2.2.2. Теоретические расчеты
Расчет процессов периодической или непрерывной ректификации предполагает определение следующих параметров: рабочего давления (атмосферного или вакуума), требуемого числа теоретических ступеней разделения, минимального флегмового числа, температуры исходной смеси, количества затрачиваемого тепла.
Обычно перед непрерывной ректификацией для определения ее основных параметров проводят периодическую пробную перегонку (или еще лучше аналитическую ректификацию). Рекомендации по выбору давления приведены в разд.5.4. Требуемое число
230
Рис. 164.
Зависимость к. п. д. тарелок от нагрузки при циклическом методе работы с тремя различными периодами цикла (1—3) и непрерывном методе (4) [76 ].
теоретических ступеней разделения и минимальное флегмовое число рассчитывают по методикам, описанным в разд.4.7.2, а рабочую температуру исходной смеси выбирают как можно ближе к температуре кипения. Необходимые размеры ректификационной ко-
лонны вычисляют по формулам, представленным в разд. 4.11, тепловые расчеты проводят по методике, изложенной в разд.4.12. При расчете диаметра колонны принимают во внимание, что в исчерпывающей части колонны количество стекающей жидкости больше, чем в укрепляющей части, вследствие подачи исходной смеси. Если количество дистиллята невелико (около 10—20% от количества исходной смеси), то рекомендуется выбирать колонну с диаметром исчерпывающей части большим, чем получается по расчету, исходя из нагрузки укрепляющей части колонны.
Следует указать на возможность проведения процесса ректификации циклическим методом, исследованным Гельбиным [74]. Например, Каннон [75] предложил подавать пар в ректификационную колонну циклически с периодом 3 с, для этого на паропроводе, соединяющем испаритель с колонной, устанавливают соответствующее регулирующее устройство. Мак-Виртер и Ллойд [76] для реализации циклического метода разделения применяли тарельчато-насадочную колонну, на пяти тарелках которой размещались небольшие слои насадки. При разделении этим методом смеси метилциклогексан—толуол они добились значительного повышения производительности ректификационной колонны. Были определены оптимальный период цикла и характер зависимости нагрузки от времени. Из графика, приведенного на рис. 164, отчетливо видно, что к.п.д. тарелок со слоями насадки при циклическом методе работы значительно выше, чем при непрерывном процессе.
5.2.2.3. Аппаратура для непрерывной ректификации
В лабораторных и промышленных ректификационных установках непрерывного действия используют в основном насадочные и тарельчатые колонны. Тарельчатые колонны, изготовленные из стекла, применяют для специальных целей, в том числе для разделения агрессивных веществ.
240
В 1931 г. Бурстин и Винклер [77 ] разработали установку с трубчатой колонной для непрерывной дистилляции при атмо-s' сферном давлении и под вакуумом, изготовленной из чугуна и никелевой стали (рис. 165). Установка предназначена для разде-? ления кубовых остатков перегонки сырой нефти. Она оказалась 1 пригодной также и для дистилляции легко разлагающихся и пе-нящихся жидкостей, поскольку в ней использован принцип пле-L ночной дистилляции. По конструктивному выполнению установка полностью соответствовала промышленному агрегату, поэтому Г все опытные данные, полученные при работе на ней, можно ИС-S. пользовать при разработке промышленного процесса.
i Коллинг и Трамм [78] описали регулирующее устройство для
f‘ лабораторной колонны непрерывного действия, обеспечивающее I автоматическое ведение процесса ректификации. На рис. 166 показана схема установки Коллинга и Трамма, предназначенной !для непрерывной ректификации при атмосферном давлении.
О вакуумной ректификационной установке непрерывного действия, выполненной по нормалям «Дестинорм» (см. рис. 162) сообщалось в разд. 5.2.2. На рис. 167 показана непрерывно дей-s ствующая ректификационная установка типа «Лабодест», пред-
, назначенная для работы при атмосферном давлении и под ваку-
' умом с остаточным давлением до 20 мм рт. ст. Ректификационная
колонна снабжена стеклянными колпачковыми тарелками с отра-« жательными перегородками для пара (см. разд. 7.3.3), обладаю-» щими к. п. д. от 80 до 100%. Подобные тарелки аналогичны по
Рис. 165.
Установка Бурстина—Винклера для непрерывной дистилляции с обогреваемой трубчатой колонной:
1 — напорный бак; 2 — игольчатый клапан; 3 — стеклянный шар для измерения расхода исходной смеси; 4 — штуцеры для присоединения к вакуумной линии; 5 — холодильники; 6 — карманы для термометров; 7 — металлический блок; 8 — газовые горелки; 9 — приемники дистиллята; 10 — дефлегматор; // — кожух с изоляцией из асбеста.
241
Рис. 166.
Автоматическая установка Коллиига— Трамма для непрерывной ректификации при атмосферном давлении:
1 — реле; 2 — контактный манометр; 3 — сосуд для холодной смеси; 4 — электромагнитные клапаны; 5 — электрические контакты реометра; 6 — клапан; 7, 13 — капилляры; 8 — реометр; 9 — линия сжатого воздуха; 10 — контактные термометры; 11 — термометр; 12 — холодильник полной конденсации; 14 — игольчатые клапаны; 15 — приемник дистиллята; 16 — кран для слива дистиллята; 17 — переливной патрубок; 18 — обогревающий кожух; 19 — теплоизолирующий кожух; 20 — электронагреватель.
Рис. 167.
Непрерывно работающая ректификационная установка Штаге модели «Ла-бодест» с колонной из стекла, содержащей колпачковые тарелки с отражательными перегородками для пара:
1 — насос; 2 — циркуляционный испаритель; 3 — предохранительное уст-стройство; 4 — регулятор давления с дифференциальным манометром; 5, 8 — исчерпывающая и укрепляющая части колонны с обогревающими кожухами; 6 — подогреватель исходной смеси; 7 — сосуд для исходной смеси; 9 — головка колонны; 10 — конденсатор; 11 — расширительный сосуд; 12 — буферный сосуд;. 13 — холодильник дистиллята; 14 — коллектор с кранами; 15 — охлаждаемая ловушка; 16 — шкаф управления; 17 — сменный приемник дистиллята; 18 — расходомер жидкости, стекающей из колонны; 19 — холодильник кубового продукта; 20 — промежуточный приемник кубового продукта.
Изготовитель: фирма Фишера «Лабораторная и исследовательская техника», Бонн — курорт Годесберг.
242
принципу работы промышленным тарелкам, поэтому на основе экспериментальных данных, полученных с использованием этой колонны, можно непосредственно разработать промышленную установку. Данная ректификационная установка особенно пригодна для демонстрационных целей, поскольку она позволяет визуально наблюдать за ступенчатопротекающим процессом массообмена. Установка поставляется с колоннами высотой 600— 1300 мм, диаметром 30—120 мм и числом тарелок 5—28 при расстоянии между тарелками в колонне 35—150 мм. Следует отметить также, что циркуляционный испаритель по конструктивному выполнению аналогичен промышленному аппарату. Все основные измерительные и регулирующие приборы сосредоточены в специальном шкафу управления.
Таким образом, на примере данной ректификационной установки отчетливо видна тенденция к созданию лабораторных и пилотных установок, наиболее приближенных к промышленным. Возможность сборки установок из серийных деталей и узлов, разработанных Штаге (серия приборов «Лабодест»), предприятием Фишера «Лабораторная и исследовательская техника» (Бонн— курорт Годесберг) и различными другими фирмами, способствовали упрощению конструктивного оформления установок.
Необходимо отметить также приборы, выполненные по нормалям «Дестинорм», выпускаемые народным предприятием «Комбинат технического стекла» (Ильменау), и детали для ректификационных установок, изготовляемые Народным предприятием «Завод стекла Шотт» (Иена). Поскольку данные узлы и детали стандартизованы, при выходе из строя какого-либо элемента установки можно быстро заменить его. При дальнейшей разработке серийных деталей и аппаратуры ректификационных установок Необходимо более широко моделировать их промышленные образцы.
5.2.2.4. Примеры непрерывной лабораторной ректификации
В лабораторных ректификационных установках непрерывного действия, описанных выше, можно разделять жидкие смеси как при атмосферном давлении и под вакуумом, так и при небольших избыточных давлениях (см. разд. 5.4.5). Кривые разгонки продуктов непрерывной ректификации смеси жирных кислот С4 — С]9, представленные на рис. 168, показывают достигаемую'при этом степень разделения.
При использовании регулирующих устройств на ректификационной установке, показанной на рис. 162, можно непрерывно разделять на основные компоненты смеси фенолов. На рис. 169 приведены результаты, полученные на первой стадии разгонки, при которой выделяют о-крезол и смесь м- и п-крезолов. Для аналитического контроля было отобрано 120 проб; определяли плотность кубового продукта и температуру затвердевания дистиллята. Как видно из диаграммы, в течение 22 ч работы значения температур исходной смеси, дистиллята и кубовой жидкости, а также их физико-химических свойств изменялись незначительно. Вакуум в системе регулировали с помощью автоматического стенда с вакуумным насосом (см. разд. 8.3).
243
Выход дистиллята, °/о(масс.) Рис. 168.
Диаграммы пробной разгонки продуктов непрерывной лабораторной ректификации смеси жирных кислот С4—С8 (а) и С10 — С1а (б).
Результаты непрерывной ректификации смеси фенолов.
Давление 30 мм рт. ст.; продолжительность испытаний 22 ч;
Дистиллят: смесь фенол—о-крезол; Кубовый продукт: смесь м- и га-кре-зол —ксиленол—высококипящий остаток. /, 2, 3 — температура кубовой жидкости, исходной смеси и дистиллята соответственно; 4 — плотность кубовой жидкости; 5 — температура затвердевания дистиллята.
Таким же способом непрерывно ректифицируют сырой фенол с температурой затвердевания 37,0 °C, получая при флегмовом числе 10 и давлении 60 мм рт. ст. чистый фенол (99,2%) с температурой затвердевания 40,3 °C. Высококипящий продукт непрерывно выводят из куба, поэтому его время пребывания в кубе значительно меньше, чем при периодическом методе работы. Благодаря этому существенно уменьшается термическое разложение получаемого вещества.
Применение непрерывного метода при азеотропной и экстрактивной ректификации, расширительной и пленочной перегонке, а также молекулярной дистилляции подробно рассмотрено ниже (см. разд. 5.4.2, 5.4.3, 5.4.4 и 6.2).
5.2.2.5 Выведение ректификационных установок на стационарный режим
Работа отдельных регулирующих устройств ректификационных установок рассмотрена в разд. 5.2.2.3, здесь обсуждены дополнительно некоторые вопросы.
Для уменьшения времени установления стационарного режима работы разделяемая смесь, вводимая в куб 15 (см. рис. 162), в момент подачи ее в колонну должна иметь состав, соответствующий составу кубового продукта. Для обеспечения хорошего смачивания насадки жидкость, введенную в куб, необходимо предвари-244
тельно перегнать периодическим методом, отбирая дистиллят ожидаемого состава. Затем в тройник 10 подают исходную смесь, которая предварительно нагревается до требуемой температуры в подогревателе 3. С помощью мерных бюреток 4 и 6 устанавливают соответственно расход исходной смеси и флегмовое число. Нагрузку укрепляющей части ректификационной колонны, зависящую от скорости подачи исходной смеси, дополнительно регулируют с помощью контактного манометра 12. В соответствии с изложенным в разд. 4.7.2 установка должна работать таким образом, чтобы расходы отбираемых дистиллята и кубового продукта соответствовали расходу исходной смеси. Поэтому краны на мерных бюретках головки колонны 6 и приемника кубового продукта 8 необходимо устанавливать в таких положениях, чтобы в единицу времени через них проходили соответствующие количества жидкости.
В качестве примера приведем оптимальные параметры процесса непрерывного разделения смеси бензол—толуол, содержащей 20% (об.) бензола. При подаче 500 мл/ч исходной смеси необходимо отбирать 100 мл/ч дистиллята и 400 мл/ч кубового продукта. При флегмовом числе 2 нагрузка должна быть равна 300 мл/ч. Время выхода установки на стационарный режим составляет от 0,5 до 1 ч, что определяется колебанием температур в головке и кубе колонны (см. рис. 169) *. После того как температура подогрева исходной смеси отрегулирована, установка работает с постоянными технологическими параметрами, и необходимое обслуживание установки ограничивается лишь контролем за расходами исходной смеси и отбираемых продуктов, а также наблюдением за показаниями контрольно-измерительных приборов.
5.2.3. РАЗДЕЛЕНИЕ МЕТОДОМ ПАРЦИАЛЬНОЙ КОНДЕНСАЦИИ
Под парциальной конденсацией (дефлегмацией) понимают частичную конденсацию движущихся паров. Ее проводят в специально предусмотренной для этого части колонны—дефлегматоре. Самопроизвольно дефлегмация может происходить при утечке тепла от стенок колонны в случае неудовлетворительной теплоизоляции. Ее называют также краевой конденсацией (рис. 170а).
Метод ступенчатой конденсации паров в дефлегматоре, состоящем из системы труб, при понижающейся температуре охлаждения позволяет разделять и получать фракции компонентов с большой разницей температур кипения (рис. 1706). Примеры расчетов для подобной чистой частичной конденсации, при которой образующийся конденсат сразу же выводится из системы, представ-
* Указанный период времени выхода на стационарный режим не относится к процессам ректификации изотопов и близкокипящих веществ, для которых пусковой период значительно больше. — Прим. ред.
245
Рис. 170.
Дефлегматор (/) с колонной (2) для краевой конденсации (а) н прибор для ступенчатой парциальной конденсации (б) Приборы Штаге модели «Лабодест», тип III.
лены в работе Зигварта [81 ]. Степень разделения при частичной конденсации можно значительно увеличить, обеспечивая
противоточное движение потоков пара и жидкости [82]. При ректификации пары, выходящие из колонны, должны быть разделены в заданном соотношении, вытекающем из задачи разделения. В промышленности дефлегматор часто применяют для получения флегмы, а отбираемый дистиллят конденсируют в продукционном конденсаторе. Таким образом используют укрепляющее действие дефлегматора для концентрирования высококипящей фракции *. Метод парциальной конденсации применяется прежде всего для разделения газовых смесей в технике глубокого холода. Отто [83 ] сообщает о проведении парциальной конденсации в вертикальных трубах для разделения бинарных смесей.
Эффективность дефлегматора зависит от природы разделяемой смеси [84] и обычно не превышает одной теоретической ступени разделения. Преимущество дефлегматора заключается прежде всего в его незначительной удерживающей способности по жидкости. Ниже приведены два примера, иллюстрирующие эффективность дефлегматоров при разделении смесей этанол—вода и бен-ьол—толуол:
Этанол— Веизол— вода толуол
Концентрация низкокипящего компонента перед
дефлегматором, % (мол.).................... 67
Концентрации низкокипящего компонента после дефлегматора, % (мол.)..................... 78 80
Флегмовое число................................ 5 5
Число теоретических ступеней разделения . . 3 1
* В современных промышленных установках парциальные конденсаторы применяют редко, так как это затрудняет регулирование работы ректификационных колонн и делает последние более громоздкими из-за необходимости сооружении крупногабаритных дефлегматоров. Кроме того, укрепляющее действие дефлегматора становится относительно малым в колоннах с большим числом теоретических ступеней разделении. — Прим. ред.
246
Разделяющую способность дефлегматора можно рассчитать по формуле Фабуса:
1g О-ви + 1) =^-Г(а1б-7- + 1й4^-) (176)
где уви — внутреннее флегмовое число, равное отношению количества конденсата (моль/ч) к количеству входящего пара (моль/ч); уг — концентрации низко-кнпищего компонента в паре на входе в дефлегматор (мол. доли); — кон-центрация ннзкокипищего компонента в ларе на выходе из дефлегматора (мол. доли).
Значения уг и у2 можно определить из этого выражения с помощью специально разработанной номограммы [85]. Применительно к многокомпонентной смеси Фишер [86] предложил уравнение для расчета концентрации паров, выходящих из дефлегматора, и состава конденсата. Герман [87] попытался разрешить некоторые сложные проблемы парциальной конденсации. Исследования проводили на промышленных парциальных конденсаторах, однако полученные результаты применимы также к лабораторным и пилотным дефлегматорам. Дальнейшая разработка методов определения разделяющей способности парциальных конденсаторов проведена Трестером [88].
При аналитической и препаративной перегонке в лаборатории обычно проводят процесс с полной конденсацией паров. Метод парциальной конденсации используют только при проведении сравнительной ректификации, аналогичной промышленному процессу. В этом случае дефлегматор устанавливают в верхней части колонны (см. рис. 170а). Преимущество метода с полной конденсацией паров состоит в том, что этим методом сравнительно просто разделять конденсат в определенном соотношении, в то же время устанавливать постоянной скорость подачи флегмы с помощью дефлегматора очень затруднительно, поскольку даже незначительные колебания расхода и температуры охлаждающей воды вызывают изменение составов флегмы и паров дистиллята, а также их количеств. В промышленности скорость подачи флегмы при перегонке методом парциальной конденсации обычно не измеряют, а регулируют степень охлаждения дефлегматора по температуре в головке колонны. Количество образующейся флегмы рассчитывают приблизительно, измеряя расход и температуру охлаждающей воды на входе и выходе дефлегматора с учетом удельной теплоты испарения дистиллята. Поскольку в промышленности обычно работают с одними и теми же продуктами, такой метод вполне пригоден. Однако при разделении многокомпонентной смеси определение количества подаваемой флегмы подобным образом становится слишком неточным.
Для лабораторных исследований с целью моделирования промышленного процесса, основанного на парциальной конденсации, можно применять разработанную автором специальную головку колонны для частичной конденсации, выполненную”по нормалям «Дестинорм» (рис. 171). Поднимающиеся пары частично конденси-
247
6
Рис. 171.
Головка колонны для парциальной конденсации с капилляром для счета капель по нормалям «Дестинорм»:
1 — теплоизолирующие полуцилиндры из стекловолокна; 2 — воронка для приема дистиллята; 3 — дефлегматор; 4,5 — термометры; 6 — конденсатор; 7 — перфорированная воронка; 8 — кран; 9 — капилляр для счета капель дистиллята; 10, 12 — мерные бюретки для дистиллята и флегмы соответственно; 11 — приемная бюретка для дистиллята.
Рис. 172.
Ректификационная колоииа с тремя парциальными конденсаторами [92].
руются в дефлегматоре 3. Образовавшийся конденсат через приемную воронку 2 отводится в мерную бюретку 12 с капилляром для счета капель, позволяющую в любой момент времени точно измерять скорость подачи флегмы. Участок головки между приемной воронкой и ректификационной колонной изолируют полуцилиндрами 1 из стекловолокна. Температуру охлаждающей воды на входе и выходе дефлегматора измеряют термометрами 4 со стандартными шлифами. Пары, не сконденсированные в дефлегматоре, полностью конденсируются в конденсаторе 6. Конденсат стекает в перфорированную воронку 7 и при открытом кране 8 проходит через капилляр для счета капель 9 в мерную бюретку 10. Между мерной 10 и приемной 11 бюретками можно дополнительно установить холодильник.
Головки для частичной конденсации применяют в основном при проведении сравнительной ректификации, поскольку разделяющая способность дефлегматора сравнительно невелика, и точное регулирование флегмового числа в лаборатории сопряжено со значительными трудностями. Метод ступенчатой парциальной конденсации преимущественно используют для предварительного разделения смесей компонентов с большой разницей в температурах кипения, и прежде всего для отделения воды или другой низкокипящей фракции. В этом случае колонну заменяют системой труб со встроенными конденсаторами и обеспечивают понижение температуры охлаждения потока паров по ходу его движения (см. рис. 1706). С помощью метода парциальной конденсации можно 248
сравнительно быстро и точно разделить, например, смесь жирных кислот С4—С20 на фракции С!—С4, С4—С9 и С9—С20. В промышленности этот метод применяют для разделения низкокипя-щих углеводородов с четырьмя и менее атомами углерода.
По данным Юнге [89] правильно проведенная парциальная конденсация в колонне может повысить ее разделяющую способность. При этом имеется в виду краевая конденсация на стенках колонны, обусловленная потерями тепла, нарушающими адиабатичность работы аппарата. Аналогичные способы ступенчатого разделения описаны также Трение [90]. Однако Кун [91] на основе обширных теоретических исследований и расчетов показал целесообразность проведения частичной конденсации именно в верхней части колонны.
Вебер [92] установил, что при разделении методом парциальной конденсации можно использовать ректификационные колонны с меньшим диаметром верхней части (рис. 172). Возможность уменьшения объема верхней части колонны обусловлена возрастанием в ней концентрации низкокипящего компонента и снижением требуемого флегмового числа. При этом поперечное сечение колонны следует уменьшать в соответствии с ростом концентрации. Кроме того, необходимо устанавливать промежуточные дефлегматоры, пропускная способность которых снижается по ходу движения потока паров. Фойгт [93 ] на основе теоретических исследований показал, что разделительную способность ректификационной колонны можно существенно повысить, если отводить тепло не от определенных участков колонны, а от всей ее поверхности. Метод парциальной конденсации позволяет обогащать пары низкокипящим компонентом и, следовательно, не пригоден, например, для обогащения стабильных изотопов, являющихся в основном высококипящими компонентами. В этих случаях необходимо, наоборот, подводить тепло к стенкам исчерпывающей части ректификационной колонны, чтобы уменьшить ее пропускную способность по жидкости (см. разд. 5.1.4; [93а, б]).
5.3. КЛАССИФИКАЦИЯ МЕТОДОВ ПЕРЕГОНКИ ПО ТЕМПЕРАТУРЕ ПРОЦЕССА
Нормальной температурной областью для проведения процессов ректификации считают интервал от 20 до 250° С. Если температуры кипения разделяемых веществ лежат ниже комнатной температуры, то проводят низкотемпературную ректификацию с использованием специальных хладоагентов для конденсации паров дистиллята. Процессы перегонки, протекающие при 250—400° С, относят к высокотемпературной ректификации. Возможна также изотермическая перегонка, при которой температуру в кубе поддерживают постоянной, а изменяют рабочее давление.
249
5.3.1. НИЗКОТЕМПЕРАТУРНАЯ РЕКТИФИКАЦИЯ
Смеси низкокипящих углеводородов и газов На, Na, Оа и СО можно разделять путем перегонки как при атмосферном давлении с применением специальных хладоагентов, так и при повышенном давлении. Если разделение проводят при повышенном давлении, то стремятся повысить температуру головки колонны до такого значения, чтобы можно было использовать обычные охлаждающие средства (см. разд. 5.4.5). Из-за того, что для перегонки под давлением необходима более сложная аппаратура, чаще применяют лабораторные и пилотные установки низкотемпературной ректификации. Методика проведения низкотемпературной ректификации разработана очень подробно. Созданы полностью автоматизированные установки для проведения низкотемпературной ректификации в интервале от —190 до 20° С. В этих установках применяют как насадочные, так и полые спиральные колонны. Во многих случаях отбираемые пробы дистиллята и кубового продукта анализируют методом газовой хроматографии (см. разд. 5.1.2).
Низкотемпературную ректификацию используют для очистки газов, а также как сравнительную ректификацию, аналогичную промышленному процессу. Это относится прежде всего к очистке отходящих промышленных газов без концентрирования в них водорода и, главным образом, к очистке природного газа, например выделение гелия и азота из природного газа, что по-прежнему является трудной проблемой.
Основные исследования газовых смесей, содержащих наряду с газами жидкие низшие углеводороды, проведены Подбильня-ком [94]. Он разработал колонну для точного фракционирования с насадкой из проволочных спиралей, навитых с малым шагом (так называемая насадка «Хэли-грид», которую применяют при обычной перегонке, см. разд. 7.3.4). В обзоре способов низкотемпературной ректификации Гроссе-Етрингхауз [95] рассмотрел технику проведения этих процессов, использовав экспериментальные данные Вустрова [96 ]. В этом обзоре также указано, что пробы дистиллята и кубовой жидкости следует отбирать очень тщательно с использованием полуавтоматических и автоматических устройств для моментального и непрерывного отбора. Процесс разделения следует проводить следующим образом. Сначала с помощью жидкого азота (—195,8° С) отделяют несконденсировавшуюся часть паров и анализируют её на аппарате Орса. Конденсирующуюся часть исходной смеси необходимо освободить от СОа, На и NH3 в промывном аппарате и сконденсировать. Для ректификации применяют насадочную колонну с посеребренным высоко вакуумированным кожухом; колонна снабжена спиралью, компенсирующей температурные напряжения. Дефлегматор с конической трубой припаивают (рис. 173) или присоединяют с помощью шлифов.
Дефлегматор представляет собой сосуд с изолирующей вакуумированной рубашкой, который предназначен для загрузки 250
хладоагента. Колонна и дефлегматор заполнены насадкой из металлических спиралей (сталь V2A) размерами 2x2x0,2 мм. Установка в значительной степени автоматизирована. Она снабжена такими регулирующими устройствами, как манометр с автоматическим регулированием давления и приспособление для стабилизации температуры в холодильнике.
Шток и Гауптшайн, а также Драун [97] предложили очень удобные устройства для поддержания постоянной температуры в холодильнике головки колонны, основанные на использовании
Рис. 173.
Колонна Гроссе—Етрингха-уза для низкотемпературной ректификации с припаянным дефлегматором.
Рис. 174.
Схема весов Штока для измерения плотности газа.
Рис. 175.
Установка Коха—Гильберата для низкотемпературной ректификации:
1 — спиральная колонна; 2 — кран на шлифе; 3 — термометр; 4 — конденсатор; 5 — капилляр; 6, 10 — пробки на шлифе; 7 — регулировочный кран для отбора дистиллята; 8, 9 — градуированные приемники для газообразного и жидкого Дистиллята соответственно; 11 — электронагреватель куба; 12 — сосуд Дьюара; 13 — куб.
251
жидкого азота или кислорода. Плотность газа можно определять с помощью весов Штока (рис. 174) [98]. Приборы, измеряющие давление паров (см. разд. 4.4.1), используются для определения содержания н- и изобутана. Подробное описание техники работы с газами дал Миллер [99]. В книге «Чистые газы» [100] Мюллер и Гнаук подробно рассмотрели приборы и аппаратуру для получения и транспортировки газов, а также методы их анализа, разделения и очистки.
Принцип работы установок для низкотемпературной ректификации рассмотрен ниже на примере установки Коха и Гильберата [101 ]. Колонна (рис. 175) выполнена в виде стеклянной трубчатой спирали (подобно колонне Янцена, см. рис. 76). Она обладает низкой удерживающей способностью по жидкости (3—4 мл), поэтому для загрузки колонны достаточно 15—25 г жидкости. Колонна работает так же, как и аппараты для разделения при обычных температурах. Благодаря реализации метода полной конденсации можно устайавливать любое флегмовое число. Дистиллят можно отбирать в газообразном или жидком состоянии [103].
Термометр 3 подвешен на тонкой проволоке. Отверстие капилляра 5 закрыто пицеином. Пробка 6 снабжена штуцером для соединения с небольшой осушительной трубкой.
Перед проведением ректификации проверяют качество герметичности кожуха колонны с помощью высокочастотного течеиска-теля. При герметичности кожуха (в затемненном рабочем помещении) в его полости не должно возникать свечения, возможна зеленая флюоресценция стеклянных стенок кожуха. Если возникает свечение, проводят повторное вакуумирование. Для этого используют трехступенчатый диффузионный ртутный насос. Пары ртути вымораживают в глубокоохлаждаемом адсорбере с активным углем или силикагелем, установленном между насосом и колонной. Для смазки кранов применяют высоковакуумную смазку (см. разд. 9.4). При достижении высокого вакуума, соответствующего остаточному давлению 10~5 мм рт. ст. и ниже, кран закрывают.
Куб 13 охлаждают до требуемой температуры с помощью бани, заполненной смесью метанола с сухим льдом или другим хладо-агентом. Одновременно хладоагент загружают в конденсатор 4. Если по каким-либо причинам нежелательно поддерживать постоянную температуру конденсации с помощью криостата с охлаждающим рассолом, то в качестве хладоагентов можно применять жидкий воздух или азот. Затем в кубе 13 конденсируют высушенную и, при необходимости, освобожденную от СО2 пробу газа. После этого вместо охлаждающей бани используют сосуд Дьюара 12. При правильной установке верхний край сосуда Дьюара должен соприкасаться с держателем штатива, поддерживающим куб. Содержимое куба 13 испаряют, как обычно, с помощью электронагревателя 11. Неперегретые пары поступают в спиральную колонну 1, изолированную посеребренным высоковакууми-рованным кожухом и дополнительно стекловолокном. Преду-252
смотрена возможность измерения температуры в верхней части колонны с помощью термометра <3, помещенного в защитный карман, который исключает переохлаждение термометра стекающим конденсатом. Дистиллят отбирают через регулировочный кран 7, расположенный под конденсатором.
Для сбора жидкого дистиллята предназначен градуированный приемник 9, куда дистиллят поступает в виде капель через припаянный спиральный капилляр. Спиральный капилляр и приемник помещены в сосуд Дьюара. Вследствие конденсации паров дистиллята в спиральном капилляре, расположенном ниже регулировочного крана, возникает постоянное разряжение, которое позволяет отсасывать пары из колонны. Для сбора дистиллята в газообразном состоянии предусмотрен градуированный сборник 8 с запорной жидкостью, также устанавливаемый за регулировочным краном. Требуемое постоянное разряжение в этом слу-
Рис. 176.
Установка Штаге с насадочной колонной для препаративной низкотемпературной ректификации:
/ — колонна; 2 — приемник дистиллята; 3 — сосуд для поддержания постоянного напора.
Изготовитель: фирма Фишера «Лабораторная и исследовательская техника», Бонн—курорт Годесберг.
Рис. 177.
Установка Гроссе—Етриигхауза для низкотемпературной перегонки.
Изготовитель: фирма Нормаг, Хофхайм.
253
Рис. 178.
Автоматизированная установка Подбильияка для низкотемпературной ректификации
Изготовитель: фирма Подбильияка, Департамент обрабатывающей промышленности, Фраиклни парк, Иллинойс, США.
чае получают, изменяя высоту уровня запорной жидкости.
Если температура кипения дистиллята ненамного превышает температуру его затвердевания, то необходимо точно..регулировать температуру в конденсаторе. Штаге [102] разработал для препаративных целей установку низкотемпературной ректификации, в которой температуры куба и конденсатора регулиру
ются автоматически (рис. 176). В качестве хладоагента используется жидкий воздух. В тех случаях, когда температура хладоагента наименьшая, открывается предварительно установленный на контактном термометре магнитный клапан и в поток хладоагента подается дополнительный воздух. Дистиллят отбирается также с помощью магнитного клапана. Низкотемпературный холодильник Гроссе-Етрингхауза (рис. 177) предназначен для перегонки низкокипящих жидкостей или жидкостей с растворенными газами. Пары или газы, несконденсировавшиеся в первом холодильнике, конденсируются в низкотемпературном холодильнике приемника дистиллята, который заполнен подходящей охлаждающей смесью.
Ректификационная установка Подбильияка (рис. 178) модели «Термокон» серии 8700 работает автоматически в интервале температур от —200 до 20° С [104] (см. разд. 8.1).
Видоизмененная ректификационная установка Подбильияка модели «Рургаз» рассчитана на загрузку 3 -4 л газа при атмосфёр,-ном давлении.
Ректификация в установке продолжается примерно' 2 й; столько же времени требуется для предварительной конденсации газа. Встроенный автоматический термограф непрерывно записывает температуру верхней части колонны. ; ‘ .
На рис. 179 показана диаграмма перегонки, проведенной на этой установке [105].
254
Для низкотемпературной ректификации небольших количеств сжиженных газов пригодна колонна Симонса [106], помещаемая в сосуд Дьюара (рис. 180).
Минимальное флегмовое число для процессов низкотемпературной ректификации можно приближенно рассчитать по формуле Подбильияка [107]; эта формула получена в предположении, что концентрация кубовой жидкости составляет 50% (мол.):
( а \ ( 1 \ ( хр \ [ 1
VMHH-^£-а_1 J [*в(1-*в)
где хЕ — концентрация низкокипящего компонента в дистилляте, мол. доли; хв — концентрация никзокипящего компонента в кубе, мол. доли.
Для определения минимального числа теоретических ступеней разделения при низкотемпературной ректификации с бесконечным флегмовым числом служит номограмма Подбильияка, показанная на рис. 181 [107]. Номограмма составлена для концентрации кубовой жидкости 50% (мол.) с учетом относительной летучести компонентов смеси и требуемой концентрации дистиллята. Номограммой пользуются следующим образом. Сначала по давлениям паров определяют относительную летучесть а компонентов разделяемой газовой смеси (см. разд. 4.4). Затем от точки, соответствующей найденному значению а, проводят вертикальную прямую до пересечения с кривой, соответствующей определенной
Рис. 179.
Диаграмма низкотемпературной ректификации, проведенной иа установке модели < Рургаз > .
Рис. 180.
Микрбколоииа Симонса для низкотемпературной ректификации.
255
Рис. 181.
Номограмма Подбильняка для определения минимального числа теоретических ^ступеней разделения пмин при низкотемпературной ректификации с V — ОО . '
концентрации дистиллята [80-99,9% (мол.) ]. От точки пересечения проводят горизонтальную прямую до оси ординат и таким образом находят число теоретических ступеней разделения. В табл. 38 для некоторых смесей низкокипящих углеводородов приведены минимальное число теоретических ступеней разделения при и = оо и минимальное флегмовое число, которые могут служить оценкой для аналогичных процессов разделения [108].
Из данных табл. 38 видно, что разделение низкокипящих смесей является сравнительно простым процессом, который может быть осуществлен в колоннах с небольшой разделяющей способностью. Следует иметь в виду, что приведенные значения чисел теоретических ступеней разделения получены при v — оо, и при конечных флегмовых числах пмии должно соответственно измениться.
256
Таблица 38.
Минимальное число теоретических ступеней разделения и минимальное флегмовое число для процессов разделения некоторых бинарных низкокипящих смесей
Смесь а А в с D
Метан—этилен 61,2 1,32 1,82 0,03 0,164
Метан—этан 107,0 1,14 1,60 0,018 0,093
Этилен—этан 2,3 6,5 9,2 1,60 7,95
Этан—пропилен 8,1 2,52 3,57 0,27 1,4
Этап—пропан 9,3 2,38 3,36 0,23 1,2
Пропилен—пропан 1,3 20,2 28,6 6,65 33,0
Пропан—изобутан 3,3 4,6 6,5 0,89 4,5
Изобутан—«-бутан 1,5 13,0 18,4 3,99 20,0
Изобутан—изобутилен 1,2 28,8 40,8 10,0 50,0
Изобутилен—1 -бутилен 1,03 25,5 36,0 8,98 43,5
н-Бутан—изопентан 2,8 5,3 7,5 1,16 5,8
Изопентан—н-пентан 1,33 18,6 26,3 6,25 30,1
н-Пентан—н-гексан 3,0 4,9 6,95 1,04 5,25
А — необходимое число теоретических ступеней разделения смеси при Xg — 50% (мол.), Xg — 99,5% и v == оо; В — то же при Xg = 10%, Xg = 99,5% и v = оо; С — минимальное флегмовое число для разделения смеси при Xg = 50% и Xg — 100% *; D — то же при Xg ~ 10% и Xg = 100% *.
* Концентрация дистиллята 100% (мол.) указана с соответствующим округлением, поскольку абсолютно чистый дистиллят получить принципиально невозможно. — Прим, ред. ---------------------------------------------—------------
5.3.2. ВЫСОКОТЕМПЕРАТУРНАЯ
И ИЗОТЕРМИЧЕСКАЯ ПЕРЕГОНКА
При температурах выше 250° С органические вещества обычно разлагаются, поэтому процессы перегонки в лаборатории редко проводят в интервале температур от 250 до 400° С. Вместе с тем ( иногда бывает необходимо определить интервал температур кипе-( ния при атмосферном давлении, например у смол, высокомолеку-; лярных парафинов, воска. Горный воск часто подвергают деструк-тивной перегонке, чтобы одновременно посредством крекинга !; выделить низкомолекулярную часть [109]. В других случаях удается избежать разложения и полимеризации с помощью ин-! гибиторов [ПО].
( Для высокотемпературной перегонки высокомолекулярных веществ при атмосферном давлении или вакууме применяют только невысокие колонны, насадка которых служит в основном для предотвращения брызгоуноса. Насадку (обычно из крупных элементов) размещают на опорной решетке из проволочной сетки. Тарельчатые колонны для высокотемпературной перегонки непригодны. Для предотвращения частичной конденсации колонну
9 э. Крель 257
Таблица 39.
Нагревательные бани для высокотемпературной ректификации
Теплоноситель Максимально достижимая температура, °C
Глицерин — 160
Серная кислота —250
Полнгликоли —300
Твердые парафины с температурами плавления 30— 60 °C —300
Минеральное масло —330
Солевая баня, состоящая из 40% (масс.) NaNO2, 53% KNO3 н 7% NaNOg 150—500
снабжают сплошным обогревающим кожухом, простирающимся от куба до конденсатора. Приемник дистиллята также необходимо снабдить обогревающим кожухом или электронагревательной обмоткой. Целесообразно нагревать кран отбора дистиллята инфракрасным или обычным светом во избежание заедания его сердечника. Преимущественно применяют магнитные клапаны.
Для обогрева кубов при высокотемпературной ректификации в лаборатории обычно применяют масляные и солевые бани [111], температуру которых регулируют с помощью контактных термометров. В табл. 39 приведены значения температур, достигаемые в таких банях (см. разд. 7.7).
Большое преимущество солевых бань состоит в том, что они работают без выделения отходящих газов в широком интервале температур. Недостатком этих бань является трудность стабилизации температуры и необходимость их охлаждения. По окончании перегонки необходимо сразу же вынуть куб из бани и смыть водой оставшуюся на нем соль.
При охлаждении дистиллята в условиях высокотемпературной перегонки иногда требуется следить за тем, чтобы не затвердевал дистиллят (сублимированная его часть); в противном случае возможно уменьшение поперечного сечения холодильника вплоть до его закупоривания. Для предотвращения затвердевания дистиллята требуемые температуры конденсации можно устанавливать и регулировать посредством циркуляционных термостатов с водой или гликолем в качестве термостатирующей жидкости. Другой простой метод установки и регулирования температуры конденсации основан на использовании холодильника с кипящим хладо-агентом. Этот холодильник состоит из двух концентричных труб. По внутренней трубе пропускают охлаждающую жидкость, температура кипения которой несколько выше температуры плавления дистиллята, но ниже его температуры кипения. Пары дистиллята, проходящие по кольцевому каналу между трубами, нагревают ох-
258
вакуум-насосу
Шкаф управления
' К форвакуум-насос у
Рис. 182.
Дистилляционная установка Штаге для йерегоики веществ с высокой температурой плавления.
Установка 'снабжена холодильником дистиллята с кипящим хладо-агеитом. Изготовитель: фирма Фишера «Лабораторная н исследовательская техника*, Бонн — курорт Годесберг.
Рис. 183.
Устройства для перегонки металлов с боковым отводом паров (а) и с нижним отводом паров (<>): 1 — печь; 2 — сосуд с расплавом; 3 — приемник; 4 — конденсатор; 5 — конденсат.
лаждающую жидкость и испаряют ее. При этом дистиллят конденсируется на наружной стенке внутренней трубы.
На рис. 182 показана дистилляционная установка Штаге [112], снабженная холодильником дистиллята с кипящим хладоагентом. В работе [112] рассмотрены также другие установки для перегонки сублимирующихся веществ с высокой температурой плавления и подобных веществ в смеси с растворителями.
В последнее время перегонку стали широко применять для очистки металлов, особенно в тех случаях, когда низкокипящую часть сплава можно отделить в виде паров от высококипящей [113]. Это условие соблюдается при перегонке бинарных сплавов А1—Zn, Al—Mg, Pb—Zn, Ag—Zn и Ag—Pb. Значения давлений паров различных металлов можно найти в справочнике Лейбольда [114]. Для реализации процесса разделения металлов в лаборатории, необходимо сначала освоить соответствующую аппаратуру для получения небольших количеств веществ. На рис. 183 показаны два лабораторных устройства для перегонки металлов с боковым и нижним отводами потока паров [115].
Подробный обзор о лабораторной перегонке под вакуумом металлов и сплавов, не содержащих железа, приведен в работе Шпендлеве [116]. Хорслей [117] описал аппаратуру для разгонки щелочных металлов. В соответствии с этими работами металл расплавляют в вакууме, фильтруют и затем перегоняют преимущественно при давлении до 10~4 мм рт. ст. Пары металла конденсируют в конденсаторе, охлаждаемом циркулирующим маслом. Для получения чистого тантала Паркер и Вильсон [118] использовали хлорид тантала ТаС15 (температура кипения 240° С при 760 мм рт. ст.). Безобразов с сотр. [118а] разработал кварцевый аппарат диаметром 40 мм и высотой разделяющей части 1250 мм для аналитической перегонки высококипящих веществ с температурой кипения до 1000° С (сера, селен, теллур, цинк, кадмий, сульфид мышьяка и Др.).
Одним из наиболее часто применяемых в лаборатории процессов перегонки является дистилляция ртути с целью отделения олова, кадмия и благородных металлов. Обычно перед дистилляцией проводят предварительную химическую очистку, при этом в ртути могут оставаться вещества, используемые при очистке.
Прибор непрерывного действия, выполненный из кварца или тугоплавкого стекла, работает на принципе объемного насоса;
260
после предварительного вакуумирования с помощью водоструйного насоса прибор в ходе эксплуатации самостоятельно вакуумируется [119]. Принцип работы подобного прибора наглядно показан на рис. 184. Для работы прибора необходимо, чтобы сливная труба 7 была выполнена в виде капилляра внутренним диаметром 2,0 мм. Перед началом дистилляции целесообразно вакуумировать прибор с помощью ртутного диффузионного насоса и после одно- двухчасовой перегонки закрывать клапан 4. При необходимости повторного вакуумирования клапан снова открывают.
Для получения больших количеств очищенной ртути можно использовать аппаратуру для двукратной дистилляции ртути (рис. 185). После испарения ртути на первой ступени образуется конденсат, поступающий по трубчатой перемычке на вторую ступень, где он снова испаряется. Полученный дистиллят через запорную трубу, работающую на барометрическом принципе, стекает в приемник. С использованием электронагревателей мощностью 300 Вт при остаточном давлении до 1 мм рт. ст., можно получать на каждой ступени около 2 кг/ч дистиллята ртути. По данным спектрального анализа перегнанная ртуть имеет очень высокую степень чистоты. Прибор снабжен сферическими шли-
Рис. 184.
Автоматический прибор Робесона для дистилляции ртути:
/ — сосуд со ртутью; 2 — испаритель; 3, 10 — трубы с внутренним диаметром 20 и 6 мм соответственно; 4 — ртутный поплавковый клапан со штуцером для ртутного диффузионного вакуумного насоса; 5,9— капилляры с внутренним диаметром 2 и 4 мм соответственно; 6 — приемник с переливным устройством; 7 — сливная труба; 8 — промежуточный сосуд; 11 — блок из алюминия; 12 — электронагревательная обмотка. *
Рис. 185.
Аппарат для двукратной дистилляции ртути.
Изготовитель: народное предприятие «Йенский завод стекла Шотт», Иена.
261
фами с прецизионными соединительными поверхностями, не требующими применения вакуумной смазки.
Если перегонку проводят при постоянном давлении, то количество отбираемого дистиллята регулируют, меняя температуру. При изотермической перегонке, наоборот, температуру в кубе стабилизируют с помощью термостата, а давление непрерывно снижают. В этом случае на диаграмме разгонки давление (ось ординат) и количество отбираемого дистиллята (ось абсцисс) указаны при постоянной температуре. Изотермическую перегонку применяют, когда необходимо подобрать степень разрежения, требуемую для испарения определенного количества многокомпонентной смеси, если, например, для обогрева имеется в распоряжении пар определенной температуры. Эхолс и Гелус [120] использовали изотермическую перегонку для того, чтобы определить количества кубовых остатков исходной смеси, которые соответствуют определенному давлению при постоянной температуре. Возможно также построение изотермической кривой равновесия *. Теоретические основы изотермической и изобарической дистилляции обсуждаются Улусоем и Закалоцем [121]. Согласно опытным данным, полученным ими, изотермическая дистилляция пригодна и в других случаях.
5.4. КЛАССИФИКАЦИЯ МЕТОДОВ ПЕРЕГОНКИ ПО РАБОЧЕМУ ДАВЛЕНИЮ
Перед проведением перегонки следует решить, при каком давлении наиболее целесообразно проводить разделение. В тех случаях, когда это возможно, следует работать при атмосферном давлении, поскольку пропускная способность аппаратов, работающих в условиях вакуума, сильно снижается. Кроме того, перегонка под вакуумом или под давлением требует применения дополнительных устройств.
Газовые смеси, содержащие низкокипящие углеводороды, разделяют методом низкотемпературной ректификации при атмосферном давлении или ректификацией под давлением-, а легко разлагающиеся и высококипящие органические вещества перегоняют под вакуумом при остаточном давлении 760—1 мм рт.ст. Высокая производительность ректификационных установок может быть достигнута при использовании расширительной перегонки под вакуумом, соответствующем остаточному давлению 20—1 мм рт.ст. Термически нестойкие вещества нельзя перегонять непосредственно из куба; их разделяют в мягких условиях методом пленочной перегонки при остаточных давлениях 20—10’1 мм рт.ст. Вещества с низкими давлениями паров и большой молекулярной массой в пределах 250—1200 разделяют методом молекулярной
* Изотермические данные по равновесию пар—жидкость более удобны для термодинамического анализа. — Прим. ред.
262
дистилляции под вакуумом соответствующем 10"3—10~6 мм рт. ст., поскольку при этих остаточных давлениях средняя длина свободного пробега молекул соизмерима с размерами аппарата. Техника дистилляции и ректификации термически нестойких веществ под вакуумом подробно рассмотрена Франком и Куче в книге «Перегонка в мягких условиях» [122].
5.4.1. ВАКУУМНАЯ ДИСТИЛЛЯЦИЯ И РЕКТИФИКАЦИЯ
Под вакуумной перегонкой понимают процесс дистилляции и ректификации, который проводят при давлении ниже давления окружающей среды. Обычно остаточное давление в колоннах составляет примерно 0,5 мм рт. ст. Для перегонки при более низких давлениях используют специальную аппаратуру.
Основное преимущество вакуумной перегонки по сравнению с перегонкой при атмосферном давлении заключается в снижении точки кипения, что позволяет проводить процесс в условиях ниже температур разложения разделяемых веществ. Вакуумную перегонку применяют прежде всего для разделения термически нестойких и, особенно, полимеризующихся веществ. В качестве примеров можно назвать промышленную дистилляцию минеральных масел и ректификацию сырой смеси фенолов, которые проводят при 20—60 мм рт. ст., а также перегонку синтетических жирных кислот при давлении 1—20 мм рт.ст. Вакуумную перегонку применяют и в тех случаях, когда нет опасности разложения веществ, однако температуры их кипения при атмосферном давлении настолько высоки, что по теплотехническим соображениям ректификация при пониженном давлении становится предпочтительной. Это имеет место, например, при работе с различными эфирными маслами. Преимущество вакуумной перегонки состоит не только в снижении температуры кипения, но и в увеличении относительной летучести компонентов разделяемой при разрежении смеси.
Если разделяемая смесь образует при атмосферном давлении азеотроп, то с понижением давления азеотропная смесь обогащается низкокипящим компонентом и при некотором остаточном давлении азеотроп может исчезнуть *. Например, смесь этанол— вода при 70 мм рт.ст. азеотропа не образует (см. разд. 6.2.1). Следовательно, вакуумной ректификацией при давлении ниже 70 мм рт. ст. можно получать абсолютно чистый спирт без примесей **. Следует обратить внимание на то, что при таком разрежении температура кипения спирта сравнительно низка (примерно
* Смещение составов азеотропов с изменением давления зависит от соотношения скрытых теплот испарения компонентов. Следует также иметь в виду, что снижением давления не всегда удается разрушить азеотроп, поскольку при этом может раньше произойти кристаллизация разделяемой смеси. — Прим. ред.
** Как уже указывалось в примечаниях выше, путем ректификации получить абсолютно чистый дистиллят невозможно. — Прим. ред.
263
Рис. 186.
Влияние степени предварительного смачивания насадки на разделяющую способность ректификационной колонны по данным Гельбе [124], полученным прн 20 мм рт. ст.
28° С), поэтому при выборе вакуума следует учитывать имеющиеся в распоряжении хладо-агенты для конденсации паров.
Чаще всего в промышленности используется греющий пар
низких параметров, поэтому достаточную разность температур между температурой греющего пара и температурой кубовой жидкости можно обеспечить только при пониженном рабочем давлении. Следует учитывать также и возможность коррозии. Часто повышение температуры выше определенного предела является нежелательным из-за опасности коррозии куба и нижней части колонны. Следовательно, ряд факторов приводит к необходимости применения вакуумной перегонки. Экономические и технические соображения с учетом перечисленных выше факторов позволяют выбрать оптимальный вакуум. Биллет и Райхле [123 ] описали метод расчета рабочего давления ректификации, обеспечивающий минимальный перепад давления потока паров при вакуумной перегонке. В разд. 4.6.2 и 4.10.6 уже обсуждались различные точки зрения о влиянии вакуума на разделительную способность ректификационных колонн. Гельбе [124] указал, что выводы многих работ очень противоречивы. На основе экспериментов, проведенных с использованием колонны диаметром 45,7 и высотой разделяющей части 500 мм с насадкой из пружинных спиралей размером 4 мм, он установил, что при постоянной массовой производительности разделяющая способность колонны не зависит от давления в интервале от 10 до 100 мм рт. ст. При повышении абсолютного давления до 740 мм рт. ст. число единиц переноса постепенно возрастает вследствие увеличения относительной скорости потока паров. Так, при разделении смесей н-декан — транс-декалин число единиц переноса увеличивается примерно на 15% (см. рис. 97). Гельбе предположил, что противоречивость опубликованных экспериментальных данных обусловлена различной степенью эмульгирования газо-жидкостного слоя, имевшую место в опытах разных исследователей. В разд. 4.10.8 отмечался этот факт, там же обсуждались методы работы в режиме эмульгирования.
Гельбе проводил ректификацию при рабочих давлениях ниже 100 мм рт. ст., причем процесс сначала протекал при более высоком давлении, необходимом для увеличения количества флегмы. Это приводило к тому, что в начальный период ректификации барботажный слой, образующийся в нижней части колонны, постепенно поднимался по слою насадки до головки колонны. Слой 264
жидкости, образовавшийся при этом в головке, многократно проходил через насадку. После снижения давления до рабочего колонна продолжала работать в режиме эмульгирования. Степень смачивания насадки, достигнутая таким образом, отчетливо иллюстрирует рис. 186; верхняя прямая на рисунке соответствует оптимальному режиму эмульгирования.
Методика расчета размеров ректификационных колонн, в том числе работающих под вакуумом, описана в разд. 4.11. Следует отметить, что очень важно правильно определять размеры вакуумной коммуникации. Потери давления в трубопроводе диаметром менее 200 мм при умеренном вакууме рассчитывают по формуле Пуазейля [113]:
Др = 8VriZ/(r4n:i;) (178)
где Др — потеря давления; V — объем потока газа; Ц —динамическая вязкость газа; I—длина трубопровода; г—радиус трубопровода; т—время.
Харрис [125] разработал удобную номограмму (рис. 187), с помощью которой можно определить размеры вакуумных линий и производительность насоса. При разработке номограммы предполагалось, что скорость вакуумирования изменяется не более чем на 30 %.
Рис. 187.
Номограмма Харриса для определения размеров вакуумных коммуникаций.
265
Правила пользования номограммой поясняются на примерах 1 и 2.
Пример 1 Пример 2 р — давление в вакуумированном сосуде, мм рт. ст.................................. 1,0 1,0
d — диаметр трубопровода, см .... 1,0 3,0
S — производительность вакуумного насоса, м3/ч................................. 2,0 50,0
I — длина трубопровода, см .............. 350,0 450,0
В примере 2 точка пересечения прямой р — S со шкалой А лежит выше точки А 2 иа 6,5 делений шкалы, поэтому коэффициент полезного действия вакуумного насоса превышает 60%.
а) Определение м-аксимальной производительности вакуумного насоса (см. пример 1). Соединяют прямой линией точки на шкалах d и I, соответствующие значениям диаметра и длины трубопровода, и получают в точке пересечения этой прямой со шкалой А значение гидравлического сопротивления вакуумного трубопровода Аг. Затем проводят прямую через точки А х и р (определяемой на шкале р по значению давления в вакуумированном сосуде), продолжают ее до пересечения' со шкалой S и получают в этой точке пересечения максимальную производительность вакуумного насоса, б) Определение минимально допустимого диаметра трубопровода (см. пример 1).
Соединяют прямой линией точки на шкалах р и S, соответствующие выбранным значениям р и S, и получают в точке пересечения этой прямой со шкалой А величину Аг. Затем проводят прямую через точки и I (определяемой на шкале I по значению длины трубопровода), продолжают ее до пересечения со шкалой d. Точка пересечения показывает значение минимально допустимого диаметра трубопровода.
в) Определение максимальной длины трубопровода (см. пример 1).
Соединяют прямой линией точки на шкалах р и S, соответствующие выбран- ным значениям р и S, и получают в точке пересечения этой прямой со шкалой А величину А^. Затем проводят прямую через точку At и d (определяемой на шкале d по значению диаметра трубопровода), продолжают ее до пересечения со шкалой /и получают в этой точке пересечения значение максимальной длины трубопровода.
Точка пересечения А, обеих прямых для примера 1 соответствует значению коэффициента полезного действия вакуумного насоса (41%), определяемого по формуле:
N = /1 + (Ро/р)2 — Ро/Р (179)
где р0 — нижний предел давления, который можно обеспечить в разрабатываемой вакуумной установке с допустимым коэффициентом полезного действия.
Для проверки правильности выбора вакуумной коммуникации в уже готовой ректификационной установке (см. пример 2) необходимо соединить прямыми линиями точки на шкалах р и S, а также d и I. Точка пересечения прямой d — I со шкалой А дает значение Л2. Если точка пересечения прямой р—S со' шкалой А лежит выше точки Л2, то размеры вакуумной коммуникации достаточны.
Более точный метод расчета параметров вакуумных коммуникаций приведен в оригинальной работе [125]. Для лабораторных и пилотных ректификационных установок пригодны стеклянные трубопроводы диаметром 20—30 мм, собираемые из участков труб длиной 1—2 мм, которые соединяют короткими отрезками вакуумных резиновых шлангов.
Установки для вакуумной дистилляции и ректификации по размерам отличаются от соответствующих установок, работающих при атмосферном давлении (см. разд. 4.11). Необходимы также 266
дополнительные элементы, такие как устройство для присоединения к вакуумной линии, которое обычно совмещают с охлаждаемой ловушкой (рис. 188), и соответствующий вакуумный приемник дистиллята (см. разд. 7.6). Наряду с насадочными колоннами для вакуумной перегонки пригодны полые колонны (см. разд. 7.3.1), а также колонны с неподвижными (см. разд. 7.3.4) и вращающимися (см. разд. 7.3.5) контактными устройствами. Наиболее подходящее отношение диаметров наружной и внутренней труб в охлаждаемых ловушках составляет 1,6. Рекомендуется измерять остаточное давление непосредственно за устройством для присоединения к вакуумной линии, чтобы между точкой измерения давления и точкой измерения температуры не было заметной потери давления. С целью контроля в каждом отдельном случае следует измерять перепад давления между точками измерения давления и температуры (см. разд. 8.3).
Для вакуумных работ предпочтительны стандартные шлифы ряда 1. В тех случаях, когда смазка шлифов легко растворяется, а также для работ при среднем и высоком разрежении применяют высоковакуумные шлифы ряда 0 (см. разд. 3.1). Поскольку обычные краны при работах под вакуумом часто разуплотняются, созданы специальные краны для вакуумных работ. На рис. 189 показан вакуумный кран с закрытой снизу муфтой, а на рис. 190— тот же кран с дополнительным ртутным затвором. Другие специальные краны и клапаны для вакуумных работ описаны в разд. 7.2.1.
Для создания вакуума применяют водоструйные, диффузионные и водокольцевые насосы, а также электрические насосы различных типов. Для получения высокого вакуума необходимы ртутные и масляные диффузионные насосы. Более подробные данные о вакуумных насосах можно найти в соответствующей специальной литературе по вакуумной технике [ИЗ, 114, 119, 122, 126, 127] *.
* По вопросам выбора отечественного высоковакуумного оборудования см. Савинский К., Зав. лаб., № 9, 1111—1127/1955/. — Прим. ред.
Устройства для присоединения вакуумной линии с припаянными охлаждаемыми ловушками к аппаратам без регулирования потерь давления (а) и с регулированием потерь давления (б).
Рис. 189.
Вакуумный кран с закрытой снизу муфтой.
Рис. 190. Вакуумный кран с ртутным затвором.
267
Рис. 191.
Экранированный высокочастотный течеискатель с электродом в виде щетки.
При выборе вакуумного насоса можно 1| также руководствоваться справочником
J Лейбольда по вакуумной технике [114].
В обзоре Арденне [128] рассмотрены области применения и характеристики важнейших типов насосов. После сборки вакуумной установки необходимо испытать ее на герметичность. Сначала проверяют вакуум, создаваемый насосом, путем присоединения его к буферной ёмкости на 5—10 л. Затем проверяют герметичность кранов, шлифовых соединений и мест спаев. Целесообразно размещать краны или клапаны на установке таким образом, чтобы можно было отдельно испытать на герметичность различные ее части. Для проверки герметичности применяют высокочастотный течеискатель типа «Тесла» с электродом в виде щетки (рис. 191). Принцип работы прибора основан на возникновении искры от электрода в месте пропускания воздуха. Можно также проверить герметичность аппарата с помощью стетоскопа или смазать предполагаемые места пропусков мыльным раствором и создать в установке избыточное давление около 0,5 кгс/см2. Изящный метод проверки герметичности состоит в том, что на поверхность вакуумированной установки наносят кисточкой слабощелочной раствор флоуресцина или эозина в метаноле, затем ее облучают в темноте ультрафиолетовым светом, при этом в герметичных местах будет отчетливо наблюдаться флуоресценция. Специальные методы испытаний установок, работающих в условиях высокого вакуума, описаны Лаппорте [119] и Мён-хом [126].
Промышленность освоила выпуск течеискателей модели «Лек-зухэр», предназначенных для проверки герметичности установок, работающих в условиях высокого вакуума и избыточного давления.
Подобный галогенный течеискатель использует в качестве хладоагента фреон 12 (CF2C12). В основу работы прибора положено известное явление, заключающееся в том, что отожженная платина при контакте с галогенами испускает большое число ионов. При проверке герметичности вакуумной аппаратуры трубу тече-искателя присоединяют к местам предполагаемых пропусков. При наличии дефектов уплотнения в испытуемую установку засасывается эталонный газ из течеискателя, что регистрируется указывающим прибором или появляется акустический сигнал. При испытании установок, находящихся под избыточным давлением, газ через неплотности поступает из приборов в трубу течеискателя. Наименьшая течь газов, регистрируемая данным тече-искателем, составляет 10 “ л-мм рт. ст./с.
268
Другие виды течеискателей, использующие в качестве эталонного газа водород или светильный газ, обладают такой же чувствительностью. Для проверки герметичности применяют также прибор, использующий гелий, который подобен масс-спектрометру. Этот прибор может измерять утечки газов до 10“10 л - мм рт. ст./с [129 ].
Если термические нагрузки небольшие, то негерметичные места можно уплотнить пицеином или замазкой. Однако лучше заменить дефектную деталь или запаять негерметичное место.
Герметичность установки Dt характеризуют уменьшением давления Ар в единицу времени, умноженным на суммарный объем ректификационной установки:
D[ = V \p/t (л-мм рт. ст./с) (180)
Значения D{ при удовлетворительной герметичности должны лежать в интервале от 10 3 до 10 6 л-мм рт. ст./с.
5.4.2. РАСШИРИТЕЛЬНАЯ ПЕРЕГОНКА
Расширительная перегонка относится к непрерывным промышленным методам перегонки. Несмотря на большие преимущества, этот метод еще не нашел широкого применения в лабораторной и промышленной практике.
Стеклянная установка автора [130], разработанная для реализации подобного процесса, показана на рис. 192. Основной частью этой установки является хорошо теплоизолированная расширительная колба 8 объемом 250—500 мл с термометром на стандартном шлифе. Температуру обогрева колбы регулируют с помощью контактного термометра. К центру колбы подведен сменный патрубок 2, служащий для впрыскивания перегоняемой жидкости. За операцией впрыскивания можно постоянно наблюдать через небольшое смотровое окно. Расширительная колба освещается небольшой лампой накаливания. Исходную смесь из бутыли 1 передавливают в напорный бак 4, работающий по принципу сосуда Мариотта. В баке 4 с помощью термостатирую-щего кожуха поддерживают определенную температуру. Дальнейший подогрев происходит в теплообменнике 3, в который подают термостатированную жидкость или пар. Для регулирования температуры теплоносителя предназначен контактный термометр 6. Мерной бюреткой5 контролируют, а краном 7 регулируют расход исходной смеси, поступающей в колбу 8.
Подогретую исходную смесь непрерывно с определенной скоростью впрыскивают в вакуумированную колбу 8. При этом жидкость распределяется по стенкам колбы в виде тонкой пленки. В результате дополнительного разрежения при впрыскивании низкокипящая фракция сразу же испаряется. Пленка, образованная на нагретых стенках колбы, имеет большую поверхность испарения, поэтому в течение небольшого периода времени испа-
269
— Рис. 192.
Установка Креля для расширительной перегонки:
1, 13, 17 —бутыли для исходной смеси, дистиллята и кубового продукта соответственно; 2 — патрубок для впрыскивания исходной смеси; 3 — подогреватель исходной смеси; 4 — напорный сосур, Мариотта; 5, 16 — мерные бюретки для исходной смеси и кубового продукта соответственно; 6, 14 — контактные термо-
метры; 7 — регулировочный край для подачи исходной смеси; 8 — расширительная колба; 9 — трубчатая перемычка;
10 — конденсатор; И — штуцер для присоединения вакуумной линии; 12 — приемник Аншюца — Тиле &ля дистиллята; 15 — колонна.
Изготовитель: народное предприятие «Комбинат технического стекла», Ильменау.
Рис. 193.
Диаграмма .пробной расширительной разгонки головной (?) и кубовой (2) фракций сырой смеси жирных кислот.
Рабочее давление перегонки для головного продукта 15 мм рт. ст., для кубового продукта 6 мм рт. ст.
Выход дистиллята , % (масс.)
ряется и оставшаяся часть низкокипящей фракции. Пары дистиллята проходят по изолированной трубчатой перемычке 9 с термометром к конденсатору 10, где конденсируются. Конденсат через' приемник Аншюца—Тиле 12 поступает в бутыль 13. Вакуумную линию присоединяют к штуцеру 11. Вакуум стабилизируют с помощью регулятора давления (см. разд. 8.3). Высококипящая фракция стекает из расширительной колбы вниз и с помощью перфорированной воронки распределяется по поперечному сечению короткой колонны 15, снабженной обогревающим кожухом. Если высококипящая фракция содержит низокипящие компоненты, то их отделяют в неадиабатически работающей колонне, к стенкам которой дополнительно подводится тепло. Температуру обогревающего кожуха колонны 15 регулируют с помощью контактного термометра 14. Кубовый продукт непрерывно отводят в бутыль 17 через мерную бюретку 16.
270
260
2<Ю
КО
120
80
Концентрация} °/о(о6.)
800—1500 г/ч.
Рис. 194.
Диаграмма пробной расширительной разгонки головной и кубовой фракций фенольного масла.
/—5 _ кривые трех разгонок дистиллята; j 4 — 6 — то же кубового продукта.
Непрерывная расширительная перегонка особенно пригодна для отделения низкокипящей фракции при давлении 1 — 20 мм рт. ст. Производительность колонны состав-получении низкокипящей
ляет фракции кислот С4—С10 из сырой смеси жирных кислот методом расширительной перегонки под вакуумом, соответствующем 15 мм рт. ст., дистиллят отбирают со скоростью 1200 г/ч.
На рис. 193 показаны результаты анализа дистиллята и кубовой жидкости, полученные при пробной перегонке. Граница разделения соответствует компоненту С10- Предварительное фракционирование дистиллята фенольного масла также осуществляли путем расширительной перегонки. При атмосферном давлении смесь разделяется в интервале температур 210—230 °C. Полученные результаты представлены на рис. 194. Кривые, соответствующие трем пробным перегонкам дистиллята и кубовой жидкости при атмосферном давлении, накладываются друг на друга (точность около 15 °C). Особенно высокой производительности до 3 л/ч можно достигнуть при большой разности температур кипения компонентов смеси, например при перегонке смеси глицерина с растворителем, кипящим при 150 °C [130].
Смесь фенолов, содержащую 9% воды, обезвоживали путем расширительной перегонки, проводимой на аналогичной установке с расширительной колбой, не имеющей колонны. При абсолютном давлении 23 мм рт. ст. и температуре исходной смеси 80 °C оптимальная производительность установки составляла 4 л/ч. Такую высокую производительность вряд ли могут иметь обычные лабораторные установки периодического действия. Серьезным препятствием для использования метода расширительной перегонки является возможное вспенивание смесей.
Данный метод наиболее пригоден для препаративного разделения многокомпонентных смесей на отдельные фракции. Этим методом можно быстро и в мягких условиях фракционировать смеси объемом до 100 л с последующей аналитической ректификацией отдельных фракций на установках периодического действия.
271
5.4.3. ПЛЕНОЧНАЯ ПЕРЕГОНКА
(ПЛЕНОЧНОЕ ИСПАРЕНИЕ)
Пленочная перегонка осуществляется из пленки жидкости в мягких температурных условиях. Этот метод соответствует одноступенчатой дистилляции, для которой разделяющее действие установки максимально приближается к одной теоретической ступени разделения. Степень разделения можно повысить путем последовательного соединения большого числа пленочных аппаратов. Опыты автора показали, что как при атмосферном давлении, так и при остаточных давлениях 5—0,1 мм рт. ст. пленочная дистилляция имеет существенные преимущества [130], заключающиеся в малом времени пребывания жидкой фазы в нагретой зоне и в отсутствии необходимости нагревания больших слоев жидкости, как это имеет место при перегонке из куба.
Поясним данные преимущества на примере. Предположим, что требуется разделить при абсолютном давлении 1 мм рт. ст. смесь насыщенных жирных спиртов С1о—С18 нормального ряда. Если в качестве куба использовать круглодонную колбу объемом 1 л, то высота слоя жидкости составит 54 мм. При этом в колбе получается следующее соотношение давлений (рис. 195п): на поверхности жидкости давление равно 1 мм рт. ст.; вследствие гидростатического давления слоя жидкости (54 : 13,5 = 4 мм рт. ст.) давление на дне куба повышается до 5 мм рт. ст. В начале перегонки фракция С1о отгоняется при давлении 1 мм рт. ст. и температуре 76 °C, при этом у дна куба температура составляет 142 °C.
При пленочной же перегонке очень тонкая пленка жидкости, находящаяся в нагретой зоне очень малое время, практически не вызывает увеличения давления. Поэтому для получения первой фракции достаточна температура пленконесущей поверхности, равная 78 °C (рис. 1956). Мягкие температурные условия пленочной перегонки наиболее пригодны для переработки термически нестойких веществ. Пленочную перегонку целесообразно применять для предварительного разделения исходной смеси в узком температурном интервале.
Если высококипящая смесь содержит незначительное количество низкокипящих компонентов, то даже при высокой температуре часто бывает невозможно целиком выделить низкокипящую фракцию перегонкой из куба. При перегонке под вакуумом (для предотвращения разложения высококипящей фракции) возникают трудности при конденсации паров низкокипящих компонентов. Этих затруднений можно избежать, если проводить пленочную перегонку при атмосферном давлении.
Различают следующие области применения непрерывной пленочной перегонки (см. также [122] и гл. 5 и 6): перегонка термически нестойких веществ в мягких температурных условиях; перегонка смеси высококипящих веществ, которую нельзя разделить обычной перегонкой из куба; разделение смесей с неболь-272
шим содержанием низкокипящей фракции и высоким содержанием высококипящей фракции; дегазация жидкостей; выпаривание жидкостей; перегонка легко вспенивающихся веществ.
При пленочной перегонке, или точнее, при пленочном испарении, применяют испарители следующих типов.
1) Испарители с падающей (гравитационно стекающей) пленкой. В состав этих испарителей входят неподвижные обогреваемые вертикальные трубы или трубчатые змеевики, по наружной поверхности которых стекает пленка жидкости (см. рис. 196, 198, 199, 212), вращающиеся контактные устройства для обеспечения циркуляции пленки жидкости, выполненные в форме щеток (см. рис. 201), стеклянных спиралей (см. рис. 210) или скребковых роторов со щетками, лопастями или роликами (см. рис. 201, 202, 211). 2) Проточные испарители, расположенные горизонтально или наклонно. Эти испарители применяют обычно для молекулярной дистилляции (см. рис. 205, 209). 3) Испарители с диспергированием жидкости. Эти испарители применяют для расширительной перегонки (см. рис. 192). 4) Роторные испарители, имеющие вращающийся куб (см. рис. 203), барабан для перемешивания пленки жидкости (см. рис. 200) и испарительные диски, обеспечивающие распределение жидкости под действием центробежных сил (см. рис. 213).
Геммекер и Штаге [132] дали подробный обзор пленочных испарителей и испарителей мгновенного действия с разбрызгиванием
Рис. 195.
Соотношение давлений в обычном перегонном кубе (а) н в пленочном аппарате (б).
Рис. 196.
Пленочный дистиллятор Креля:
/ — электронагреватель внутренней трубы;
2 — обогреваемая внутренняя труба; 3 — устройство для точной установки расхода исходной смеси с поплавком 6\ 4 — напорный <5ак; 5 — фотоэлемент; 7 — стеклянная воронка для подачи исходной смеси; 8 — передвижное металлическое кольцо; 9 — бутыль для „ исходной смеси.
/ Изготовитель и разработчик: народное предприятие «Комбинат технического стекла», Ильменау.
273
жидкости, а также пленочных ректификационных колонн. Сведения о пленочных аппаратах можно найти также в монографии [122].
На рис. 196 показан испаритель с падающей пленкой, в котором жидкость стекает по неподвижной вертикальной трубе. В аппарате имеются две концентричные трубы. Внутренняя сменная труба с нагревателем является поверхностью испарения, а наружная труба служит поверхностью конденсации.
Утцингер [133] указал на трудность обеспечения равномерного распределения пленки жидкости (см. разд. 4.2). Крель предложил для уменьшения образования струй на обогреваемом цилиндре исходную смесь подавать в углубление, находящееся в его верхней части; при этом жидкость равномерно стекает через края цилиндра. Кроме того, штуцер для ввода исходной смеси целесообразно погружать в это углубление, что уменьшает капле-образование. Для дополнительного улучшения распределения жидкости поверхность обогреваемого цилиндра выполняли шероховатой с помощью пескоструйного аппарата. Передвижные металлические кольца 8 (см. рис. 196), насаженные на обогреваемый цилиндр, обеспечивали циркуляцию пленки, а расположенная над углублением для подачи исходной смеси стеклянная воронка 7 предотвращала попадание исходной жидкости на поверхность конденсации.
Аппарат снабжен прибором для автоматической установки расхода исходной смеси с помощью фотоэлемента и автоматическим регулятором температуры нагревателя. Дистиллятор заключен в охлаждающий кожух, что позволяет разделять методом пленочной перегонки низкокипящие вещества.
Регулирование работы дистиллятора и всех вспомогательных аппаратов проводится с центрального пульта управления. Исходная смесь заливается в бутыль 9, вмещающую около 10 л. Из бутыли смесь передавливается воздухом по трубопроводу в напорный бак 4, расположенный над дистиллятором. Расход исходной смеси регулируется с помощью поплавка 6, который посредством фотоэлемента 5 включает или выключает воздушный насос. Таким образом гарантируется непрерывная подача подогретой исходной смеси в напорный бак. Устройство 3 для точной установки расхода обеспечивает точное и надежное дозирование разделяемой смеси из напорного бака на наружную поверхность внутренней обогреваемой трубы 2 дистиллятора. Дистиллят и кубовый остаток могут отводиться в отдельные приемники без нарушения технологического режима процесса дистилляции. Дистиллятор предназначен для работы при атмосферном давлении и под вакуумом.
Как показали испытания дистиллятора, на результаты разделения при постоянных условиях (вакуум и температура) значительное влияние оказывает скорость подачи исходной смеси. При повышении скорости подачи увеличивается количество кубового продукта, а при ее уменьшении возрастает количество отби-274
раемого дистиллята при одновременном повышении четкости разделения. Изменяя рабочую температуру при постоянном вакууме, можно определить оптимальные условия процесса.
Если известен состав исходной смеси, то устанавливают такую скорость ее подачи, чтобы количества головной и кубовой фракций соответствовали необходимой степени разделения. При этом всегда целесообразно при первой пленочной разгонке отобрать возможно большее количество дистиллята. Такое «превышение» позволит получить кубовый продукт с минимальным содержанием низкокипящей фракции. Высококипящие продукты, попавшие в первичный дистиллят, путем повторной пленочной дистилляции переводят к кубовый остаток, причем после второй перегонки обычно получают очень чистый дистиллят. С помощью градуированных приемников можно непрерывно контролировать необходимое соотношение скоростей отбора дистиллята и кубового продукта (рис. 197).
Методом непрерывной пленочной дистилляции можно выделять фракции жирных кислот до кислотного числа 90, так что в дистиллят попадают кислоты с числом атомов углерода, меньшим 30. Пленочная перегонка предпочтительна также для разделения воска и силиконовых масел, которые особенно хорошо смачивают греющую поверхность. Гутвассер и Мюллер [22] разработали установку с пленочным испарителем, предназначенную для разделения смеси спермацета и жирных кислот. На рис. 198 показана схема этой установки и пленочного испарителя.
Аппараты фирмы Лейбольд—Хэраеуз (Кёльн), предназначенные для работы при 1 —10~3 мм рт. ст., имеют особенно развитую поверхность испарения и обеспечивают постоянное обновление «активной поверхности» жидкости путем интенсивного перемешивания пленки. Эта фирма выпускает ректификационные аппараты следующих типов.
1) Колонны с перемешивающейся пленкой (рис. 199), служащие для дистилляции жидких смесей, кубовый остаток которых при рабочей температуре может отбираться только в виде капель. Такими смесями могут быть высокомолекулярные фракции нефти и сложные эфиры, ароматические вещества, моноглицериды и растительные экстракты. В этих колоннах восходящий поток паров движется перпендикулярно к потоку жидкости. Пленочный испаритель
Рис. 197.
Диаграмма разгонки кубовой (/) и головной (2) фракций смеси жирных кислот С8—С25 методом непрерывной пленочной дистилляции. Выход дистиллята отнесен к суммарному количеству обеих фракций.
275
Рис. 198.
Установка Гутвассера—Мюллера [22], предназначенная для пленочного испарения жидкостей под вакуумом.
а — Схема установки:
1 — дистилляционный куб; 2 — предохранительный холодильник; 3 — пленочный испаритель; 4 — обогревающий кожух; 5 — термостат; 6,7 — штуцеры для присоединения термостата; 8, 10 — термометры; И — распределитель пара и жидкости; 12 — конденсатор головки испарителя; 13, 14 — охлаждаемые ловушки; 15 — ионизационный вакуумметр; 16 — ртутный диффузионный насос; 17 — масляный манометр.
б — Пленочный испаритель со стандартными шлифами NS70: /<
13 — запорные карманы; 2 — сферический шлиф для присоединения приемника дистиллята; 3,6 — штуцеры для установки термометров; 4 — конденсатор паров дистиллята; 5 — штуцер для присоединения вакуумной линии; 7, 8 — штуцеры для присоединения термостата; 9 — распределитель конденсата нз головки испарителя; 10 — конденсаторы, совмещенные со вторичными испарителями; И — стеклянная пластина распределителя; 12 — основная испарительная труба.
Изготовитель: народное предприятие «Комбинат медицинской и лабораторной техники», завод V, Лейпциг.
• - обогрев
& - конденсации вещество
Рис. 199.
Схема колонн с перемешивающейся пленкой жидкости.
Рис. 200.
Барабан для перемешивания пленки жидкости.
Обозначения см. на рис. 199.
276
модели 11 пригоден для получения клейких продуктов.
2) Барабаны с перемешивающейся пленкой жидкости (рис. 200), предназначенные для перегонки смесей веществ, имеющих вязкие или твердые кубовые остатки. К таким смесям относятся каменноугольный пек, натуральный или синтетический воск и битуминозный (горючий) сланец.
3) Колонны с вращающимися щетками (рис. 201), служащие для дистилляции смесей веществ, дистиллят и кубовый остаток которых при рабочей температуре в колонне имеют вязкость до 800 сП, например талловое масло, ди- и трихлорфенилы, ланолин [146].
Для этих аппаратов выпускаются стандартные взаимозаменяемые детали, с помощью которых можно собирать многоступенчатые пленочные дистилляторы любых типов, предназначенные для дегазации и перегонки жидкостей при различных температурах. Дальнейшим развитием данных аппаратов являются дистилляционные устройства для перегонки «на коротком пути» со скребковыми роторами и внутренним охлаждающим устройством, из которых также могут собираться многоступенчатые дистилляторы (см. разд. 5.4.4).
Испарители с падающей пленкой, снабженные вращающимися контактными устройствами, аналогичны по конструктивному выполнению роторным ректификационным колоннам (см. разд. 7.3.5).
рвогрев о —
- дещестдо
& —конденсация
Колонна с вращающимися щетками.
Рис. 202.
Дистилляционная установка со стеклянным испарителем модели Самбай:
1 — воронка для подачи исходной смеси;
2 — игольчатый клапан; 3 — регулируемый приводной механизм ротора; 4 — магнитная муфта; 5 — змеевик для подогрева исходной смеси; 6 — калиброванная стеклянная труба; 7 — скребковый ротор с лопастями; 8 — конденсатор; 9, 12 — промежуточные вставки со штуцером для вакуумметра; 10 — дезодорирующая промежуточная вставка; 11, 13 — приемники для кубового продукта и дистиллята соответственно; 14 — термостат.
Изготовитель: фирма «Стеклянная техника», Висбаден --Ширштайи.
277
Основное отличие состоит в том, что в роторных колоннах жидкая смесь испаряется в кубе и массообмен происходит при противоточном движении паровой и жидкой фаз, за счет чего обеспечивается высокая степень разделения. Однако как в роторных испарителях, так и в роторных ректификационных колоннах с помощью ротора обеспечивается циркуляция пленки жидкости и тем самым предотвращается обеднение ее поверхности низкокипящим компонентом. Проблемы механической турбулизации тонких слоев жидкости обсуждены в работе Яносфиа [134].
Испарители с падающей пленкой могут работать как при прямо-или противотоке паровой и жидкой фаз, так и на принципе скоростной дистилляции (см. разд. 5.4.4). Подвод тепла обычно осуществляется снаружи (см. рис. 198, 200—203, 211), однако есть аппараты и с внутренним подводом тепла (см. рис. 196, 199, 210, 212).
Пленочные испарители хорошо зарекомендовали себя в промышленности [135]. Производительность и массообменные характеристики различных испарителей сравнил Дитер [136]. Математическое моделирование и методы расчета роторных пленочных испарителей разработал Биллет [137].
На рис. 202 показана дистилляционная установка с пленочным испарителем модели Самбай. Испаритель снабжен скребковым ротором. Эта установка выпускается фирмой QVF «Стеклянная техника».
Исходная смесь поступает в испаритель из воронки 1 с капельницей, снабженной обогревающим кожухом. Точное дозирование жидкости обеспечивается с помощью игольчатого клапана 2 воронки, которая работает как сосуд Мариотта. Перед входом в испаритель смесь нагревается в обогреваемом змеевике 5 до температуры кипения, вследствие чего рабочая зона испарителя полностью используется только для испарения жидкости. Одновременно в змеевике происходит дегазация исходной смеси, что исключает образование брызг в рабочей зоне испарителя.
Пленка образуется на внутренней поверхности калиброванной стеклянной трубы 6 при вращении скребкового ротора 7 с лопастями (для уменьшения коррозии ротор выполняется из тантала или нержавеющей стали марки VA). Регулируемый приводной механизм 3 со ступенчато изменяющейся скоростью вращения соединяется с ротором магнитной муфтой 4, которая лишена всех недостатков сальникового уплотнения. Нижний конец ротора снабжен самоустанавливающимся шарикоподшипником в виде тефлонового шара, размещенного в стеклянной опоре. Смазкой подшипника служит стекающий кубовый продукт. Источником тепла является циркуляционный термостат 14 мощностью электрообогрева 1,5 или 2 кВт. При температурах до 200° С в качестве теплоносителя используют парафиновое масло, а при более высоких температурах — силиконовое масло. Эти масла полностью прозрачны.
278
В комплект установки входят также приемник кубового продукта 11, служащий для отбора высококипящей фракции, и промежуточная вставка 9 со штуцером для вакуумметра. При дезодорации жидкостей или дистилляции смесей с небольшим содержанием низкокипящей фракции промежуточную вставку 9 заменяют на дезодорирующую вставку 10. Через боковой штуцер этих вставок 9, 10 можно вводить паровой поток низкокипящей фракции для увеличения скорости движения паров в направлении конденсатора и уменьшения возможного испарения дистиллята. Наличие сифона способствует предотвращению конденсации паров в приемнике кубового продукта.
Низкокипящая фракция конденсируется в конденсаторе 8 и собирается в приемнике дистиллята 13. Штуцер промежуточной вставки 12 служит для присоединения установки к вакуумной линии.
Обогревающая поверхность испарителя составляет 0,016 м2, а охлаждающая поверхность конденсатора — 0,2 м2. Максимальная рабочая температура равна 220° С. При этой температуре и при давлении 25 мм рт. ст. наибольшая производительность установки составляет 480 г/ч.
Лабораторные и промышленные испарители с вращающимся кубом, применяющиеся в различных ректификационных установках, стандартизированы. Их применяют как в пилотных дистилляционных установках, так и в лабораторных приборах, предназначенных для микроперегонки. Данные испарители имеют вращающийся куб в виде трубы с шаровым расширением (см. разд. 5.1.1) или круглодонной колбы, емкость которых может изменяться в интервале от 1 мл до 100 л. Наряду с дегазацией масел и смол испарители с вращающимся кубом используют для отделения растворителей и пенящихся веществ в мягких температурных условиях. На рис. 203 показана принципиальная схема данного испарителя. Конструкции таких испарителей и области их применения подробно рассмотрены Эгли [138]. Частота вращения колбы 4 может ступенчато изменяться и регулироваться в интервале от 10 до 220 об/мин. Для удобства эксплуатации установка снабжена механическими и автоматическими
Рис. 203.
Испаритель «Ротавапор» с вращающейся колбой модели R-10:
/ — приемник дистиллята; 2 — конденсаторы; 3 — штуцер для присоединения вакуумной линии; 4 — вращающаяся перегонная колба; 5 — обогревающая баня; 6 — пульт управления.
Изготовитель: фирма В. Бюхи, Флавиль, Швейцария.
279
подъемными приспособлениями для куба. Система взаимозаменяемых деталей для сборки испарителей с вращающимся кубом и ее возможности рассмотрены в работе Крамера [138а].
5.4.4. МОЛЕКУЛЯРНАЯ дистилляция
Методом молекулярной дистилляции перегоняют высококипя-щие, часто термически нестойкие вещества при абсолютном давлении, не превышающем 10“3 мм рт. ст. При таких давлениях расстояние, проходимое молекулами между испаряющей и охлаждающей поверхностями, меньше средней длины свободного пробега молекул. Таким образом достигают того, что большинство молекул испаряющейся жидкости попадает на поверхность конденсации без столкновения с молекулами другого газа. Под средней длиной свободного пробега молекулы понимают теоретически рассчитанное расстояние, которое молекула может пройти, не столкнувшись с другими молекулами. Например, для триглицеридных жиров нормального ряда с молекулярной массой 800 эта величина имеет следующие значения (в мм) [141]:
При остаточном давлении 8-Ю-3 мм рт. ст...................... 7
» » » 3-10'3 мм рт. ст................... 25
» » » 1 • 10'3 мм рт. ст................. 50
При молекулярной дистилляции не происходит кипения жидкости. Этот процесс можно определить как «молекулярное испарение». Равновесное состояние между жидкостью и ее паром постоянно нарушается из-за конденсации. При этом должно устанавливаться новое равновесие, т. е. новые молекулы должны испариться с поверхности жидкости. Таким образом, молекулярная дистилляция является ярким примером прямоточной дистилляции, которую также можно назвать «однократным испарением» [141].
Области применения молекулярной дистилляции рассмотрены в монографиях Барроуза и Холо с сотр. [139]. Ридгвэй-Уатт [140] дал подробный обзор установок молекулярной дистилляции различной производительности (от микроаппаратов до промышленных установок). В разд. 1.5 и в работе [1221 можно найти ссылки на ряд других обзорных статей по данному методу перегонки [108, 131, 145, 156, 157].
Скорость испарения зависит от давления паров вещества р° при температуре поверхности испарения, а также от молекулярной массы перегоняемого продукта. Формальное математическое описание этой зависимости предложил Лэнгмюр [142]:
D = 0,0583/?° К М/Т (181)
где D— максимальная удельная скорость испарения, г/(с-см2); /?“—давление паров вещества при температуре Г, мм рт. ст.; М — молекулярная масса вещества; Т — температура дистилляции, К.
280
Эта формула получена при допущении, что испарению не препятствуют молекулы другого вещества. Поскольку в реальных процессах испарившиеся молекулы при движении к поверхности конденсации сталкиваются с молекулами другого газа, значение D в большинстве случаев не достигается. С целью его коррекции вводят коэффициент испарения а, который учитывает остаточное давление другого газа. Для процессов молекулярной дистилляции, осуществляемых в современных промышленных установках, а равен 0,9. Очевидно, что значение а будет тем ближе к 1, чем ниже остаточное давление в дистилляторе. Барроуз [143] исследовал применимость формулы (181) и получил ряд полуэмпирических уравнений, которые лучше соответствуют различным условиям процесса молекулярной дистилляции.
Теоретически достижимые скорости молекулярной дистилляции очень малы, что наглядно видно из данных, приведенных в табл. 40 [108 ].
Если принять коэффициент испарения равным 0,8, то из данных таблицы можно определить количества дистиллята, получаемые за 1 ч с испаряющей поверхности 100 см2. Эти количества лежат в интервале от 15 до 22 г/ч.
Для достижения высокой производительности установок молекулярной дистилляции следует работать при абсолютных давлениях выше 10 :| мм рт. ст. по методам Утцингера [133] и Яекеля [144], последний из которых предпочтительно применять в полупромышленных и промышленных установках. В большинстве случаев бывает достаточно, если остаточное давление приближается к 10 мм рт. ст. При этом давление паров перегоняемой жидкости может быть во много раз больше и достигать порядка 1 мм рт. ст. Молекулы оставшегося газа при столкновении с поверхностью конденсации отражаются от нее, а молекулы паров жидкости, напротив, задерживаются на этой поверхности [145].
Области применения молекулярной дистилляции весьма разнообразны. Прежде всего этим методом перегоняют термически не-
Таблица 40.
Теоретически достижимые скорости молекулярной дистилляции при остаточном давлении 10-3 мм рт. ст.
Вещество Молекулярная масса D . 104 по формуле (181) р° при 120 °C, мм рт. ст. К м/т при 10-3 мм рт. ст. ро/1/~М При 120 °C
г/(с. см2) МОЛЬ
(с см2)
Стеариновая кис- 284 0,52 0,0021 35,0 0,90 2,07
лота Холестерин 387 0,56 0,0014 0,5 0,97 0,025
Тристеарин 891 0,76 0,00С9 10~4 1,32 —
281
стойкие вещества с молекулярной массой от 250 до 1200. В качестве примера укажем следующие области применения молекулярной дистилляции (см. [122, 158] и гл. 5, 6): получение масел для вакуумных насосов, а также высоковязких смазочных масел, вязкость которых мало зависит от температуры; исследование триглицеридных масел и жиров, а также восков, смол и высокомолекулярных жирных кислот и спиртов; получение витаминов и концентратов гормонов; очистка пластификаторов и других веществ с низким давлением паров; дезодорация высокомолекулярных веществ; очистка эфирных масел и ароматических веществ.
В последнее время молекулярную дистилляцию применяют для исследования высокомолекулярных веществ, содержащихся в нефтяных и других кубовых остатках, которые получают при обычной перегонке. Методом многократной дистилляции получают вещества максимально возможной степени чистоты. Обзор аппаратуры и методов молекулярной дистилляции жирных кислот и липидов представил Перри [145]. Франк [146] рассмотрел специальные вопросы, касающиеся данного способа, и описал конструкцию многоступенчатых испарителей.
Важным преимуществом молекулярной дистилляции является возможность разделения смеси веществ с одинаковыми давлениями паров, но разными молекулярными массами.
Из формулы Лэнгмюра
D =-p/]^2nMRT (182)
где D—максимальная удельная скорость испарения, моль/(с-см2); р—давление пара, дин/см2; М — молекулярная масса; R—универсальная газовая постоянная, равная 8,3-10’ эрг/(К-моль); Т — абсолютная температура, К-следует, что при постоянной температуре количество образующихся паров зависит от отношения р/1/ М. Относительную испаряемость компонентов бинарной смеси при молекулярной дистилляции по аналогии с относительной летучестью можно представить в виде [1081:
О!,/О2= (Л КОМКАЛ) (183)
где pj и р2 — парциальные давления паров компонентов.
Из этой формулы следует, что два вещества с разными молекулярными массами можно разделить путем молекулярной дистилляции даже при равных парциальных давлениях их паров.
. Конструкции аппаратов для проведения молекулярной дистилляции должны удовлетворять следующимтребованиям. Они должны иметь устройство для присоединения к вакуумной линии с большим диаметром условного прохода, чтобы потери давления были минимальными (см. разд. 5.4.1). В этих аппаратах должно обеспечиваться равномерное распределение пленки жидкости по поверхности испарения и малое время ее пребывания на этой поверх-282
кости (см. разд. 5.4.3). Расстояние между поверхностями испарения и конденсации должно быть примерно равно средней длине Свободного пробега молекул (1—2 см), разность температур между Йтими поверхностями должна составлять 50—100 °C. Установку Следует оборудовать устройствами для предварительной дегазации перегоняемых веществ с целью удаления растворенных инертных газов.
Имеющиеся колонны для молекулярной дистилляции по принципу действия можно разделить на следующие группы: аппараты с тонким слоем жидкости в плоскодонном кубе; дистилляторы с поверхностью испарения в виде горизонтальных или наклонных лотков; колонны с вертикально стекающей (падающей) пленкой жидкости; аппараты центрифужного типа.
Чтобы более полно отделить низкокипящий компонент, в колоннах этих групп можно проводить многократную перегонку путем циркуляции разделяемой смеси. Отметим также, что в данных дистилляторах испарение происходит только с поверхности пленки жидкости и молекулы селективно покидают эту поверхность без какого-либо механического воздействия [147].
Наиболее простыми по конструкции являются аппараты с тонким слоем жидкости в плоскодонном кубе. На рис. 204 показан современный дистиллятор подобного типа. С помощью таких аппаратов можно успешно разделять вещества с молекулярными массами до 300. Дистилляцию из куба с плоским дном и тонким слоем жидкости обычно применяют для получения предваритель-
ных данных о температурных пределах перегонки и способности веществ к разложению.
Микромолекулярная дистилляция, осуществляемая по методам «холодного пальца» и падающей пленки жидкости и предназначенная для перегонки 0,5—5 г исходной смеси, описана в разд. 5.1.1 (см. рис. 131—133). Наиболее распространенным аппаратом с испаряющей поверхностью в виде лотка является дистиллятор Ут-цингера [133, 147]. Утцингер
Рис. 204.
Высоковакуумный молекулярный дистиллятор. Условия работы: температура 20 — 200 °C, загрузка исходной смеси 2,5 или 10 мл. Снабжен регуляторами остаточного давления до Ю“* мм рт. ст. и регуляторами мощности электрообогрева.
Изготовитель: фирма Фишера «Лабораторная и исследовательская техника», Бонн —курорт Годесберг.
283
Рис. 205.
Аппарат Утцингера для получения трех фракций методом дистилляции на «коротком пути» .
Изготовитель: фирма «Завод стекла Шотт», Майнц.
ввел понятие дистилляции «на коротком пути» *, обозначающее однократную перегонку при остаточных давлениях более 10'3 мм рт. ст. Аппарат, созданный им еще в 1943 г., в дальнейшем был видоизменен применительно к процессам непрерывного фракционирования. На рис. 205 показана новейшая модель этого дистиллятора. Установка работает следующим образом.
Исходная смесь из колбы для дегазации поступает по капилляру в дистиллятор, в котором она стекает тонким слоем по наклонному лотку, обогреваемому циркулирующим высококипя-щим минеральным маслом. Вдоль лотка возникает градиент температур, поскольку температура повышается в направлении течения дистиллируемой жидкости. Пары, окружающие лоток, конденсируются в холодильнике, наклоненном под тем же углом, что и лоток. Установка имеет три зоны конденсации и четыре штуцера для отбора трех фракций дистиллята и кубового остатка.
Незначительный перепад давления между колбой для дегазации и дистиллятором (оба сосуда перед проведением перегонки подключаются к одному и тому же вакуумному насосу) облегчает ввод исходной смеси в лоток дистиллятора и позволяет измерять скорость ее подачи. Предусмотрена возможность регулирования скорости течения перегоняемой жидкости по обогреваемому лотку путем соответствующего изменения наклона дистиллятора с помощью сферического шлифа, а также возможность изменять скорость течения во время дистилляции. Регулирование этих параметров необходимо для установления оптимального соотношения между количеством отбираемых фракций.
* В отечественной технической литературе этот термин не применяют. — Прим. ред.
284
Циркулирующее обогревающее масло нагревается с помощью горелки Бунзена или электронагревательной обмотки, размещенной на входном штуцере циркуляционного контура. Продольный температурный градиент в обогреваемом лотке зависит от скорости подачи исходной смеси, скорости течения ее по лотку, состава перегоняемой жидкости, а также от типа вакуумного насоса. В описанном стеклянном дистилляторе температурный градиент между началом и концом лотка составляет 10—20° С. Рабочую температуру дистилляции определяют косвенным путем, измеряя температуру кипения небольшой пробы дистиллята при стабильном вакууме. Термометр, погруженный в циркулирующее обогревающее масло, обычно показывает температуру на 60—80° С выше измеренной температуры кипения. Это означает, что пленочное испарение приводит к сильному охлаждению испаряющей поверхности лотка. Средняя пропускная способность установки составляет около 100 мл/ч, что соответствует количеству отбираемого дистиллята 50—70 мл/ч.
Дистиллятор Утцингера был усовершенствован народным предприятием «Йенский завод стекла Шотт», на котором были созданы макро- и микрокаскадные аппараты (рис. 206). На этих аппаратах, собранных из стандартных взаимозаменяемых деталей,
^Рис. 206.
I Мнкрокаскадный аппарат для молекулярной дистилляции н дистилляции на «коротком пути» .
№: Изготовитель: народное предприятие «Йенский завод стекла Шотт», Иена. ,
ЕрРис. 207.
К Дегазатор Утцингера.
285
0 20 ОО 60 80 100
Выход дистиллята,У а
Рис. 208.
Разделяющая способность одноступенчатого (/) и многоступенчатого (2) испарителей.
Молекулярная дистилляция смеси диизооктилсебацииат (ДИОС), молекулярная масса 426, — днизооктилфталат (ДИОФ), молекулярная масса 391 ‘[146].
Рис. 209.
Установка Малюсова—Жаворонкова [149] для многоступенчатой моле-кулярной дистилляции:
1 — штуцеры для присоединения вакуумной линии; 2 — устройство для отбора дистиллята; 3 — штуцеры для подачи охлаждающей воды; 4 — труба для подачи исходной смеси; 5 — электронагреватели.
можно получать любое необходимое число фракций. В каждом элементе каскада путем циркуляционного способа нагревания можно устанавливать любую температуру. Предварительную дегазацию исходной смеси проводят отдельно в устройстве, показанном на рис. 207. При этом учитывают то обстоятельство, что процессы дегазации и дистилляций протекают с существенно различными скоростями.
Мельпольдер с сотр. [148] разработал двадцатиступенчатый аппарат для молекулярной ректификации, предназначенный для разделения высококипящих фракций нефти. В работе [140] предложен десятиступенчатый испаритель, который медленно вращается в наклонном положении. Разделение исходной смеси в этом случае происходит на всей поверхности испарителя. Образовавшийся на первой ступени охлаждения конденсат поступает во вторую ступень испарения и таким образом продвигается далее до места отбора дистиллята. Сравнение разделяющих способностей одноступенчатого испарителя (см. рис. 204) и десятиступенчатого испарителя [140] показано на рис. 208 [146]. Методы расчета чисел теоретических ступеней разделения, достигаемых в подобных многоступенчатых аппаратах, представлены в работах Жаворонкова и Малюсова с сотр. [149, 150]. На рис. 209 показана установка для молекулярной дистилляции, использованная этими исследователями.
Ректификация по принципу «падающей пленки» нашла наиболее широкое применение. Большую часть работающих на этом прин-286
ципе аппаратов, описанных в разд. 5.4.3, можно использовать также и для молекулярной дистилляции. Благодаря малому времени пребывания гравитационно стекающей пленки жидкости на поверхности испарения, составляющему для лабораторных приборов 3—4 с и для промышленных аппаратов до 0,001 с [92], перегонка веществ протекает в исключительно мягких температурных условиях. В лабораторных приборах расчетная толщина слоя жидкости лежит в пределах 0,1—0,2 мм, что соответствует слою из 50 000 молекул, а в промышленных аппаратах толщина слоя может составлять 0,001—0,005 мм, что соответствует слою из 400 молекул. Теоретически идеальным условием перегонки являлся бы мономолекулярный слой.
Очень важными факторами для аппаратов, работающих по методу молекулярной дистилляции, являются форма выполнения поверхности испарения и, конечно, расстояние между поверхностями испарения и конденсации. Хорошее смачивание поверхности испарения и оптимальная циркуляция пленки жидкости обеспечиваются в аппаратах «с падающей пленкой» жидкости, описанных в разд. 5.4.3 [122].
На рис. 210 показана установка для молекулярной дистилляции с испарителем, снабженным спиральным ротором. Стеклянный спиральный ротор 8 испарителя вращается вокруг испарительной свечи 10. Такое конструктивное решение обеспечивает получение тонкослойной жидкой пленки толщиной около 0,1 мм и хорошую циркуляцию жидкости. Время пребывания жидкости в аппарате составляет всего несколько секунд. Установка имеет следующие технические данные: условная производительность — 1000 г/ч; производительность* — 250—2000 г/ч; максимальная скорость испарения * — 18 000 г/ч; частота вращения ротора — около 40—90 об/мин; площадь поверхности испарения — около 600 см2; максимальная температура дистилляции — 300 °C; рабочее напряжение электросети 380—220 В; потребляемая мощность — 2 кВт; расход охлаждающей воды — около 350 л/ч. В качестве вымораживающих хладоагентов рекомендуется использовать жидкий воздух или азот, а в подходящих случаях смесь СО2 — ацетон.
При разработке лабораторной установки модели КДБ4, ;в основу работы которой положен метод дистилляции на «коротком пути», фирма Лейбольд—Хэраеуз (Кёльн) исходила из того, чго первой стадией молекулярной дистилляции является дегаза-?ция исходной смеси, за которой следует большое число стадий дистилляции, проводимых при различных температурах. Уста-сновка снабжена испарителями с роликовым скребковым ротором, j/изготовленным из тефлона или тефлоновой керамики. Ротор размещается между внутренним охлаждаемым цилиндром и наружным обогреваемым корпусом. Для обеспечения многоступенчатой
* Зависит от разделяемой смеси.
287
Рис. 210.
Установка для молекулярной дистилляции модели MDRR600.
1 — пульт управления; 2 — охлаждаемая ловушка для дегазатора; 3 — клапан для точной дозировки исходной смеси; 4 — привод ротора; 5 — дегазатор; 6 — охлаждаемая ловушка дистиллятора; 7 — сосуд для исходной смеси; 8 — спиральный ротор; 9 — дистиллятор с конденсационным кожухом; 10 — испарительная свеча; 11 — термопара; 12 — высоковакуумный масляный диффузионный насос; 13 — электромагнитные клапаны сменных сборников дистиллята и кубового продукта; 14 — сборник дистиллята; 15 — сборник кубового продукта; 16 — двухступенчатый шиберный вакуумный насос; 17 — одноступенчатый шиберный вакуумный насос; 18 — вакуумный запорный клапан; 19 — универсальный лабораторный термостат.
Изготовитель: народное предприятие «Йенский завод стекла Шотт», Иена.
дистилляции испарители, выполненные из стекла или нержавеющей стали, устанавливают друг за другом. Схема лабораторной установки KDL4 непрерывного действия показана на рис. 211. Давление на различных ступенях дистилляции может достигать 1(Г3 мм рт. ст. Минимальная производительность установки составляет 100 г/ч. Хабендорфф [140а] представил рекомендации по монтажу и вводу в эксплуатацию промышленных установок для дистилляции «на коротком пути», а также технологические параметры процесса.
288
; Ю Э. Крель
Некоторые аппараты «с падающей пленкой» снабжают устройством для циркуляции перегоняемой жидкости (рис. 212). Циркуляцию осуществляют с помощью электромагнитного насоса. J Обычно в месте ввода жидкости размещают кольцо из проволочной сетки для обеспечения равномерного распределения исходной смеси [152].
Основной проблемой при разработке аппаратов, работающих ; по принципу «падающей пленки», является предотвращение воз-• никновения струйного течения на поверхности испарения. Ее решают одним из следующих способов: поверхность испарения выполняют шероховатой путем обработки ее пескоструйным аппа-
Рис. 211.
Лабораторная установка модели KDL4, работающая по методу дистилляции «на коротком пути», с роторными испарителями.
Ротор испарителя снабжен скребками в виде роликов. 1 — распредели-ельные электрощиты со штепсельными розетками; 2 — дозирующий со-£УД‘, 3 — привод ротора; 4 — штуцер для присоединения вакуумного на-са 1 ступени; 5 — охлаждаемые ловушкн; 6 — стеклянный дистиллятор; __ — металлический дистиллятор; 8 — циркуляционные термостаты; 9 — ^Мерный сосуд (в разрезе); 10 — штуцер для присоединения вакуумного асоса 2 ступени, зготовитель: фирма Лейбольд—Хэраеуз, Кельн.
Рис. 212.
ппарат Хикмана для молекулярной дистилляции с циркуляционным уст-йством.
289
ющую поверхность подают
Рис. 213.
Аппарат Хикмана для молекулярной дистилляции с центробежным распределением жидкости: 1 — насос для подачи исходной смеси; 2 — устройство для отвода кубового продукта; 3 — поверхность конденсации; 4 — вращающийся обогреваемый диск; 5 — сборник кубового продукта; 6 — емкость для исходной смеси.
ратом, травления или наплавлива-ния стеклянного порошка (см. рис. 196, 205, 206); в месте ввода исходной смеси устанавливают кольцо из проволочной сетки либо плотно прилегающий цилиндр из проволочной сетки или стеклоткани (см. рис. 196, 212); поверхность испарения выполняют волнистой или в виде спирали (см. рис. 210); жидкость на испаря-с помощью ротора со скребковыми
устройствами или при вращении всей поверхности испарения (см. рис. 200—203, 211).
Центробежный принцип получения наиболее тонких слоев жидкости развит Хикманом 1153), он применил его для разработки промышленных аппаратов большой производительности [151 ]. На рис. 213 показан лабораторный аппарат с вращающимся диском. Разделяемая смесь из емкости 6 подается насосом 1 на вращающийся обогреваемый диск 4, с которого под действием центробежной силы жидкость распределяется в виде тонкой пленки. Поверхность конденсации 3 расположена на небольшом расстоянии от центрифуги. Дистиллят можно либо отбирать с поверхности конденсации, либо возвращать в емкость 6. Кубовый остаток с помощью устройства 2 отводится в сборник 5, откуда он может быть отправлен в емкость 6 для дальнейшей переработки.
Хикман [152] и Эмбре [154] ввели для молекулярной дистилляции понятие «дистилляционная способность», под которой понимают отношение числа молекул вещества, покидающих в единицу времени поверхность испарения, к числу молекул того же вещества, остающихся при данных условиях в пленке жидкости. Многократной циклической перегонкой можно полностью получить вещество в виде дистиллята. При этом продолжительность времени дистилляции удается сократить путем повышения температуры испарения. «Кривую выделения» находят следующим образом. Смесь перегоняют при стабилизированном вакууме и постоянной скорости повышения температуры (например, последовательно повышая температуру на 10 °C) и определяют концентрацию низкокипящего компонента в дистилляте. Типичные «кривые выделения» показаны на рис. 214. Как видно из рисунка, концентрация вначале растет до максимума, а затем снижается
290
г до нулевого значения. Форма кривой зависит, конечно, от физико- химических свойств веществ и прежде всего от удельной энталь-5 пии испарения, а также от времени пребывания жидкости в испа-рителе (которое для возможности сравнения экспериментальных г данных должно быть постоянным). Максимум на «кривой выделения» соответствует температуре кипения при обычной дистилляции. При большем времени пребывания продукта в горячей зоне «кривая выделения» смещается таким образом, что ее максимум соответствует более низкой температуре (см. кривую 2, рис. 214) [159]. Для достижения оптимальной производительности установок молекулярной дистилляции необходимо предварительно тщательно исследовать условия ее проведения путем определения «кривых выделения». Такое теоретическое исследование проведено Горрицом с сотр. [155].
Данный метод получил дальнейшее развитие благодаря применению стандартных веществ, в большинстве случаев красителей, » которые удобно анализировать колориметрическим методом [108].
Если стандартного вещества мало, то для получения хорошей • пленки жидкости добавляют другое вещество — носитель.
Хикман и Тревой [141 ] при исследовании процессов высоковакуумного испарения и молекулярной дистилляции получили интересные данные. Оказалось, что коэффициент испарения чистых жидкостей может иметь значение 1. Отклонения чаще всего обусловлены незначительными загрязнениями. При определении • относительной летучести смеси ди-2-этилгексилфталат (ЭГФ)— ' ди-2-этилгексилсебацинат (ЭГС) было установлено, что она в значительной степени зависит от условий определения (рис. 215).
Малюсов с сотр. [160] при исследовании процессов перегонки под вакуумом, соответствующем 10~4 мм рт. ст., определял раз-
Рис. 214.
«Кривые выделения» , полученные при кратковременном (/) и длительном (2) пребывании продукта на поверхности испарения.
Рис. 215.
Зависимость относительной летучести для смеси дн-2-этнлгекснлфталат (ЭГФ) — ди«2-этилгекснлсебацннат (ЭГС) от температуры.
Температура кипения ЭГФ 183—181 °C н ЭГС 199 — 202 °C при 0,5 мм рт. ст. Кривые получены при работе на приборе для исследования фазового равновесия, снабженном кубом (/), н на приборе <с падающей пленкой» (2).
Г— 10*
291
ницу между неравновесным анравн и равновесным аравн факторами разделения. С повышением температуры отношение аирави/арави приближается к 1, что соответствует графику на рис. 215.
Уеха и Хагихара [163] исследовали эталонную смесь ди-2-этилгексилфталат—ж-трикрезилфосфат при остаточном давлении менее 1,5-10”® мм рт. ст. (2-10-3 мбар) в пределах температур 130—160 °C. Оказалось, что в области низких температур эта смесь в интервале концентраций ди-2-этилгексилфталата от 20 до 80% (мол.) ведет себя как идеальная смесь.
5.4.5. РЕКТИФИКАЦИЯ ПОД ДАВЛЕНИЕМ
Под давлением выше атмосферного можно перегонять смеси, которые при комнатной и более высокой температурах находятся в газообразном состоянии. В данном случае в качестве хладоагента можно применять воду, что делает процесс перегонки более экономичным. Однако для получения избыточного давления необходима дополнительная аппаратура.
Ректификацию под давлением целесообразно использовать в тех случаях, когда относительная летучесть компонентов смеси увеличивается с повышением температуры. Например, для смеси 2,4-диметилпентан — 2,2,3-триметилпентан разность температур кипения при атмосферном давлении составляет лишь 0,2 °C, а значение а возрастает с 1,006 при 80 °C до 1,055 при 200 °C (см. разд. 4.6.2).
Известно лишь небольшое число лабораторных установок для ректификации под давлением. Для давлений выше 5 кгс/см1 2 в качестве конструкционного материала для колонн стекло не-
Рис. 216.
Установка Симонса для ректификации под давлением до 5 кгс/см2:
1, 16 — клапаны для автоматической - стабилизации давления; 2 — коиус поплавка клапана; 3 — седло клапана; 4, 5 — штуцеры для подачи сжатого воздуха и вывода его в атмосферу соответственно; 6 — поплавок клапана; 7, 15 — груши клапанов; 8,9 — конденсаторы дистиллята; 10 — капилляр с электронагревательной обмоткой; 11 — воронка для подачи флегмы; 12 — колонна; 13 — куб; 14 — электронагреватель куба; 17 — приемник дистиллята; 18, 19 — трубы для подачи дистиллята; 20 — край.
Рис. 217.
Кран для работы под давлением с предохранительным устройством.
292
►применимо. Шнайдер описал металлический ректификационный (аппарат, предназначенный для работы под давлением до 45 кгс/см2, "который состоит из колонны диаметром 25 мм и куба емкостью 3 л [161 ].
Ректификационную установку Симонса [162], полностью изготовленную из стекла, можно применять до давлений, не превышающих 5 кгс/см2 (рис. 216). Автоматическая стабилизация давления сжатого воздуха, подаваемого по штуцеру 4, осуществляется (с помощью клапана 1, заполненного ртутью. Клапан имеет поплавок 6, оканчивающийся конусом 2. Этот конус запирает отверстие в седле клапана 3. Уровень ртути в клапане устанавливают ’С помощью груши 7, таким образом поплавок 6 поддерживает в системе необходимое давление. Если давление превысит заданное, то поплавок 6 опустится вниз и избыток воздуха через штуцер 5 будет сбрасываться в атмосферу до тех пор, пока снова не (установится равновесие сил давления и выталкивания, и клапан (опять закроется. Таким образом в системе поддерживается постоянное давление.
Разделяемую смесь загружают в куб 13, обогреваемый электронагревателем 14. Колонну 12 заполняют необходимой насадкой. Пары, выходящие из колонны, конденсируются в холодильнике 9. (Конденсат в виде капель поступает в воронку 11, снабженную небольшим отверстием, через которое часть конденсата — фле-Гма — стекает назад в колонну. Дистиллят, протекая по капилляру 10 с электронагревательной обмоткой, вновь испаряется, Тйатем вторично конденсируется в холодильнике 8 и по трубе 19 втекает в клапан 16, идентичный клапану 1. Поднятием и опусканием груши 15 клапан 16 открывают или закрывают. При этом часть дистиллята собирается в слое, расположенном над )тутью в клапане 16, а остальная его часть постоянно перетекает . Цо трубе 18. Если закрыть клапан 16 и открыть кран 20, то давле-йе под клапаном 16 уменьшится и отобранная низкокипящая фракция испарится. Ее конденсируют в приемнике дистиллята 17, Охлаждаемом жидким воздухом. Таким образом отбирают пробы,. ie изменяя давления в установке.
При соответствующих мерах предосторожности обычные стек-|1янные аппараты можно применять в ректификационных уста-овках, работающих под давлением до 2 кгс/см2 *. В этих случаях все шлифовые соединения должны быть укреплены прочими стягивающими пружинами или, еще лучше, кольцевыми айбами. После внесения соответствующих изменений обычные >аны могут быть использованы для работы под давлением ЙИс. 217).
* В этих случаях необходимо выполнять все требования техники безопас-' ОсТи, предъявляемые при работе со стеклянной аппаратурой под вакуумом едохранитедьные ксжухи, защитные очки и пр.). — Прим-, ред........
6
Селективные методы перегонки
К селективным методам перегонки относятся такие методы дистилляции и ректификации, разделяющая способность которых обеспечивается или дополнительно повышается при добавлении в исходную смесь какого-либо вещества в газообразном, жидком или твердом состоянии. Сюда же относят и методы, связанные с образованием новых веществ в результате каких-либо реакций, протекающих в процессе перегонки. К селективным методам можно также причислить комбинированные методы, дополняющие перегонку другими методами разделения, например газовой хроматографией.
Согласно правилу фаз Гиббса, двухфазная система, состоящая из двух компонентов с неограниченной взаимной растворимостью, может быть обогащена ректификацией в отличие от трехфазной системы, содержащей два взаимно нерастворимых компонента (см. рис. 29 а—г). С другой стороны, известно, что трехкомпонентная система с ограниченной взаимной растворимостью компонентов, т. е. система с двумя жидкими фазами и одной паровой фазой, может быть разделена ректификацией. Типичный пример такого процесса разделения — получение абсолютного спирта азеотропной ректификацией с бензолом.
6.1. ПЕРЕГОНКА В ПАРАХ НОСИТЕЛЯ
Наиболее известным примером метода перегонки в парах носителя является перегонка высококипящих веществе водяным паром. Носитель вводят в паровую фазу, во-первых, чтобы извлечь из нелетучего кубового остатка более летучие компоненты смеси, и, во-вторых, чтобы снизить температуру кипения и смягчить термический режим перегонки. При этом достигаются условия, близкие к тем, которые имеют место при вакуумной перегонке. Давление паров летучего вещества рх меньше, чем общее давление системы р. Разность р — рг соответствует парциальному давлению водяного пара р2. При вакуумной перегонке по мере уменьшения содержания легколетучего компонента в кубе необходимо либо увеличить температуру, либо повышать вакуум. При перегонке с водяным паром требуется лишь постепенно увеличивать расход водяного пара [2].
Перегонку с водяным паром применяют, главным образом, для очистки и разделения нераствормых или мало растворимых в воде 294
J веществ, например, эфирных масел, жирных кислот, спиртов жирного ряда, анилина, таллового масла и восков, а также бензинов и фракций смол различного состава. Водяной пар вытесняет из установки воздух и предохраняет тем самым загруженный материал от окислительных процессов. Перегонка с водяным паром
дает хорошие результаты при разделении смазочных масел и при отгонке веществ из сильных оснований в препаративных целях.
Как показано в разд. 4.3 и 4.5, при перегонке с водяным паром оба компонента и их пары ведут себя так, как если бы при той же температуре присутствовал только один компонент. Общее давление над кипящей смесью складывается из парциальных давлений ’t обоих компонентов, один из которых вода. Парциальные давления и температуру кипения смеси можно определить графически с по-; мощью суммирующей формулы (39), как описано в разд. 4.5. ‘ Целесообразно использовать диаграмму Бэджера—Кэба [2], на
которую нанесены кривые давления паров перегоняемых веществ (рис. 218). Кривые, соответствующие давлению паров воды для общего давления 760, 300 и 70 мм рт. ст., показывают разность между общим давлением над смесью и парциальным давлением водяного пара. Например, давление паров воды при 60,1 °C равно 150 мм рт. ст., тогда ордината точки кривой А с абсциссой 60,1 °C
800
100
600
В 500 Cj
в ^600
, 300
200
100
О 20 60 60 80 100 120 1<Ю 160 180 200
t,°C
'Рис. 218.
(^Диаграмма Бэджера—Мак-Кэба для определения температуры кнпення н Парциальных давлений при перегонке с водяным паром;
f —• простой эфир; 2 — сероуглерод; 3 — ацетон; 4 — метанол; 5 — че-Ыреххлористый углерод; 6 — бензол; 7 — этанол; 8 — муравьиная кислота; 9 — толуол; 10 — уксусная кислота; 11 — терпентин; 12 — фенол; !8 — аннлнн; 14 — крезол; 15 — нитробензол; 16 ~ нитротолуол. А — •Ода прн 760 мм рт. ст.; Б — вода при 300 мм рт, ст.; В — вода при >0 мм рт. ст.
295
Рис. 219.
Взаимосвязь температуры кипения и давления перегонки для насыщенных жирных кислот ряда с не-разветвленной углеродной цепью и четным числом атомов углерода в присутствии водяного пара и без него.
будет равна разности 760—150 = 610 мм рт. ст. Точки пересечения кривой 80 КО 160 200 2W 280 320 360 для ВОдЫ с крИВОЙ ДЭВле-’ ния паров любого компо-
нента соответствуют температуре кипения смеси и парциальному давлению этого компонента. Диаграмму можно построить также и в логарифмических координатах.
Исходя из парциальных давлений, по формуле (35) рассчитывают состав паров над смесью. Соотношение массовых количеств отгоняемого компонента и носителя — водяного пара определяют по формуле
Gx/G2 = рМ(р2Мг) (184)
Например, для смеси толуол — вода получаем:
Gi/Ga =337-92/(423.18)= 4,1 (185)
Это означает, что для выделения 4,1 кг толуола потребуется 1 кг водяного пара. По формуле (35) содержание толуола в парах составит 80,3% (масс.), температура кипения смеси в соответствии с диаграммой на рис. 218 равна 84,4 °C.
Перегонку легко разлагающихся веществ с водяным паром можно проводить также и под вакуумом. Согласно диаграмме, представленной на рис. 218, температура кипения смеси, содержащей анилин (98 °C при 760 мм рт. ст.), снижается до 75 °C при остаточном давлении 300 мм рт. ст. При этом предполагается, что водяного пара подводят ровно столько, сколько нужно для взаимного насыщения компонентов.
Перегонку с водяным паром выгоднее проводить под вакуумом также для выделения веществ с очень низким давлением паров, например, жирных кислот с длинной углеродной цепью, или при разделении смесей, один из компонентов которых содержится в незначительных количествах, например следы растворителя в маслах.
Вопросы применения перегретого водяного пара при перегонке обстоятельно рассмотрены Штаге. Перегретый пар широко используют в промышленности при перегонке смол, минерального масла и жирных кислот, а также при очистке глицерина. На примере гомологического ряда насыщенных жирных кислот С4—С20 с прямой углеродной цепью показано, что при перегонке с насыщенным паром температуры кипения снижаются приблизительно 296
на 160—240 °C, однако при этом разность температур кипения кислот с различным числом углеродных атомов настолько уменьшается, что разделить компоненты уже невозможно (рис. 219). Если же работать при остаточном давлении 10 мм рт. ст. с 10%-ной добавкой перегретого водяного пара, то температура процесса снизится только на 20 °C. При этом сохранятся различия в температурах кипения, а также такие важные преимущества перегонки с водяным паром, как хорошее перемешивание кубовой жидкости и уменьшение опасности ее перегревания [31. Следует указать, что подобные смеси жирных кислот и спиртов сравнительно хорошо разделяются в присутствии водяного пара также методом многоступенчатой парциальной конденсации [4 ].
Водяной пар вследствие низкой молекулярной массы, большой теплоте парообразования и хорошей конденсируемости широко применяют в качестве носителя. Для снижения парциального давления перегоняемой смеси в промышленности используют также и другие газы. Например, при перегонке каменноугольных смол для получения твердого пека в качестве газа—носителя применяют отходящие газы, которые содержат СО2, N2 и водяной пар [6 ].
Не следует опасаться значительного снижения эффективности колонны в результате введения в нее инертного газа 171. Алгер-миссен (81 разработал графический мётод расчета процесса конденсации паров из смеси с газами.
Аппарат для перегонки с водяным паром можно собрать из обычных стандартных деталей. На рис. 220 представлена схема установки для перегонки с насыщенным водяным паром при атмосферном давлении и под вакуумом. Перегонную колбу 1 тщательно изолируют стекловатой или шлаковатой. Колбу рекомендуется обогревать, чтобы исключить конденсацию в ней водяного пара.
Патрубок для ввода водяного пара 2 снабжен 'краном для слива водного конденсата, и он может служить также для подачи другого газа—носителя. На рис. 221 показана установка для ректификации с перегретым водяным паром. Для получения водяного пара применяют металлический куб 1 с во-
Рис. 220.
Установка для перегонки с насыщенным водяным паром:
1 — перегонная колба; 2 — патрубок для ввода водяного пара;
3 — предохранительный клапан;
4 — капельница.
297
Рис. 221.
Установка для ректификации с перегретым водяным паром:
1 — металлический куб для" получения водяного пара; 2 — металлический змеевик для перегрева водяного пара; 3 — предохранительный клапан.
Рис. 222.
Прибор Поццн—Эскота для дистилляции с водяным паром малых количеств вещества.
домерной стеклянной трубкой. Пар перегревается в металлическом змеевике 2, размещенном в коническом корпусе, за которым установлен брызгоуловитель с термометром. Целесообразно в обеих описанных установках предусмотреть предохранительный клапан 3.
Для получения перегретого пара очень удобен пароперегреватель конструкции Тропша [7]. Для получения воспроизводимых результатов при перегонке в колонну необходимо подавать постоянные количества пара. Достаточно равномерного дозирования пара можно достигнуть, если над колбой с водой установить градуированный цилиндр с капельницей 4 (см. рис. 220), с помощью которой регулируют подачу воды при ее постоянном уровне в колбе. Более надежным является приспособление, описанное Меркелем [9 ], в котором количество пара регулируется по перепаду давления, контролируемого с помощью манометра. Штаге с сотр. [10] разработал устройство, обеспечивающее точную дозировку пара за счет того, что вода из измерительной бюретки непрерывно подается в генератор водяного пара, который почти наполовину засыпан крупным песком для интенсификации теплопередачи.
Для перегонки малых количеств вещества удобен прибор Поцци—Эскота [11], в котором колба для получения водяного пара служит одновременно баней для перегонной колбы (рис. 222). Для микродистилляции пригоден прибор Парнасса и Вагнера [12]. 298
| Оборы методов перегонки с водяным паром можно найти I у Бернхауэра и Торманна [14]. Вебер [15] подробно рассматри-I вает теорию и практику процессов перегонки с водяным паром на I примере ректификации эфирных мас1ел. Ригамонти [16] разра-Е ботал номограмму для расчета расхода водяного пара при задан-I ной степени обогащения.
I 6.2. АЗЕОТРОПНАЯ И ЭКСТРАКТИВНАЯ
I ПЕРЕГОНКА
I Методы азеотропной и экстрактивной перегонки широко приме-I няют в промышленности в тех случаях, когда обычное ректифика-I ционное разделение связано со значительными трудностями. В ла-I бораториях же для разделения близкокипящих компонентов не-L идеальных смесей без азеотропа, а также азеотропных смесей I обычно используют другие методы, например химические методы, | экстракцию или хроматографию. Ниже показано, что для разделе-I ния указанных выше смесей селективные методы перегонки, та-г кие как азеотропная и экстрактивная ректификация, имеют су-I щественные преимущества. Общий отличительный признак этих I обоих методов перегонки заключается в том, что они основаны на I влиянии специально подобранного вещества на отношение коэф-I фициентов активности разделяемых компонентов [17]. Кюммерле К [18] показал, что возможна также комбинация обоих методов — I азеотропноэкстрактивная ректификация. Герстер [19] сравнил | эти методы и обычную ректификацию с экономической точки В зрения.
t Мюллер [19а] сопоставил между собой экстракцию и экстрак-| тивную перегонку применительно к выделению ароматических к соединений из продуктов пиролиза бензина и риформинга. Он fc показал, в каких случаях экстракция и экстрактивная ректи- фикация имеют определенные преимущества одна перед другой, К и привел экономические расчеты. Вопросы азеотропной и экстрак-Ц тивной перегонки подробно рассмотрены в монографии Хофф-К мана, которая содержит многочисленные примеры расчетов для В бинарных, тройных и многокомпонентных систем.
Ц Неидеальные смеси, не образующие азеотропа, характери-Ц зуются тем, что их кривая равновесия асимптотически прибли-Ц жается к диагонали в верхнем или нижнем углу диаграммы Ц (например, см. рис. 29, е и з). Разделение подобных смесей требует К значительного числа теоретических ступеней разделения даже при К сравнительно большой разнице в температурах кипения компо-Е центов. Неидеальные смеси с одной азеотропной точкой (их кратко Е называют азеотропными) при ректификации образуют (см. разд. Е 4.6.2), либо азеотроп с минимальной температурой кипения Е (положительный азеотроп), либо азеотроп с максимальной темпе-к ратурой кипения (отрицательный азеотроп).
299
Если азеотроп положительный, то в головном продукте (дистилляте) получают смесь, обоих компонентов, а в кубовом остатке перегонки содержится чистый компонент. Если азеотроп отрицательный, то дистиллят представляет собой чистый компонент, а кубовый остаток — смесь обоих компонентов. Какой из компонентов получится в чистом виде, зависит от состава исходной смеси. После получения азеотропного состава дальнейшее обогащение смеси легколетучим компонентом не может быть достигнуто методом обычной ректификации. Положительные азеотропы (см. пример на рис. 43, тип 3, ряд III) встречаются значительно чаще, чем отрицательные азеотропы (см. рис. 43, тип 5, ряд III). По таблицам Леката [20], которые содержат сведения о 6287 азеотропных и 7003 неазеотропных смесях, количественное соотношение между положительными и отрицательными азеотропами примерно составляет 9:1.
Хорслей [51] приводит данные по азеотропам, опубликованные до 1962 г. В справочнике по азеотропным смесям Когана с сотр. [21 ] содержатся сведения о 21069 смесях, в том числе о 19735 бинарных, 1274 тройных и 60 многокомпонентных смесях. В справочнике наряду с таблицами также дано введение в теоретические основы и технику экспериментального исследования явления азеотропии. Здесь же приводятся кроме того сведения о влиянии температуры на состав азеотропных смесей, о методах расчета азеотропного состава и о методах исследования свойств азеотропных смесей.
Вопросы теории азеотропных смесей рассмотрены с термодинамической и молекулярно-кинетической точек зрения в ряде работ [22], [23], [27] и [28]. Эти работы содержат сведения о тройных и четверных азеотропах. В работе Отмера [28а] приводится подробный обзор достижений в области теории и практического использования азеотропной ректификации.
В первом томе своей фундаментальной монографии Светослав-ский [27] излагает теорию азеотропии, обсуждает методику экспериментального исследования этого явления, приводит классификацию разновидностей азеотропов, а также анализирует возможности использования явления азеотропии и полиазеотро-пии в промышленности. Во втором томе монографии явление азеотропии рассмотрено с термодинамической точки зрения.
Превосходное введение в азеотропную и экстрактивную ректификацию в лабораторных условиях как с теоретической, так и с практической точки зрения дает Рёк [17]. Азеотропии бинарных систем обсуждается в первом томе книги Шуберта [29] по термодинамике растворов.
При перегонке смесей близкокипящих компонентов обычно следует считаться с возможностью образования азеотропа. Азеотропии проявляется не только при наличии специфического взаимодействия между молекулами, но также наблюдается у смесей неполярных веществ, например бензол—циклогексан или бен-300
зол—четыреххлористый углерод. В. Кун и Г. Дж. Кун [24] объясняют распространенность азеотропии у смесей близкокипящих компонентом тем, что в таких смесях эффекты, основанные на различии в давлениях паров чистых компонентов, компенсируются, а тепловые эффекты смешения компонентов суммируются. Циборак [30] показал, что при добавлении в смесь этанол—вода углеводородов в процессе для обезвоживания образуется ряд четырехкомпонентных гетероазеотропов и трех компонентных гомоазеотропов. Орзаг [31 ] исследовал различные полиазеотропные жидкие смеси и влияние образовавшихся азеотропов на процесс перегонки. Малесинский [32] и Штеки [33] дают классификацию гомо- и гетероазеотропных систем. В обзоре, посвященном выбору добавок для азеотропной и экстрактивной перегонки, Берг [34 ] рассматривает классификацию жидкостей по водородным связям их молекул.
Разумеется, для разделения близкокипящих компонентов и неидеальных смесей, не образующих азеотропа, можно подобрать ректификационные колонны эффективностью в 100 и более теоретических ступеней разделения, поскольку насадка с ВЭТС, равной 1—2 см, сейчас не является уже редкостью. Однако вместо применения колонн с 200 или даже 300 теоретическими ступенями разделения (относительная летучесть а = 1,03 — 1,02) такие смеси можно разделить, если воздействовать на фазовое равновесие в направлении повышения значений а и достижения более благоприятных условий разделения. В качестве примера рассмотрим экстрактивную ректификацию смеси близкокипящих компонентов w-гептан — метилциклогексан, для которых разность температур кипения составляет 2,7 °C (а = 1,075). При обычной ректификации с бесконечным флегмовым числом требуется 48 теоретических ступеней, чтобы сконцентрировать смесь от 15,3 до 95,4% (мол.). Если же в смесь добавить 70% (масс.) анилина, то такого же обогащения можно достигнуть при числе теоретических ступеней 12,4 и флегмовом числе v = 35. При этом относительная летучесть возрастает с 1,07 до 1,30 [35]. Если смесь является азеотропной, то чистые компоненты можно получить только с помощью селективного метода разделения.
С одной стороны, явление азеотропии крайне нежелательно, например при концентрировании смеси этанол—вода и других азеотропных смесей. С другой стороны, это явление дает возможность разрушить уже имеющийся азеотроп и разделить смесь весьма близкокипящих компонентов. Введением дополнительного компонента * сознательно вызывают образование азеотропа между этим компонентом и одним из компонентов бинарной смеси * ** и легко отделяют новый азеотроп от второго компонента благо-
* В советской литературе добавочный компонент называют разделяющик
агентом. — Прим. ред.
** При азеотропной перегонке. — Прим. ред.
301
Рис. 223.
Схема установки для азеотропной ректификации а периодическом режиме (а) и в непрерывном режиме (<Г):
1 — гетероазеотроп; 2 — разделяющий агент (возврат на орошение); 3 — компонент А; 4 — в начале перегонки исходная смесь разделяющий агент; в конце — компонент В; 5 — исходная смесь -|- разделяющий агент; 6 — компонент В.
даря увеличившейся разнице в температурах кипения. Конечно, необходимо условие, чтобы азеотропный конденсат легко расслаивался на составные части. Это достигается охлаждением, высаливанием, химическим выделением разделяющего агента, экстракцией выделяемого компонента или также повторной азеотропной перегонкой.
302
Если после полной конденсации паров азеотропного состава образующийся дистиллят не расслаивается, то говорят о гомогенном азеотропе (например, смесь этанол—вода), в то время как азеотроп, расслаивающийся в условиях конденсации на две фазы, называют гетерогенным (например, смесь бензол—вода).
При экстрактивной перегонке благодаря введению в исходную жидкость смесь высококипящего разделяющего агента с более высокой (на 50—100 °C) температурой кипения добиваются того, чтобы разделение вообще стало возможным (если в исходной
Рис. 224.
Схема установки для экстрактивной ректификации в периодическом режиме (а) и в непрерывном режиме (£):
1 — дистиллят А н В; 2 — разделяющий агент (ввод); 3 — экстракционная зона; 4 — в начале перегонки исходная смесь, в конце — разделяющий агент; 5 — дистиллят А; 6 — исходная смесь (ввод); 7 — дистиллят В; 8 — разделяющий агент (вывод).
303
смеси разность равновесных концентраций Ах между паром и жидкостью равна 0) или стало более легким за счет увеличения Ах [17].
Разделяющий агент не должен образовывать азеотропа с компонентами разделяемой смеси, должен легко отделяться от разделяемой смеси и специфически воздействовать на один из компонентов, вызывая тем самым увеличение относительной летучести компонентов. Обычно используют те же разделяющие агенты, которые применяют при жидкостной экстракции, либо подобные им вещества. Принцип азеотропной и экстрактивной ректификации в периодическом и непрерывном режимах пояснен на рис. 223 и 224.
6.2.1. АЗЕОТРОПНАЯ ПЕРЕГОНКА
Азеотропную перегонку применяют для разделения смесей близкокипящих компонентов, которые в большинстве случаев уже сконцентрированы обычной ректификацией, и для разделения азеотропных смесей, которые близки по составу к азеотропу.
Как правило, с помощью подобранного разделяющего агента добиваются образования с одним из разделяемых компонентов положительного азеотропа. Однако возможно также, что разделяющий агент образует двойные или даже тройные положительные азеотропы с обоими разделяемыми компонентами. Конечно, при этом необходимо, чтобы состав новых азеотропов отличался от состава исходной смеси.
Характерным примером разделения смесей близкокипящих компонентов азеотропной ректификацией является разделение смеси индол—дифенил с применением диэтиленгликоля в качестве разделяющего агента. При атмосферном давлении разность температур кипения указанных веществ составляет всего 0,6 °C. Благодаря добавке диэтиленгликоля разница в температурах кипения образовавшихся азеотропов достигает уже 12,2 °C. Кипящие соответственно при 230,4 и 242,6 °C азеотропные смеси дифенил—диэтиленгликоль и индол—диэтиленгликоль, которые содержат почти по 60% диэтиленгликоля, можно легко разделить, даже используя малоэффективные колонны, при небольшом флегмовом числе. Так как дифенил в отличие от индола мало растворим в диэтиленгликоле, то больших количеств гликоля не требуется. Отгоняемый в первую очередь азеотроп дифенил—диэтиленгликоль расслаивается в приемнике дистиллята, и гликоль непрерывным потоком возвращают на стадию ректификации. Из полностью отогнанной смеси индол—диэтиленгликоль индол осаждают, разбавляя смесь водой [361.
Имеющийся в продаже Р-пиколин представляет собой смесь а-пиколина, Р-пиколина и 2,6-лутидина. Разница в температурах кипения Р-пиколина и 2,6-лутидина при атмосферном давлении составляет 0,15 °C. Методом азеотропной ректификации с до-304
бавлением уксусной или пропионовой кислоты удается сконцег трировать отдельные основания до 95—98% [37]. Другие при ложения азеотропной ректификации в промышленности описан] Думметом [38]. Следует коротко остановиться на некоторы: примерах, представляющих интерес для промышленности.
Согласно Хунсманну и Суммроку [39] при разделении трои ной смеси вода—муравьиная кислота—уксусная кислота следуе ожидать образования бинарного (В) высококипящего азеотроп. 4ип = 107,65 °C, состоящего из 56,7% (мол.) муравьиной кислоте и 43,3% воды и тройного (Т) азеотропа (107,1 °C) состоящей из 39,3% (мол.) воды, 48,2% муравьиной кислоты и 12,5% уксус ной кислоты. Весь интервал концентраций трехкомпонентно! смеси можно разделить на четыре отдельных области перегонк! (рис. 225). Смесь обезвоживают азеотропной перегонкой с однш из высших эфиров.
Закономерности ректификации трехкомпонентных смесей с азео тропными точками были изучены Петликом и Аветьяном [39а ] Лино с сотр. [40] методом азеотропной ректификации с добавкоЁ бромистого этилена удалось удалить из ацетона следы воды вплоп до концентрации менее 1400 рргп.
Обзор всех известных приемов азеотропной перегонки был бь слишком громоздким. Техническая литература, в том числе е патентная, по данному вопросу исключительно обширна. Уже приведенные примеры показывают, насколько велики возможности этого метода перегонки. Поэтому целесообразно указать лишь классы веществ, которые особенно выгодно разделять азеотропной перегонкой. Азеотропную перегонку широко применяют для обезвоживания органических веществ, таких как муравьиная
кислота, уксусная кислота и пиридин, а также для выделения угле-
водородов из спиртов, очистки ароматических углеводородов, разделения моно- и диолефинов и т. д. Мэйр, Глазгов и Россини [41, 42], как и Берг [34], провели систематическое исследование процесса разделения углеводородов азеотропной ректификацией. Разделение изомеров метилнаф-
талина в присутствии ундеканола показывает, что при азеотропной перегонке имеет значение также правильный выбор рабочего давления. Разделение происходит наилучшим образом при 200— 300 мм рт. ст. В кубовом остатке
Муравьиная кислота
Рис. 225.
Схематическое изображение четырех областей перегонки трехкомпонентной смеси вода—муравьиная кислота—уксусная кислота с,б (парным (В) и тройным <Т> азеотропом 139].
R — состав исходной смеси.
305
Рис. 226.
Зависимость состава азеотропа от давления для смесей этанол—вода (/) и вода—фенол (2).
Рис. 227.
Графический метод Наттинга—Хорсли для определения интервала давлений, для которых существует азеотроп.
/ — положительный азеотроп; II — отрицательный азеотроп; Л, В — чистые вещества; С — бинарный азеотроп.
получают чистый а-метилнафталин, а в дистилляте — смесь, содержащую 80% р-метилнафталина [43].
Если азеотроп не стабилен, то его можно исключить, изменяя внешние термические условия (температура или давление) путем проведения перегонки под вакуумом или при избыточном давлении, не прибегая к азеотропной или экстрактивной перегонке. Для гомоазеотропных двухкомпонентных систем Шуберт [44] приводит приближенные зависимости, по которым можно ориентировочно рассчитать условия, необходимые для устранения азео-тропии за счет изменения давления, температуры или путем подбора определенной минимальной концентрации разделяющего агента (применительно к азеотропной или экстрактивной перегонке).
В общем случае понижение давления сказывается таким образом, что азеотропная смесь обогащается легкокипящим компонентом. Во многих случаях в конце концов при некотором вакууме, азеотропный состав исчезает. В качестве примера можно указать на разделение смесей этанол—вода и вода—фенол (рис. 226). Вакуумной перегонкой при 70 мм рт. ст. получают абсолютный спирт и без разделяющего агента. Азеотропная точка на кривой равновесия смеси вода—фенол исчезает при остаточном давлении 32 мм рт. ст. Шнайнкер и Пересслени [45] установили, что азеотропная точка смещается следующим образом: азеотроп смеси муравьиная кислота—вода при 55 мм рт. ст. содержит 66% (масс.) муравьиной кислоты, а при 200 мм рт. ст. — 72% (масс.) кислоты; азеотроп смеси бутанол—бутилацетат при 50 мм рт. ст. содержит 37% (мол.) бутанола, а при 760 мм рт. ст. — 79% (мол.) спирта. 306 **
Рютер и Лю [46] разработали методы расчета азеотропного состава, которые были экспериментально проверены на системах этилацетат—этанол, этанол—вода, метанол—бензол.
Можно привести примеры смесей, для которых применение избыточного давления перегонки также оказывает такое влияние на фазовое равновесие, что азеотропная точка на кривой равновесия исчезает.
В соответствии с графическим методом Нуттинга и Хорслея [47 ] можно очень просто определить интервал давлений, в котором еще существует азеотроп. На рис. 227 показаны кривые давления насыщенных паров чистых исходных компонентов и азеотропа, нанесенные на диаграмму Кокса в координатах 1g р — 1/(Т + 230). Вследствие прямолинейности зависимости такого рода для ее построения нужны только две точки. При давлениях вне интервала, ограниченного ординатами точек пересечения Р и Р' прямой для азеотропа с прямыми для чистых веществ, азеотроп уже не существует. Если указанные прямые не пересекаются, то это означает, что азеотроп сохраняется при всех давлениях. Согласно методике Иоффе [48 ] достаточно знать состав азеотропа при какой-либо одной температуре (давлении), чтобы вычислить состав азеотропа при других температурах (давлениях) методами экстраполяции или интерполяции. Малесинский [49] предлагает зависимости, по которым можно рассчитывать температуры кипения тройных азеотропов.
Если нельзя достигнуть эффективного разделения исходной смеси за счет изменения рабочего давления перегонки, то подбирают подходящее вещество, которое образует с одним из компонентов смеси гетеро- или гомоазеотроп, легко разделяемый на составные части [34].
Рассмотрим следующий пример. Смесь вода—пиридин образует при 92 °C положительный азеотроп, содержащий 54% (масс.) пиридина. Воду можно удалить в виде азеотропа, подобрав подходящий разделяющий агент (выноситель). К такому разделяющему агенту предъявляются следующие требования [34, 50]. Он должен образовывать с водой положительный азеотроп с температурой кипения ниже 92 °C и не должен образовывать азеотропа с пиридином, а также тройного азеотропа. При этом он должен связывать в виде азеотропа возможно большее количество воды и не должен практически растворяться в воде (чтобы обеспечить разделение гетероазеотропа при охлаждении).
Замечено, что положительный азеотроп образуется, когда силы притяжения между неодинаковыми молекулами меньше, чем между одинаковыми. В противном случае образуется отрицательный азеотроп. Обработав имеющиеся данные по азеотропам, собранные Легатом [20] и Хорсли [51], Эвелл, Харрисон и Берг [52 ] разработали теорию, согласно которой под силами притяжения, обуславливающими образование азеотропа, следует иметь в виду водородные связи, которые значительно превосходят
307
Таблица 41.
Классификация жидкостей по водородным связям
А. Классы по Бергу [34, 52]
Классы Тип молекул Примеры
I Молекулы, способные образовывать трехмерную структуру Н2О, многоатомные спирты, аминоспирты, оксикислоты, полифенолы, гидроксиламин
II Молекулы, содержащие как активные атомы Н, так и электроотрицательные атомы со свободными парами электронов (О, N, F) Спирты, кислоты, фенолы, первичные и вторичные амины, оксимы, нитросоединения и нитрилы с атомами Н ва-положении, аммиак, гидразин, фтористый и цианистый водо-
III Молекулы с электроотрица- род Простые и сложные эфиры, кетоны,
тельными атомами, но без активных атомов Н альдегиды, третичные амины, пиридин, нитросоединения и нитрилы, не имеющие атомов Н ва-положении
IV Молекулы с активными атомами Н, но без электроотрицательных атомов СНС13, СН2С12, СН3СНС12, СН2С1СН2С1, СН2С1СНС12 и т. д.
V Молекулы, не способные образовывать водородные связи Углеводороды, CS2> СС14, сульфиды, меркаптаны, металлоиды
Б. Смеси жидкостей с ожидаемыми отклонениями от закона Рауля
Классы Характер отклонения Водородные связи
1+ V | и+V/ Всегда положительное; смеси I + + V часто характеризуются ограниченной взаимной растворимостью компонентов Ослабляются
III + IV 1+ IV 11+ IV Всегда отрицательное Всегда положительное; смеси I + IV характеризуются ограниченной взаимной растворимостью компонентов Образуются Частично ослабляются, частично образуются вновь; первый эффект преобладает
1+ I I + II I + III 11+ II II + III Обычно положительное, но возможно и отрицательное; иногда образуется отрицательный азеотроп Частично ослабляются, частично образуются вновь
III + III III + V IV + IV IV + V v+ V Квазиидеальные системы; отсутствие отклонения или слабое положительное отклонение; изредка образуется положительный азеотроп Отсутствуют
308
другие межмолекулярные силы, например дипольные или индукционные. На основе этого представления все жидкости можно разделить на пять классов, различающихся по числу и прочности водородных связей (табл. 41).
Если оба компонента данной разделяемой смеси отнести по признаку водородной связи к соответствующим классам, то по данным табл. 41Б можно оценить предполагаемые отклонения каждого компонента от представителей другого класса и таким образом подобрать подходящий разделяющий агент. В табл. 42 указаны компоненты, которые образуют отрицательный азеотроп (см. [29]).
В табл. 43 указаны возможные разделяющие агенты для смеси вода—пиридин. Вода (класс I) проявляет сильные отклонения от закона Рауля в смесях с жидкостями классов III, IV и V, в то время как для пиридина (класс III) в смесях с теми же веществами характерны слабые положительные отклонения или вообще их отсутствие.
Из данных табл. 43 видно, что точка кипения вновь образовавшегося азеотропа тем ниже, чем меньше температура кипения чистого разделяющего агента. С учетом растворимости и содержания воды в азеотропном конденсате подбирают оптимальный разделяющий агент, который образует азеотроп с минимальной температурой кипения, которая не должна превышать 92 °C.
Берг и сотр. [34, 52] изучили ряд смесей близкокипящих углеводородов и подобрали для их азеотропной перегонки опти-Таблица 42.
Вещества, образующие отрицательные азеотропы
Компоненты
Примеры
Вода I
Сильные кислоты J
Вода |
Ассоциированные жидкости J
Жидкости класса III )
Неассоциированные жидкости J
Органические кислоты 1
Амины /
Фенолы 1
Амины J
Органические кислоты
Кислородсодержащие соединения
класса III
Фенолы
Кислородсодержащие соединения
класса III
Фенолы
Спирты
Вода с HCI, НВг, HI, HNO3
Вода с муравьиногГкислотой, с этилендиамином
Ацетон—хлороформ, циклогексанон —-бромоформ, бутилацетат — 1,2,2-три-хлорпропан
Уксусная кислота — триэтиламин, пропионовая кислота — пиридин
Фенол — анилин, о-крезол —диметил-анилин
Муравьиная кислота — диэтилкетон, масляная кислота — циклогексанон
Фенол — метилгексилкетон, о-крезол— этилоксалат
Фенол — н-октиловый спирт, о-крезол — гликоль
309
Таблица 43.
Разделяющие агенты для обезвоживания пиридина азеотропной ректификацией
Класс Разделяющий агент Темп, кип. при 760 мм рт. ст.» °C Темп, кип. азеотропа с водой, °C Количество разделяющего агента на 1 масс. ч. удаляемой воды, масс ч. Растворимость в 100 масс. ч. воды, масс. ч.
III Изобутилформиат 98,2 80,4 11,8 1,0
Этилпропионат 99,0 81,2 9,0 2,4
Метилбутират 102,0 82,7 7,7 1,5
Этилизобутират 111,7 85,2 5,6 Незначительная
Диизобутиловый эфир 122,2 88,6 3,4 То же
Амилформиат 132,0 91,6 2,5 То же
IV • Дихлорэтан 83,0 72,0 11,0 0,9
1,2-Дихлорпропан 96,8 78,0 7,4 0,3
V Бензол 80,2 69,3 10,2 0,1
Толуол 110,7 84,1 6,4 Не растворяется
Таблица 44.
Оптимальные разделяющие агенты для азеотропной перегонки углеводородов
Разделяемая смесь углеводородов Относительная летучесть без азеотропа а Разделяющий агент Изменения относительной летучести
Неогексан—циклопентан 1,006 н-Пропиламин 0,987
Метилциклогексан—изооктан 1,046 Этанол или 1,4-диоксан 1,084
З-Метилпентан—гексен-1 1,009 Метиленхлорид 1,159
З-Метилпентан-2-этил бутен-1 1,037 Этилформиат 1,156
2-Этилбутен-1-н-гексан 1,056 Хлороформ 1,094
2,2,4-Триметилпентан-2,2,4- 1,040 Изопропилацетат 1,129
тр иметил пентен-1
н-Гептан-2,4,4-триметилпентен-1 1,045 Изопропилацетат 1,129
Этилбензол-п-ксилол 1,035 2-Метилбутанол 1,079
Ксилол-.и-ксилол 1,020 2-Метилбутанол 1,029
Ксилол-о-ксилол 1,105 Метилизобутилкарбинол 1,150
мальные разделяющие агенты (табл. 44). Как видно из данных табл. 44, при введении разделяющего агента относительная летучесть компонентов а резко увеличивается.
Для экстрактивной перегонки также весьма важен вопрос о том, образует ли разделяющий агент азеотроп с компонентами или нет. При экстрактивной перегонке разделяющий агент не должен давать азеотропа ни с одним из компонентов разделяемой
310
смеси. Поэтому рассмотрим еще несколько эмпирических методов подбора разделяющих агентов, важных как для азеотропной, так и для экстрактивной перегонки.
Мэйр, Глазгов и Россини [41 ] предлагают простой графический метод определения параметров азеотропа. Рис. 228 представляет температуры кипения азеотропных смесей бензола с различными углеводородами как функцию состава смеси [53]. Если соединить точку кипения каждого углеводорода (на оси ординат) с точкой, взятой на кривой и соответствующей температуре кипе
ния азеотропа, то можно получить прямые линии с различным наклоном. На основе диаграммы можно предсказать, что углеводороды с точками кипения ниже 68 °C и выше 100 °C не дают азеотропа с бензолом. Далее, для любого углеводорода ‘можно определить температуру кипения и состав его азеотропа с бензолом, если от значения температуры кипения чистого вещества провести линию, параллельную ближайшей уже построенной наклонной прямой. Наклон прямой зависит от температуры кипе-
ния чистого углеводорода и степени разветвленности его молекулы. Если, например, на диаграмме отметить точку кипения 3-этилпентана (93,5 °C), то на кривой линии найдем в хорошем
соответствии с опытными данными точку кипения азеотропа при 80 °C, а на оси абсцисс — состав азеотропа, равный 96% (мол.) бензола. Подобные диаграммы можно строить по методу Хорсли [54], а также Мейсснера и Гринфельда [55]. Графический метод, предложенный Новиковой и Наградзе [56], основан на исполь
зовании трехмерной координатной системы, изменение параметров в которой выражается пространственной кривой. На оси абсцисс
откладывают значения состава азеотропа в % (мол.), на оси орди-
нат — значение, обратное
температуре кипения, а на аппликате (третьей оси) — логарифм давления. Если известна характеристика азеотропа при двух различных давлениях, то для бинарных азеотропных смесей можно также точно определить: характеристику азеотропа при любом давлении, давление и концентрации, при котором может появиться азеотропная смесь.
Литвинов [26] разработал графический метод для вычисления
Рис. 228.
Графический метод Мэйра, Глазгова и Россини определения параметров азеотропных смесей бензола с углеводородами:
1 — 2,2,4-триметилпентаном; 2 — н-гептаном;
3 — 2,3-диметилпеитаиом; 4 — 2,2,3-триме-тилбутаном; 5 — 2,4-диметилпентаиом; 6 — циклогексаном; 7 — метилциклопентаном; 8 —
н-гексаном.
311
Рис. 229.
Графический гметод Смольника для определения параметров азеотропных смесей бензола с углеводородами:
1 — 2,2,4’Триметилпентаиом; 2 — 2,3-диметил-пентаном; 3 — 2,4-диметилпеитаиом; 4 — 2,2,3-триметилбутаиом; кривая I — соответствует формуле (186), кривая II — формуле (187).
вероятности появления азеотропа в тройных смесях. Показано, что тройной азеотроп может также обра-
зоваться в системе, в которой отсутствуют бинарные азеотропы. По ^методу Скольника [57 ] возможно также чисто расчетное определение параметров азеотропа. Для членов одного и того же гомологического ряда (например, углеводородов) и какого-либо второго компонента (например, бензола) имеют силу следующие линейные зависимости:
1g хаз = А (273,1 + /аз) + В (186)
где хаз — содержание второго компонента в азеотропной смеси, % (мол), ta3 — температура кипения азеотропа, °C, А и В — константы, и
lg(P-fa3) =£-Л273,1 +/) (187)
где D, Е и F — константы; t — температура кипения рассматриваемого члена гомологического ряда.
На основе зависимостей (186)"и?( 187) можно получить кривые, которые представлены на рис. 229.
Г Непосредственно по кривым могут быть определены параметры азеотропов для углеводородов одного гомологического ряда. Например, 2,2-диметилпентан кипит при 79,2 °C. По кривой 2 находим, что состав азеотропа составляет 50% (мол.), а по кривой 1 определяем температуру кипения' азеотропа с бензолом (75,8 °C). Кривая 2 служит также для определения интервала температур кипения, в котором азеотроп с бензолом еще возможен. Получающийся^ при этом интервал 65—98 °C хорошо согласуется с результатами определения по графическому методу Мэйра, Глазгова и Россини. Эти температурные интервалы кипения для парафинов, дающих с бензолом гомоазеотропы, называют «азеотропным эффектом» бензола [57 ]. По данным инфракрасной спектроскопии, Наканиси и сотр. [58] разработали диаграмму, с помощью которой для бинарных смесей с метанолом можно выяснить, образуется или нет азеотроп с определенным компонентом данной смеси при заданной температуре в интервале 30—150 °C.
Брандани [58а ] сообщает о применении предельных коэффициентов активности для определения состава азеотропа при постоянной температуре и частичной взаимной растворимости компонентов в бинарных жидких смесях.
312
Расчет условий азеотропной перегонки, необходимых для разделения, например, числа теоретических ступеней разделения и флегмового числа проводят в обычном порядке, как описано в гл. 4.
Для расчета числа теоретических ступеней разделения при ректификации правильных систем с азеотропным составом, в которых абсолютная теплота смешения мала, Кун [59] приводит следующее уравнение, которое соответствует формуле Фенске (108в) для непрерывной ректификации при v = оо:
ЛМИИ —
1 2хаз -1 бо 2(1 — хаз)
ХЕ \-хЕ
(188)
где 60 = а — 1; хаз — концентрация азеотропа.
Это уравнение справедливо при условии, что хЕ < хаз, т. е. когда требуемая концентрация легколетучего компонента в дистилляте меньше, чем в азеотропе. Для хЕ = хаз необходимое число теоретических тарелок составляет пмин = оо.
Шапиро [60] привел формулы для расчета процесса разделения бинарных смесей с малым содержанием менее летучего компонента, а также для расчета почти азеотропных смесей. В качестве примера проведен расчет процессов разделения смесей вода — этиловый эфир уксусной кислоты (1,75%), бензол —толуол (4%) и этанол —вода (11%). На примере разделения смеси 1,3-бута-диена — 1-бутен азеотропной ректификацией с метиламином Хунсманн [61а] указывает последовательность расчета азеотропной колонны. Сначала исследуют фазовое равновесие двух- или трехкомпонентных систем в технически важной области давлений, результат представляют рядом уравнений и проводят последовательный расчет от нижней тарелки к верхней тарелке. Влияние температуры, а также избытка амина подтверждается вычислениями. Результаты расчетов подтверждены опытами для условий ректификации на маленькой колонне; получилось весьма удовлетворительное согласие.
Принцип работы и особенности гетероазеотропной перегонки раскрыты Вольфом и сотр. [61] на примере разделения смеси вода — уксусная кислота.
В заключение рассмотрим процесс абсолютирования спирта методом ректификации с бензолом в качестве разделяющего агента, как пример азеотропной перегонки, при которой образуется тройной азеотроп. К этанолу концентрацией 95% (масс.), находящемуся в кубе добавляют рассчитанное количество бензола. При перегонке в периодическом режиме при 64,9 °C получается тройной азеотроп, который содержит 74,1% (масс.) бензола,
313
18,5% этанола и 7,4% воды. При охлаждении до 28 °C азеотроп расслаивается на две жидкие фазы. Верхний слой, составляющий 84,7% (масс.) от всего количества тройного азеотропа, содержит 85,6% (масс.) бензола, 11,6% этанола и 2,8% воды. Нижний.слой, составляющий 15,3% (масс.) от всего количества азеотропа, содержит 8,1% (масс.) бензола, 53,1% этанола и 40,6% воды.
После удаления воды при 68,3 °C отгоняют бинарный азеотроп, содержащий 32,4% этанола и 67,6% бензола. Наконец, при 78,4 °C начинает отгоняться безводный спирт концентрацией 99,8%. Конечно, можно работать непрерывно, как обычно принято в промышленности, однако дополнительное усложнение аппаратуры в лабораторных условиях не оправдано. Целесообразнее оба слоя образовавшегося тройного азеотропа, а также бинарный азеотроп разделять перегонкой в периодическом режиме.
6.2.2. ЭКСТРАКТИВНАЯ ПЕРЕГОНКА
Экстрактивную перегонку, также как и азеотропную, применяют, если смесь необходимо обогатить до концентрации выше азеотропной или разделить близкокипящие компоненты и неидеальные смеси с относительной летучестью компонентов, близкой к 1. Однако, экстрактивная перегонка благодаря почти неограниченному числу возможных разделяющих агентов имеет намного большее распространение, чем азеотропная.
Количество добавляемого разделяющего агента следует устанавливать по возможности точнее, так как оно оказывает заметное влияние на эффективность разделения.
В качестве примера экстрактивной ректификации смеси близкокипящих компонентов уже было рассмотрено разделение смеси w-гептан-метилциклогексан с применением анилина. Система метилциклогексан—толуол имеет кривую равновесия, которая приближается к диагонали диаграммы равновесия пар—жидкость асимптотически. Чтобы получить чистый метилциклогексан, необходимо практически бесконечно большое число теоретических тарелок. Благодаря добавке 55% (мол.) такого полярного растворителя как анилин разделение сильно упрощается.
Экстрактивная перегонка удобна также и для разделения азеотропных смесей. На азеотропную смесь циклогексан—бензол можно воздействовать тем же разделяющим агентом (анилином) таким образом, что в дистилляте получится чистый циклогексан. Кортюм и Биттель [61 ] сообщили о разделении первичных, вторичных и третичных ароматических аминов с помощью таких разделяющих агентов^ как глицерин и парафиновое масло. Достойна внимания полностью автоматизированная установка этих исследователей для периодической и непрерывной экстрактивной ректификации. Экстрактивная перегонка оправдала себя и при разделении многокомпонентных смесей, содержащих вещества различных классов. Так, экстрактивной перегонкой можно извлечь
314
ароматические вещества из их смесей с неароматическими углеводородами с помощью одного из бутандиолов (например, 2,3-бу-тандиола) с добавкой октилового спирта в качестве растворителя [62].
Представляет также интерес выделение НО из азеотропа соляной кислоты экстрактивной перегонкой в присутствии H2SO4 по методу Гревера [63]. В этом случае для ректификации применяли колонну, изготовленную из графита. На рис. 230 приведена диаграмма равновесного кипения системы НО—H2SO4—Н2О при постоянном давлении.
В качестве разделяющего агента применяют также твердое вещество. Ино и сотр. [94] успешно использовали для удаления из ацетона малых количеств хлористого метилена соль ZnBr2, растворимость которой в ацетоне превышает 50% (масс.). Содержание хлористого метилена в ацетоне удалось снизить до 0,09%. Соль растворяли в ацетоне, применяя куб с мешалкой.
Принцип экстрактивной перегонки состоит в том, что благодаря разделяющему агенту увеличивается разность концентраций Ах между парами и жидкостью при равновесном испарении. Путем добавления третьего вещества можно даже преобразовать положительный азеотроп в отрицательный [63]. Обычно высококипящий разделяющий агент подбирают таким образом, чтобы относительная летучесть компонентов заметно повышалась. Агет вводят непрерывно в верхнюю часть колонны, чтобы его концентрация оставалась постоянной в жидкой фазе по всей высоте колонны.
Рис. 231 иллюстрирует, как изменяется форма кривой равно веси я азеотропной смеси ацетон—хлорофор в зависимости от ко личества добавляемого разделяющего агента (метилизобутилен кетона). Система имеет максимум температуры кипения при 34,5% (мол.) ацетона; этот максимум уже при добавке 30 % (мол.) разде ляющего агента исчезает; дальнейшее добавление агента вызывает
значительное увеличение от носительной летучести ком понентов смеси [50].
Павлов с сотр. [63а] ис следовал экстрактивную пе регонку углеводородов с 4 i 5 атомами углерода с по мощью 19 различных вспо
Рис. 230.
Диаграмма равновесного кипенн; смеси НС1 —H3SO4—Н2О прн 750 mi рт. ст.
— мольная доля H2SO< в жидко фазе. —— линии постоям
ных составов пара; — — —- — изс термы.
31
Рис. 231.
Зависимость формы кривой равновесия для смеси ацетон—хлороформ от количества введенного метнлнзооутил• кетона.
могательных веществ; найденные значения относитель-ной летучести были представлены в виде диаграмм, отражающих влияние добавки высококипящих углеводородов на относительную летучесть.
Разделяющий агент, применяемый для проведения экстрактивной перегонки,
должен отвечать следующим требованиям. Он не должен образовывать азеотропа с компонентами разделяемой смеси, должен легко отделяться от компонентов разделяемой смеси, давать с одним из разделяемых компонентов возможно большее положительное отклонение от закона Рауля, а с другим компонентом — возможно меньшее или даже противоположное (отрицательное) отклонение, причем разделяющий агент должен обеспечивать максимальное увеличение относительной летучести исходных компонентов [64].
При рассмотрении азеотропной перегонки мы уже познакомились с приемами, позволяющими установить для какой-либо смеси вероятность образования азеотропа (см. разд. 6.2.1). Дополнительные возможности в этом направлении представляет методика Шайбеля [65], основанная на использовании коэффициентов активности компонентов разделяемой смеси. Выделение экстрагированного компонента относительно высококипящего разделяющего агента перегонкой не представляет трудностей. Согласно Бергу [34, 52] в соответствии с данными табл. 41 (см. разд. 6.2.1) наиболее подходящими разделяющими агентами для экстрактивной ректификации являются вещества, принадлежащие классам I и II. Это, главным образом, вещества, которые имеют склонность к образованию прочных водородных связей и могут взаимодействовать и как доноры протонсв, и как доноры электронов. Сюда относят фенолы, ароматическе амины (анилин и его производные), высшие спирты, гликоль и т. д.
При исследовании процессов экстрактивной перегонки смесей хлористый аллил хлористый пропил и «-бутан-трц«с-бутен-2 Гарбер и Мироненко [65а ] разработали универсальный метод подбора наиболее подходящего разделяющего агента. Хакенберг и Шмидт предложили метод парсфазной газовой хроматографии для оценки избирательной способности разделяющих агентов. Согласно этому 316
методу определяют отношение площадей хроматографических пиков веществ 1 и 2, распределенных в паровой фазе над некипящими смесями в присутствии и в отсутствие разделяющего агента.
Оценка избирательной способности разделяющего агента требует проведения многочисленных опытов. Целесообразно определять кривые равновесия для образцов разделяемой смеси, в которые добавлены различные количества разделяющего агента. Роуз 135 ] описывает результаты таких опытов для смеси «-гептан—метилциклогексан, в которую добавляли различные разделяющие агенты. Относительную летучесть компонентов в присутствии растворителя обозначают аэ. Чтобы можно было сравнить действие различных разделяющих агентов, относительную летучесть компонентов, равную 1,07 в отсутствие агента, условно приравняем к 1. Тогда полезный эффект, вызванный введением разделяющего агента, будет равен N = аэ/а. Очевидно, введение 92% (мол.) анилина в жидкую фазу оказывает наибольшее влияние на фазовое равновесие. Влияние разделяющего агента на относительную летучесть разделяемых компонентов можно также оценить, исходя из интегральных теплот смешения [50].
Коган [66] показал, что при добавлении третьего компонента к бинарной смеси увеличивается относительная летучесть того компонента смеси, в котором менее всего растворяется третий компонент. Другие приемы подбора разделяющего агента рассмотрены Кафаровым и Гордиевским [67], а также Коганом [68]. Рёк [69 ], а также Портер и Джонсон [70 ] показали, что с помощью распределительной газовой хроматографии при простом аппаратурном оформлении можно подобрать разделяющие агенты, подходящие для экстрактивной перегонки.
Определение разности между температурами кипения, специально приготовленных эталонных смесей позволяет быстро оценить эффективность разделяющего агента. Для этого смешивают в равных объемах каждый из разделяемых компонентов с предполагаемым разделяющим агентом и опытным путем определяют температуры кипения обеих полученных смесей. Затем эти температуры вычисляют, исходя из мольных концентраций компонентов, при этом предполагают линейную зависимость температуры от состава. Наконец, вычисляют разность температур кипения полученных смесей, при этом используют как экспериментальные, так и расчетные значения температур кипения. Если найденная из опыта разность температур кипения смесей значительно превышает расчетную, то разделяющий агент считают пригодным [35].
Другим решающим фактором, влияющим на выбор разделяющего агента, действующего избирательно на легкокипящий компонент, является относительная полярность компонентов смеси. Жидкости с одинаковой полярностью образуют идеальные растворы; при смешении же компонентов с различной полярностью следует считаться с отклонениями от идеального поведе-
317
ния в соответствии с разностью полярностей компонентов. Например, введением неполярного высококипящего масла в сильно полярную смесь уксусная кислота—вода можно увеличить летучесть воды (по отношению к кислоте). Добавление к неполярной смеси циклогексан—бензол такого полярного разделяющего агента как анилин повышает летучесть циклогексана по отношению к бензолу. Методы подбора разделяющих агентов по относительной полярности компонентов смеси были развиты Бергом [52], а также Гильденбрандом [71].
Экстрактивную перегонку используют не только для разделения бинарных смесей, ее применяют также для выделения отдельных компонентов из многокомпонентных смесей, например бензола из нефтяных фракций [72]. Смеси насыщенных и ненасыщенных углеводородов с почти одинаковыми температурами кипения разделяются экстрактивной ректификацией в присутствии эфиров кетокислоты [73]. В последнее время большое значение приобретает разделение низших углеводородов Ci—С2 [74]. Кар-нер с сотр. [75] исследовал эффективность разделения смеси метилциклогексан—толуол в насадочных колоннах при экстрактивной ректификации с добавлением фурфурола; на основании полученных данных были выведены уравнения для расчета процесса ректификации.
При экстрактивной перегонке, как и при азеотропной, после подбора оптимального разделяющего агента необходимо построить по возможности более точные кривые паро-жидкостного равновесия для тройных или многокомпонентных смесей. Нулл и Пальмер [76 ] дают рекомендации, как можно получить необходимые данные по равновесию при минимуме экспериментального материала.
Необходимое число теоретических ступеней разделения определяют расчетным путем по формуле Фенске, подставляя вместо аэ значение относительной летучести, полученное в присутствии разделяющего агента (см. разд. 4.7.5.3). Точно также можно выполнить расчет графически по методу Мак-Кэба, учитывая количество исходной смеси и применяя кривую равновесия, которая соответствует возросшей величине аэ (см. разд. 4.7). Как показал Гельбин [7], число теоретических ступеней разделения при экстрактивной ректификации можно рассчитать с помощью метода полюсных точек Бошняковича.
Нагель и Зинн [78] предлагают модифицированный метод Мак-Кэба для расчета экстрактивной ректификации. Кортюм и Фальтуш [79 ] обсуждают проблемы, возникающие при реализации подобных селективных методов разделения. К ним относятся конструирование автоматизированной аппаратуры для непрерывной экстрактивной перегонки (из стали V4A) с подбором избирательно действующего агента, а также расчет минимального флегмового числа и необходимого количества разделяющего агента.
Требуемое количество разделяющего агента рассчитывают с учетом количества флегмы. Принимая 83% (мол.) за содержание 318
разделяющего агента (анилина) в его смеси с низкокипящим компонентом А («-гептаном), получим, что объем анилина относится к объему «-гептана как 3:1. Если согласно вычислениям оптимальными условиями ректификации являются нагрузка (расход паров) 1100 мл/ч «-гептана и флегмовое число 10, то при таком режиме разделения в головную часть колонны следует непрерывно подавать 3000 мл/ч анилина. При этом отбирают 100 мл/ч чистого «-гептана в качестве дистиллята, а 1000 мл/ч «-гептана и 3000 мл/ч анилина возвращают в колонну на орошение. Разделяющий агент (анилин) можно накапливать в кубе, пока низкокипящий компонент А не перейдет полностью в дистиллят. Только затем периодической ректификацией отделяют высококипящий компонент В, после чего в кубовом продукте остается разделяющий агент, который имеет более высокую температуру кипения, чем компоненты исходной смеси. Можно также одновременно получать второй компонент (В) в качестве головного продукта (дистиллята) на параллельно включенной колонне. При этом кубовый остаток параллельной колонны (разделяющий агент) непрерывно выводят и снова направляют в верхнюю часть первой колонны (см. рис. 224).
Таблица 45.
Примеры разделения идеальных, неидеальных и азеотропных смесей экстрактивной ректификацией
Разделяющий агент н его полярность, выраженная дипольным моментом (при 20 °C) Разделяемая смесь Разность температур кипения компонентов при 760 мм рт. ст., °C Примечания
Анилин, 1,51 н-Гептан—метилциклогексан Бензол—ци клогексан н-Гептан—толуол 2,7 0,7 12,8 Идеальная смесь (а = = 1,07) Образует азеотроп Неидеальная смесь
Фенол, 1,70 н-Гептан—толуол Изооктан—толуол Метилциклогексан— толуол 12,8 11,4 9,5 с асимптотическим приближением кривой равновесия к диагонали диаграммы
Монобутиловый эфир этиленгликоля Мети лэти л кетон — вода 20,4 Образует азеотроп
Диэтиловый эфир Этанол—вода 21,6 То же
Высшие кетоны или высшие спирты (для октанола 1,68) Ацетон—метанол 8,5 Образует азеотроп
Высшие сложные эфиры или высшие спирты Этил ацетат—этанол 1,3 То же
Высшие кетоны или внешне хлорпроиз-водные Ацетон—хлороформ 5,0
319
Рис. 232.
Головка с электромагнитным флегмоделителем и декантатором для колонн азеотропной ректификации с отбором легкой (а) и тяжелой (б) фаз.
Изготовитель: фирмы Отто Фриц, фирма Нормаг, Хофхайм (Таунус).
В табл. 45 дано несколько примеров [64 ] успешного применения экстрактивной ректификации, которые иллюстрируют приведенные выше теоретические положения.
Как для азеотропной, так и для экстрактивной перегонки в периодическом и непрерывном режимах работы можно применять традиционные лабораторные аппараты. Пригодны как насадочные, так и тарельчатые колонны. Лишь в особых случаях требуются специальные дополнительные устройства (см. рис. 223 и 224).
Рассмотрим сначала некоторые вопросы аппаратурного оформления процесса азеотропной перегонки. Если азеотроп гомогенный, то в ректификационную установку не требуется вносить каких-либо принципиальных изменений. Если азеотроп гетерогенный, то на орошение в колонну возвращают не азеотроп, а только разделяющий агент. Для этого применяют специальную «азеотропную» головку (рис. 232), которая позволяет выборочно применять в качестве орошения либо легкую, либо тяжелую фазу. Орошающую жидкость (флегму) целесообразнее подавать не в самую верхнюю часть колонны, а несколько пониже — во вторую сверху царгу. При работе в непрерывном режиме с образованием гомогенного азеотропа разделяющий агент примешивают к исходной смеси. При перегонке с образованием гетероазеотропа определенное количество разделяющего агента непрерывно возвращают в цикл с помощью азеотропной головки (см. рис. 223 и 232). Другие азеотропные головки описаны в разд. 7.5.3.
320
При экстрактивной ректификации принципиально необходима экстракционная зона, куда непрерывно подают разделяющий агент. Чтобы получить в головке колонны дистиллят, не содержащий следов разделяющего агента, последний вводят на одну царгу ниже верха колонны. При непрерывной ректификации исходную смесь подают в верхнюю треть колонны (см. рис. 224). Можно усовершенствовать метод, если кубовый продукт непрерывно выводить, а затем разделять в параллельно включенной колонне на высококипящий компонент и разделяющий агент, который затем возвращают в цикл.
Следует отметить, что в принципе не исключено применение разделяющего агента, который кипит при более низкой температуре, чем разделяемая смесь. Однако в этом случае, например, при экстрактивной ректификации смеси этанол—вода в присутствии диэтилового эфира разделяющий агент вводят в колонну несколько выше куба.
6.3. СОЛЕВАЯ ПЕРЕГОНКА
Описанные выше селективные методы перегонки основаны на смещении фазового равновесия при введении в смесь разделяющего агента в виде пара (перегонка с водяным паром) или в виде жидкости (азеотропная и экстрактивная перегонка). Смещение фазового равновесия можно вызвать также путем растворения в разделяемой смеси соли, что приводит к изменению соотношения парциальных давлений исходных компонентов.
Метод солевой перегонки до сих пор применяли для разделения смесей, один из компонентов которых вода. Кривая равновесия смеси вода—уксусная кислота (для атмосферного давления) при концентрации уксусной кислоты выше 96% (масс.) подходит очень близко к диагонали диаграммы, следовательно, полное удаление воды методом обычной перегонки затруднено. Кроме того, с практической точки зрения недостатком этого способа является (особенно при высоком содержании воды в исходной смеси) то, что энтальпия испарения для воды почти в пять раз больше, чем для уксусной кислоты. Поэтому было бы выгоднее при непрерывной ректификации в дистилляте получать уксусную кислоту, а в кубе — воду. Этого можно достичь добавкой к смеси более 8% (масс.) хлорида кальция [80]. Например, при 760 мм рт. ст. относительная летучесть компонентов смеси вода—уксусная кислота с содержанием 30% (масс.) уксусной кислоты в результате добавления 20% (масс.) хлорида кальция изменяется от 1,36 до 0,525. Таким образом, относительная летучесть воды и уксусной кислоты становится меньше 1, а это означает, что в головке колонны конденсируется уксусная кислота, а в кубе накапливается вода. Разумеется, при этом предполагается, что разделяющий агент (хлорид кальция) равномерно распределен по всей высоте колонны. В соответствии с систематическими измерениями Даубаха [81 ],
11 Э. Крель 321
Рис. 233.
Кривые равновесия смеси этанол—вода без добавки (/) и’с добавкой 10 г СаС12 на 100 мл смеси (2).
Рис, 234.
Данные для разделения смеси метанол—ацетон:
а — кривая равновесия без добавки (/) и с добавкой 2,3 М раствора CaClg (2); б — схема установки.
На осях диаграммы указаны значения концентраций ацетона в жидкости и в парах.
добавление 10 г хлорида кальция к 100 мл азеотропной смеси этанол—вода вызывает исчезновение азеотропной точки (рис. 233). Таким образом, благодаря введению солевого раствора в непрерывнодействующую колонну получают дистиллят высокой степени чистоты [82]. Аналогичные исследования с NaNO3 и K2SO4 провели Гурси и Томпсон [83]. Новелла и Тарассо [84] использовали соли, растворимые в обоих компонентах разделяемой смеси, например СиС12 и КС2Н3О2. Они установили, что благодаря этому при ректификации исключается образование азеотропа. О влиянии солей металлов на равновесие в системе жидкость— пар сообщает Шир [85].
Азеотропную смесь метанол—ацетон можно обогатить до концентрации, значительно превышающей азеотропную, если к 1 объему смеси добавить 3,5 объема водного раствора хлорида кальция плотностью 1,2 г/см3 (при 20 °C это соответствует 2,3/И раствору) [86]. На рис. 234 приведены кривые равновесия смеси метанол—ацетон с добавкой соли и без нее, дана также упрощенная схема установки.
При перегонке смеси вода—фенол путем добавления 17% NaCl достигают смещения азеотропного состава с 91% (масс.) до 84% воды; поэтому можно использовать область ограниченной растворимости системы [87]. Насыщая нитратом калия смесь этанол—вода в интервале концентраций этанола от 15 до 70%, также достигают большего обогащения, чем без добавления соли [88]. Гайер с сотр. [89] исследовал влияние солевых добавок на парожидкостное равновесие смеси муравьиная кислота—вода. При содержании в системе 35,5% соли азеотроп больше не обра-322
зуется, благодаря чему можно получить высокую концентрацию муравьиной кислоты. Систематические исследования солевого эффекта в системе этанол—вода провел также Миро с сотр. [90].
В своем обзоре Фуртер [91 ] анализирует степень влияния солевого эффекта на перегонку с точки зрения возможности использования подобных методов в промышленности, особенно при экстрактивной ректификации. Ряд работ Шуберта с сотр. [92] посвящен исследованию влияния различных хлоридов металлов и других простых солей на изотермическое фазовое равновесие систем пропанол—вода, н-бутанол—вода, а также метанол—вода.
Уравнение Жака и Фуртера [95 ] для оценки влияния соли на паро-жидкостное равновесие бинарных смесей описывает взаимосвязь температуры и концентрации жидкости для трехкомпонентной смеси этанол—вода—соль. Это уравнение имеет шесть констант. Фуртер и Меранда [96] обсуждают теорию солевого эффекта. Допустив упрощения, Сада с сотр. [97] вывел соотношение для расчета паро-жидкостного равновесия для неводных бинарных систем, в которых соль растворяется только в одном компоненте (например, бензол—этанол с добавлением хлорида лития или кальция).
Уже эти немногие примеры показывают, что для решения трудных проблем разделения все возрастающее значение приобретают селективные методы перегонки; систематические исследования позволяют открыть новые области их применения. Сочетание процессов перегонки с адсорбционными эффектами привело к разработке «адсорбционной перегонки». Фукс и Рот [93] исследовали влияние материала насадки на процесс разделения смеси вода— уксусная кислота.
11
Материалы и аппаратура
Специальные устройства, применяемые в технике лабораторной перегонки, были рассмотрены в гл. 1—6. В настоящей главе описываются новейшие детали, узлы и приборы, предназначенные для различных процессов перегонки. Путем сравнительного анализа большого числа принципиально тождественных элементов ректификационных установок автором отобраны те элементы, которые можно стандартизировать. Преимущества стандартизации проявляются как при изготовлении, так и при использовании ректификационной аппаратуры. Очевидно, что условием стандартизации отдельных элементов ректификационных установок является возможность использования .их в любых процессах перегонки, применяемых в промышленности при оптимальных условиях.
7.1. МАТЕРИАЛЫ ДЛЯ ИЗГОТОВЛЕНИЯ РЕКТИФИКАЦИОННОЙ АППАРАТУРЫ
В качестве материалов для изготовления различных аппаратов ректификационных установок применяют в основном стекло, а также металлы, фарфор и кварц. Металлы используют для изготовления установок высокотемпературной перегонки или ректификационных установок, работающих под давлением, когда прочность стекла становится недостаточной. Наиболее широкое применение находят аппараты из нержавеющей стали V2A *, которые могут быть использованы для высокотемпературной перегонки и перегонки под давлением, а также для ректификации агрессивных веществ. Фарфор применяют в тех случаях, когда металлы и стекло не могут использоваться вследствие их коррозионной неустойчивости. Из кварца изготовляют в основном аппараты для перегонки воды и высокотемпературной ректификации. Отдельные детали и узлы выполняют также из пластмасс, например тефлона.
Стекло, применявшееся уже около 3000 лет назад в древнем Египте, для изготовления косметических сосудов, амулетов и позднее приборов для алхимии, не потеряло своего основного значения в лаборатории и промышленности до настоящего времени. Широкое использование стекла обусловлено его ценными свой
* По составу близка к стали марки 12Х18Н10Т, выпускаемой в СССР. — Прим. ред.
324
ствами: этот материал успешно применяется как для изготовление лабораторной аппаратуры, предназначенной для проведения химических и физико-химических процессов, так и для производстве промышленных колонных аппаратов.
Прозрачность стекла позволяет наблюдать за ходом процесса. В адиабатических процессах, протекающих при температурах примерно до 120 °C, кожух из стекла, вакуумированный дс остаточного давления 10-в мм рт. ст., обеспечивает достаточнук термоизоляцию аппарата. При более высоких температурах, а также при использовании крупногабаритных аппаратов в качестве термоизоляционного материала применяют стекловолокно: в слое изоляции оставляют смотровую щель, предназначенную для визуального наблюдения за ходом процесса (см. разд. 7.7). Важным преимуществом стекла является его высокая коррозионная стойкость. Поэтому многие химические реакции и процессы разделения проводят в аппаратах и установках, изготовленных из стекле или других керамических материалов. Широкому примененик стекла в химической промышленности способствует высокая твердость и незначительная шероховатость поверхности стеклянных изделий. Стенки стеклянных аппаратов во время работы незначительно загрязняются и легко поддаются очистке. Ценным свойством стекла является также сравнительно небольшой коэффициент линейного расширения. Использование стеклянных аппаратов при переработке фармацевтических продуктов и однократной или двойной перегонке воды дает возможность получать продукть: без запаха, вкуса и, главное, без примесей металлов.
Основным недостатком стекла, по сравнению с металлами, является его хрупкость, а также недостаточная устойчивость к резким изменениям температуры. Прочность стекла можно повысить, если его расплав охлаждать по специальному режиму, способствующему возникновению в глубине материала определенных внутренних напряжений. Перспективным направлением развития стекольной промышленности является разработка и получение различных марок стекла, свойства которых были бы наиболее оптимальными для изготовления аппаратуры применительно к конкретным физико-химическим процессам. Стекло с универсальными свойствами, приближающееся к металлическим сплавам по возможностям его применения и обработки, по-видимому, никогда не будет получено. Однако при правильном выборе марки стекла можно в значительной мере удовлетворить необходимые требования [1, 5].
В настоящей работе автор не счел целесообразным привести физико-химические свойства стекла различных марок, так каг испытания последних проводили, как правило, в несопоставимых условиях. Все необходимые характеристики стекол можно найти в каталогах промышленности стекольных изделий [1].
Подробные данные о свойствах стекол приведены Мёнхом [2 и Айтелем с сотр. [3]. В работе центральной лаборатории сте
32J
кольной промышленности ГДР в Ильменау [4] представлена сравнительная характеристика физико-химических свойств стекла, фарфора, металлов и пластмасс. Кроме того, в данной работе подробно описаны возможные области применения стекла, а также характеристики стекловолокна различных видов и изделий из них.
Сведения об использовании полупромышленных стеклянных аппаратов в лабораторных и пилотных ректификационных установках приведены Ригером [5]. Применение аппаратов из специальных марок стекла в ректификационных установках, работающих под избыточным давлением до 4,3 кгс/см2, описано в работе [6]. Камель [7] рассмотрел вопросы, связанные с использованием фарфора в лаборатории.
7.2. СТАНДАРТНЫЕ И СБОРНЫЕ ПРИБОРЫ
Стандартизованные методы перегонки описаны в разд. 3.1. На рис. 235 показан дистилляционный прибор Энглера со стандартными шлифовыми соединениями. В этом приборе глубина погружения термометра и расположение капельницы в трубке для отвода дистиллята точно фиксированы. Автоматические дистилляционные установки, в состав которых входит прибор Энглера, подробно описаны в разд. 8.1.
Прибор Энглера целесообразно применять для скоростной
перегонки в промышленных условиях, однако он не пригоден для проведения более точных работ. Этот прибор не пригоден также и для вакуумной дистилляции, при перегонке на нем высококипящих веществ возможно частичное разложение исходной смеси,
искажающее полученные результаты.
Для определения температурных пределов выкипания при остаточных давлениях около 10 мм рт. ст. применяют дистилляционный прибор с колбой Кляйзена (рис. 236). Следует отметить,
что измерение давления за приемником дистиллята может привести к заметным погрешностям. Для получения точных результатов необходимо измерять давление и температуру по возможности в одном месте. Место установки вакуумметра при работе в интервале остаточных давлений ниже 10 мм рт. ст. показано на рис. 236 штриховой линией.
Недостаток прибора Энглера и прибора с колбой Кляйзена
в том, что они не позволяют точно определить температуру начала
перегонки. При перегонке с использованием прибора Энглера в соответствии с указанием стандарта фиксируют температуру
Рис. 235.
Дистилляционный прибор Энглера со стандартными шлифовыми соединениями.
326
Рис. 236.
Дистилляционный прибор с колбой Кляйзеиа для перегонки под вакуумом.
Рис. 237.
Дистилляционный прибор с насадочной колонной, собранной из узлов и де* талей, выполненных по нормалям «Дестинорм» .
начала перегонки в момент отбора первой капли дистиллята. В приборе с колбой Кляйзена температура начала перегонки также завышается, поскольку в первую порцию дистиллята попадает некоторая часть высококипящей фракции. Искажения температуры начала перегонки особенно сильно проявляются при загрузке небольших количеств исходной смеси (до 100 г), т. е. именно тех количеств, которые обычно используют при аналитическом контроле в производственных условиях. В последнее время широко применяются дистилляционные приборы для пробной дистилляции, снабженные небольшой насадочной ректификационной колонной (рис. 237), в куб которой можно загружать от 200 до 300 мл исходной смеси. В таких приборах можно точно определять температуру начала отгонки при работе с бесконечным флегмовым числом. Воспроизводимость результатов при вакуумной перегонке можно обеспечить, если снабдить данный прибор точными регуляторами давления, нагрузки колонны по пару и флегмового числа. Для полной отгонки исходной смеси к ней добавляют вещество-вытеснитель, температура кипения которого примерно на 50° С выше конечной температуры дистилляции. После окончания дистилляции целевого продукта в кубе колонны остается только вещество-вытеснитель [8].
В лабораторной практике обычную дистилляцию применяют в основном для регенерации растворителей и других жидкостей, а также для предварительного отделения большей части основного
327
Рис. 238.
Детали и аппараты дистилляционных установок:
1 — капилляр, применяемый для устранения пульсаций давления при кипении; 2 — приставка; 3,9 — воздушные холодильники; 4 — перемычка;
5, 11 — холодильники Либиха и Димрота соответственно; 6, 12 — форштосы; 7 — холодильник с винтовой охлаждаемой поверхностью; 8 — шариковый холодильник; 10 — змеевиковый холодильник; 13, 14, 16 — сборники дистиллята, разработанные Аншюцем—Тнле, Бредтом и Брюлем соответственно; 15 — градуированная цилиндрическая бюретка.
вещества от исходной смеси. Для проведения дистилляции используют колбу с приставкой, снабженной термометром на шлифе, устройства для конденсации пара и сбора дистиллята, а также приспособление для присоединения системы к вакуумной линии. Во многих случаях для предотвращения уноса брызг полезно
328
снабдить колбу короткой насадочной колонной высотой 5—10 см. На рис. 238 показаны стандартные детали и аппараты дистилляционных установок [9].
Приставку 2 к кубу в виде колбы применяют для дистилляции низко- и высококипящих веществ, а перемычку 4 используют в основном при работе с высококипящими и легкозатвердевающими веществами. При дистилляции высококипящих веществ обычно достаточно воздушного охлаждения паров дистиллята, поэтому в этих случаях пригодны детали 3 и 9. Если полученный дистиллят затвердевает, то его можно легко расплавить с помощью горелки Бунзена или электроспирали. Однако в целях безопасности лучше применять холодильники 7 и 8, которые охлаждаются жидкостью из термостата и легко очищаются после окончания процесса дистилляции. Особенно удобен винтовой холодильник 7, в котором деталь с винтовой поверхностью при необходимости очистки может быть вынута через шлиф.
При дистилляции низкокипящих веществ, требующих интенсивного охлаждения, часто бывает недостаточно холодильника Либиха 5, так же, как и трубки с узкой охлаждаемой рубашкой Веста. Более эффективное охлаждение обеспечивают змеевиковый холодильник 10 и холодильник Димрота 11, позволяющие, кроме того, собирать установки, более компактные по высоте.
Для вакуумной дистилляции наиболее подходящим является сборник дистиллята Аншюца—Тиле 13 (TGL 13839), предназначенный для отбора любого количества фракций без нарушения технологического процесса. Это устройство хорошо работает и при атмосферном давлении в тех случаях, когда процесс пере-
Рис. 239.
Приставки к кубам в виде колб, используемые при дистилляции вспенивающихся жидкостей:
а — приставка Райтмайра; б — приставка Фридрихса.
Рис. 240.
УстройствоХФридрихса для дистилляции легковоспламеняющихся жидкостей.
325
Рис. 241.
Детали для сборки соединительных коммуникаций:
/ — труба; 2, 3 — колена с углом изгиба 135 и 90° соответственно; 4, 5 — Т- и Y-образные тройники; 6,7 — краны.
Рис. 242.
Подвижная соединительная коммуникация из колеи со шлифами, предложенная Фридрнхсом.
гонки сопровождается выделением газов. В качестве сборников дистиллята удобно использовать градуированные цилиндрические бюретки 15, с помощью которых можно непрерывно контролировать количество отбираемого дистиллята. Следует отметить также форштосы 6 (TGL 9972) и 12 (TGL 9971), применяемые в тех случаях, когда дистиллят не разделяют на отдельные фракции. Сборник дистиллята Бредта 14 (TGL 13841), выполненный в виде «коровьего вымени», рассчитан на четыре фракции; аналогичное устройство Брюля 16 имеет семь сосудов размером 120 X х 30 мм. Для устранения пульсаций давления при кипении нередко в дистилляционный прибор через капилляр подают воздух или азот, при этом в качестве куба удобно применять колбу Кляйзена (см. рис. 236) или трехгорлую колбу, в которую на шлифе вводят капилляр 1. Для перегонки сильно вспенивающихся веществ используют специальные приставки Райтмайра (рис. 239, а) или Фридрихса (рис. 239, б). При дистилляции низкокипящих и легковоспламеняющихся жидкостей лучше использовать другое устройство Фридрихса (рис. 240), обеспечивающее безопасность работ. К трубе А можно присоединить шланг для отвода выделяющихся газов в атмосферу.
Ректификационные установки, также как и дистилляционные приборы, можно собирать из отдельных деталей. Без особых 330
трудностей можно собрать даже подобные промышленным сложны установки непрерывного действия с двумя ректификационным колоннами (см. рис. 134 и 162) [10, 11]. Комбинируя различны детали, выполненные по нормали «Дестинорм», можно собрат не только разнообразные установки для перегонки, определени давления паров и данных по равновесию систем пар—жидкость но и более сложные установки для непрерывной ректификаци при атмосферном давлении и под вакуумом (см. рис. 223 и 224J Серия деталей для установок типа «Лабодест» разработана Штаг (изготовитель: фирма Фишера «Лабораторная и исследовательска техника», Бонн—курорт Годесберг). Фирма Нормаг (Хофхайы и предприятие «Стеклянные приборы с нормализованными шли фами» (Вертхайм) также изготавливают отдельные детали, пред назначенные для различных ректификационных установок.
На рис. 241 показаны детали для сборки соединительных коы муникаций. Длинные трубопроводы целесообразно составлять и прямых труб 1 длиной 1 — 1,5 м, снабженных стандартными шли фами. С помощью колен с углом изгиба 135° (2) или 90° (3), а такж Т- или Y-образных тройников 4, 5 можно собирать любые комму никации, включающие при необходимости краны 6 и 7. Дл: обеспечения некоторой подвижности коммуникаций можно ис пользовать соединительные детали со сферическими шлифам) (см. рис. 19) или к штуцеру аппарата подсоединять коммуникации из колен со шлифами, предложенную Фридрихсом (рис. 242) Шлифы различных размеров можно соединять с помощью пере ходов (рис. 243). Ниже приведены диаметры (в мм) муфты и ко нуса переходов, выпускаемых промышленностью:
Муфта WS 12,5 14,5 14,5 24 29 29 29 29 29 29 29 29 29
Конус NS 14,5 14,5 29 29 24 29 34,5 45 55 60 70 85 100
Переход 6 имеет два стандартных конуса, а переход 7 имеет ко нус NS 14,5 и наконечник для шланга.
В табл. 46 указаны некоторые стандартные детали для ректи фикационных установок.
Трубопроводные коммуникации можно собирать также и: деталей с плоскими и сферическими шлифами. При этом макси мальная длина труб для коммуникаций промышленных установоь не превышает 3 м. Для полупромышленных и пилотных установо! стекольная промышленность изготавливает детали, подобньк
Рис. 243.
Переходы:
1—4 — переходы с диаметром конуса, равным или пре вышающим диаметр муфты; 5 — переходы с диаметроь конуса меньшим, чем диаметр муфты; 6 — переходь с двумя конусами; 7 — переходы с конусом NS 14,5 i наконечником для шланга.
Таблица 46.
Стандартные детали для ректификационных установок
Стандарт Наименование
TGL 13828 13831 13834 13835 13836 13837 13838 9966 9973 9978 Соединительные перемычки, прямые * Соединительные перемычки, изогнутые под углом 90° * Соединительные перемычки S-образной формы * Соединительная перемычка с одноходовым краном * Соединительная перемычка с пароотводной трубкой * Соединительная перемычка с каплеотбойником * Соединительная перемычка с воронкой и краном * Переходы * U-образные переходы с двумя шлифовыми муфтами Капилляры, применяемые для устранения пульсаций дав-
DIN 12404/63 ления при кипении * Капилляры со стандартными шлифами, применяемые для
12565/70 устранения пульсаций давления при кипении Капельная воронка
* Все вышеуказанные детали снабжены стандартными шлифами.
деталям, показанным на рис. 241. Типы выпускаемых деталей можно найти в каталогах [1, 5, 71. Кроме того, к настоящему времени выпущен ряд стандартов на подобные детали (TGL 10192— 10200, 11641—11644, 12023—12032, 20277 и проекты стандартов DIN 28800—28808).
7.2.1. КРАНЫ И КЛАПАНЫ
Стандартные стеклянные краны, предназначенные для лабораторных ректификационных установок, приведены в табл. 47.
На рис. 189 и 190 показаны стеклянные краны для работы под вакуумом. Многоходовой кран Кинца [12], предназначенный для работы под вакуумом, устроен так, что его сердечник вращается в одном направлении, и поэтому ошибки в его установке исключены (рис. 244).
В обзоре Вольфа [13] рассмотрены краны специальных конструкций. Один из подобных кранов — плоский кран «Вестале» — показан на рис. 245. Для полупромышленных установок этот кран может выполняться в виде проходного, выпускного, а также трех-и четырехходового крана.
Обычные стеклянные краны с жировой смазкой в качестве уплотнительного материала хорошо заменяют краны со сменными сердечниками из тефлона. Предприятие Отто Фриц Нормаг (Хоф-хайм/Таунус) выпускает различные стеклянные краны с ограничительными кольцами. Миниатюрные краны французской фирмы Гафлон выполняются полностью из тефлона. Стеклянные трубы 332
Таблица 47.
Стандартные стеклянные краны для ректификационных установок
Стандарт Наименование
TGL 21734 группа 1 группы 2—7 13843 группа 1 группа 2 группа 3 DIN 12541/67 группа 1 /70 группа 1 Краны (описание) Специальные краны Высоковакуумные краны Одноходовые краны (размеры) Двухходовые краны (размеры) Одноходовые конусные краны (масса) Одноходовые конусные краны (испытание гер-
12551/67 группа 1 метичности) Одноходовые конусные краны с иезаменяе-
12553/61 мым массивным сердечником Двухходовые краны с незаменяемым сердечником
12554/67 Трехходовые конусные краны с незаменяемым
12563/67 сердечником Трехходовые конусные краны с незаменяемым массивным сердечником, снабженные отверстиями, смещенными по окружности на 120°
присоединяют к этим кранам с помощью тефлоновых болтовых соединений с прокладками также из тефлона.
Обычно стеклянные краны снабжают устройствами, предотвращающими выход сердечника крана из его гнезда при вращении (см. рис. 217). В качестве таких устройств можно использовать резиновые шайбы или кольца, либо, еще лучше, предохранители из пружинной стали (рис. 246).
Рис. 245.
Плоский край «Вестале» (патент ФРГ 1112846):
а — детали крана; б — кран в сборке.
Изготовитель: фирма Эдмунд Бюлер. Тюбинген.
333
Рис. 247.
Схема клапана для точной регулировки расхода жидкостей и газов с седлом из стекла и шпинделем из тефлона (диаметр седла или диаметр подводящей трубы 4 илн 7 мм):
а — угловой клапан; б — проходной клапан.
Изготовитель: народное предприятие «Йенский завод стекла Шотт», Иена.
В полупромышленных и пилотных установках не рекомендуется использовать стеклянные краны с жировой смазкой, особенно при диаметрах условного прохода трубопроводов NW выше 40 мм. В этих случаях целесообразно применять клапаны современных конструкций, выполненные из стеклянных и тефлоновых деталей [1]. Вместо кранов в лабораторных установках часто используют клапаны с тефлоновыми шпинделями (рис. 247). Прецизионные клапаны Совиреля системы «Торион» предназначены для установки на трубопроводах диаметром 2,5—10 мм. Клапаны должны надежно работать при остаточных давлениях до 10-4 мм рт. ст.
В клапанах для промышленных установок в качестве запорного устройства применяется тефлоновый сильфон с запорной пластиной или запорным конусом из тефлона (рис. 248). Установка
положения клапанов может осуществляться вручную с помощью электромагнитов, электромотора или пневматического привода, что позволяет применять эти клапаны в автоматизированных ректификационных установках.
Промышленность стекольных изделий выпускает клапаны различных моделей с плоскими или сферическими шлифами, имеющими диаметр условного прохода NW до 150 мм.
Рис. 248.
Пневматический регулируемый клапан (без сервопривода).
Диаметр условного прохода NW25, 32, 40 и 50 мм.
Изготовитель; народное предприятие «Йенский завод стекла Шотт», Иена.
334
Различают клапаны следующих типов (см. разд. 5.1.3.2): проходные, выпускные, угловые, дроссельные, обратные с заслонкой или шаром, предохранительные для максимальных скачков давления, не превышающих 5 кгс/см2.
7.3. РЕКТИФИКАЦИОННЫЕ КОЛОННЫ
Физические процессы массо- и теплообмена протекают в ректификационной колонне на поверхности раздела жидкой и паровой фаз (см. разд. 4.1). Следовательно, конструкция колонны должна обеспечить протекание этих процессов в условиях противоточного движения пара и жидкости при наличии развитой поверхности контакта фаз, причем удерживающая способность колонны не должна превышать допустимую (см. разд. 4.10.5). Оптимальная конструкция колонны характеризуется минимальным значением ВЭТС и минимальной удерживающей способностью по жидкости.
Хекманн [14] предложил критерии выбора колонн с целью их систематизации. Число разработанных конструкций колонн чрезвычайно велико, однако можно выделить следующие основные типы колонн: безнасадочные, насадочные, тарельчатые, с неподвижными контактными устройствами и с вращающимися контактными устройствами.
Райхельт [14а] описал тарельчато-насадочные колонны. В этих колоннах на колосниковых решетках или тарелках размещены слои насадки, состоящие обычно из легких шариков. При работе колонны в условиях противотока фаз на тарелках образуются кипящие слои насадки, обеспечивающие повышение пропускной способности колонны.
7.3.1. БЕЗНАСАДОЧНЫЕ КОЛОННЫ
Особым преимуществом безнасадочных колонн являются малая удерживающая способность по жидкости и небольшой перепад давления. Это обусловливает их применение, главным образом, для микроперегонки (см. разд. 5.1.1), высокотемпературной ректификации (см. разд. 5.3.2) и вакуумной дистилляции (см. разд. 5.4.1). В колоннах этого типа процессы массо- и теплообмена протекают между паром, поднимающимся в свободном пространстве, и жидкостью, стекающей в виде пленки по стенкам. Области применения пленочных колонн, а также проблемы, возникающие при их эксплуатации, подробно рассмотрены Малевским [15]. Он предложил некоторые конструкции подобных аппаратов, а также указал на нерешенные проблемы.
Ректификационные колонны могут быть выполнены в форме прямой или спиральной трубы, трубы с развитой поверхностью, а также в виде канала кольцевого сечения. Подобные колонны благодаря малому гидравлическому сопротивлению имеют сравнительно высокую пропускную способность по пару, однако их
335
эффективность, особенно при больших нагрузках, невелика вследствие ограниченной поверхности контакта фаз. Исключение составляют лишь колонны, выполненные в виде канала кольцевого сечения. Вертикальные гладкостенные трубы диаметром 20—50 мм в настоящее время редко применяют в качестве ректификационных колонн, обычно их используют для отделения брызг при перегонке. Только при нагрузках ниже 30 мл/ч удается достичь ВЭТС менее 10 см. Разделяющую способность этих колонн можно повысить путем увеличения поверхности ее стенок. Например, сферическая колонна Вюрца (см. рис. 15) и грушевидная колонна Юнга имеют более высокую эффективность, однако обладают при этом большей удерживающей способностью. Характеристики безнасадочных колонн приведены в табл. 48.
Для увеличения поверхности контакта фаз на внутреннюю стенку трубы наплавляют стеклянный порошок или припаивают горизонтально и наклонно небольшие стеклянные стержни, как в известной колонне с елочным дефлегматором. Наклонные стержни служат также для подачи флегмы, стекающей по стенкам, к оси колонны (рис. 249). В модифицированной модели, предложенной Рэем [17], стержни расположены перпендикулярно к стенкам. Достигаемая при этом эффективность разделения указана в табл. 48. Колонну с елочным дефлегматором используют в несложных процессах перегонки, требующих минимального времени пребывания жидкости в колонне. Например, в таких колоннах проводят
Таблица 48.
Характеристики безнасадочных колонн
Тип колонны Диаметр, мм Рабочая длина, мм Нагрузка, мл/ч ВЭТС, см Объем удерживаемой жидкости, приходящийся на 1 теор. ступень разделения, мл Литература
Полая вертикальная труба 30 1000 25 [16]
Труба с наплавленным на внутреннюю поверхность стеклянным порошком 7 500 12,5 [16]
Колонна с елочным дефлег- 12 460 54 5,4 [18]
матором. 96 7,1 0,46
294 7,7 0,62
540 7,7 0,68
24 460 120 10,2 1,3
240 12,1
510 11,5 1,8
Модифицированная колонна с елочным дефлегматором, предложенная Рэем 6 1200 60 2,0 <0,02 [17]
Колонна Янцена 4—6 50—200 4—6 0,2—0,6 [19]
336
Рис. 249.
Колонна с елочным дефлегматором и изоляционным кожухом, заполненным воздухом.
Рис. 250.
Трубчатые змеевиковые колонны Янцена с вакуумированным изоляционным кожухом со змеевиком длиной 3 м (а) и 6 м (д’).
микродистилляцию или высокотемпературную ректификацию смол К' и других высококипящих веществ, которые необходимо разделить г под вакуумом с отгонкой до 98% от общего количества исходной смеси [18]. Для аналогичных целей применяют и змеевиковую трубчатую колонну Янцена (рис. 250), которая выполняется со змеевиками длиной 3 и 6 м. Она обладает удерживающей способов ностью, равной 0,2—0,6 мл на одну теоретическую ступень раз-91 деления [19]. При испытании подобной колонны на смеси че-тыреххлористый углерод—бензол автор получил следующие дан-ные:
К Предел захлебывания....................... 900 мл/ч
| Объем удерживаемой жидкости, приходя-щийся на одну теоретическую ступень
г разделения................................ 0,4 мл (при нагрузке
| 50 мл/ч)
I ВЭТС..................................... 4,3 см (при нагрузке
I 50 мл/ч)
I 5,4 см (при нагрузке
| 90 мл/ч)
К. 5,6 см (при нагрузке
к 190 мл/ч)
337
Рис. 251.
Схема, поясняющая принцип повышения разделяющей способности ректификационной колонии, разработанный Куном.
Рис. 252.
Миоготрубчатая колонна Куна:
1 — трубы; 2 — термостат; 3 — капилляры, обогреваемые снаружи.
Рис. 253.
Трубчатая щелевая колонна Куна:
1 — дефлегматор; 2 — штуцер для присоединения к вакуумному насосу;
3 — охлаждаемый сборник дистиллята; 4,5 — концентрические трубы;
6 — вакуумированный кожух; 7 — куб; 8 — электронагреватель.
Испытанная колонна имела змеевик с диаметром 50 мм, шагом 12 мм и длиной 6 м; змеевик был изготовлен из трубы диаметром 6 мм.
Колонну с елочным дефлегматором обычно изготавливают со съемным теплоизоляционным кожухом, а колонну Янцена — с вакуумированным кожухом. Для всех безнасадочных колонн важно поддерживать в ходе ректификации температуру стенки ниже температуры стекающей флегмы, так как в противном случае нарушается равномерное образование пленки на поверхности стенки. Следует отметить, что эффективность безнасадочных колонн возрастает при уменьшении нагрузки. Используя многотрубчатые колонны, состоящие из параллельно соединенных труб небольшого диаметра, можно получить любую необходимую производительность. Подобную многотрубчатую колонну еще в 1936 г. запатентовал Фенске [20].
Экспериментально установлено, что при нагрузке трубчатой ректификационной колонны диаметром 0,6 см, равной 10 мл/ч, ВЭТС составляет около 1,73 см [21]. Колонна указанного диа-338
метра высотой 1 м обладает при этом гидравлическим сопротивлением 2,7 мм рт, ст. и удерживающей способностью по жидкости около 0,4 мл. На основе этих данных Кун [22 ] разработал новый принцип повышения разделяющей способности ректификационной колонны (рис. 251). Этот принцип основан на многократном увеличении сравнительно небольшого эффекта разделения, имеющего место в поперечном сечении колонны (в направлении вектора а) при стационарном режиме путем создания продольного потока, изменяющего свое направление на концах трубчатой ректификационной колонны (векторы ui и и2).
Для достижения высокой эффективности разделения необходимо иметь в колонне достаточно низкую скорость потока паров при значительном флегмовом числе. Чтобы получить при этом удовлетворительную производительность колонны, нужно, как уже указывалось выше, использовать многотрубчатую колонну (рис. 252). Целесообразным оказалось образование флегмы непосредственно в трубах 1, помещенных в общий термостат 2; при этом дистиллят отбирают из верхних концов труб 1 с помощью обогреваемых снаружи капилляров 3 (см. также рис. 157).
Данные о производительности подобной колонны, содержащей 100 параллельных труб диаметром 0,4 см и длиной 2 м, полученные Куном с сотр. [22] при различных исходных и конечных концентрациях и разностях температур кипения компонентов, приведены в табл. 49.
При расчетах производительности принимали, что молекулярная масса компонентов составляет 100, а температура кипения — около 90 °C. Другая колонна Куна, содержащая 61 трубу диаметром 0,2 см с рабочей высотой 1,5 м, была использована для разделения изотопов 12С и 13С с разностью температур кипения всего 0,03 °C. В этом случае число теоретических ступеней разделения определяли по данным анализа продуктов перегонки на масс-спектрометре. Удерживающая способность одной трубы диаметром 0,2 см и высотой 2 м составила около 0,5 г [22].
Таблица 49.
Производительность колонны Куна, содержащей 100 труб диаметром 0,4 см и длиной 2 м
Разность температур кипения компонентов, °C Производительность колонны (в г/сут) при исходной и конечной концентрациях ннзкокипящего компонента (xQ -> *кон» % мол.)
40 -> 60 10 -> 90 10 -> 99 50 -> 90 1 -> 99
0,1 30
0,31 300 5 2,9 22 0,2
1,0 3 000 50 29 220 2
3,1 30 000 500 290 2 200 20
10,0 300 000 5000 2900 22 000 200
339
7
100 200 300 ЧОО 500-
Нагрузка, мл/ч
Рис. 254.
Диаграмма разделения изомеров ксилола с помощью колонны Куна высотой 0,8 м.
Рис. 255.
Зависимость числа теоретических ступеней разделения, приходящихся на
1 см высоты колонны ЯуД, от ее производительности [28]:
/ — трубчатая щелевая колонна диаметром 2,5 см и шириной щели 0,4 см;
2 — колонна диаметром 2,5 см с насадкой Стедмана; 3 — колонна диаметром 2,4 см с насадкой из проволочных спиралей 4X4 мм.
Метод перегонки, основанный на принципе многократного увеличения эффекта разделения, был разработан Куном путем обширного аналитического исследования 124, 25]. Эти теоретические расчеты были проведены для колонны с кольцевым поперечным сечением, образованной из двух концентрических труб (рис. 253). Подобные колонны называют колоннами с кольцевой щелью или трубчатыми щелевыми колоннами (см. рис. 257а).
Метод, предложенный Куном, можно успешно применять при разделении таких близкокипящих изомеров, как ксилолы (рис. 254) и изомерные амиловые спирты. Колонна Куна пригодна также для проведения процессов перегонки, требующих более 100 теоретических ступеней разделения, так как эта колонна имеет исключительно малую ВЭТС.
Методы расчета подобных колонн для разделения идеальных смесей были обсуждены в разд. 5.1.4.2. Крэйг [23] описал щелевую колонну, предназначенную для микроперегонки при загрузке исходной смеси в объеме 0,25 мл. Эта колонна представляет собой канал кольцевого сечения, образованный капилляром и соосно размещенным внутри его стержнем. Теоретические основы расчета этих колонн разрабатывали также Вестхавер [26], Доннелл и Кенеди [27 ], а также Янцен и Викхорст, проводившие сравнение данной колонны с колоннами, заполненными насадкой Стедмана и насадкой из проволочных спиралей [28 ]. Как видно из рис. 255, щелевая трубчатая колонна при производительности менее 10 мл/ч 340
имеет бдльшую эффективность, чем обе описанные выше колонны.
Перепад давления в трубчатых щелевых колоннах можно рассчитать по формуле:
Pi — Pi = Зт)1/<2/(26а2) (188а)
где рг — р2 — перепад давления, дин/см2; л — динамическая вязкость пара, П; / — высота колонны, см; Q — расход пара, рассчитанный по среднему давлению в колонне (p-i + р3)/2, см3/с; b — длина средней окружности кольцевого сечения колонны, см; а — половина ширины кольцевого сечения, см.
Гидравлическое сопротивление трубчатой щелевой колонны в сопоставимых рабочих условиях примерно на порядок меньше, чем у вышеупомянутых насадочных колонн (рис. 256). Кох и Файнд [29] определяли в изотермических условиях гидравлическое сопротивление трубчатых щелевых колонн с шириной щели, изменяющейся от 4,05 до 19,68 мм, при одинаковом диаметре наружной трубы, равном 49,96 мм. Ими был также исследован процесс теплопередачи в' подобных колоннах при внешнем обогреве наружной трубы. Бек [30 ] исследовал эффективность теплообмена и гидравлическое сопротивление в трубчатых щелевых колоннах, образованных коаксиально или некоаксиально расположенными трубами, при вынужденном движении и свободной конвекции.
Доннелл и Кенеди [27] показали, что при перегонке на трубчатых щелевых колоннах можно достигнуть ВЭТС 0,5—0,6 см при соблюдении следующих условий: обеспечение одинаковой ширины щели по поперечному сечению и высоте колонны (достигается путем применения калиброванных труб); равномерное распределение пленки жидкости по поверхности стенок (достигается путем спиралевидного травления стенок); адиабатический режим перегонки (обеспечивается хорошей теплоизоляцией колонны или использованием обогревающего кожуха); постоянная нагрузка колонны по пару (достигается с помощью автоматического регулирования скорости потока паров).
В ряде работ Фишера [31 ] описана разработанная им стеклянная трубчатая щелевая колонна новой конструкции, а также приведены полученные на ней экспериментальные данные. Эта колонна предназначена, главным образом, для ректификации высококипящих веществ; она обладает гидравлическим сопротивлением от КГ1 до 10“3 мм рт. ст. при удерживающей способности по
Рис. 256.
Зависимость гидравлического сопротивления от производительности колонны при атмосферном давлении, рассчитанная на 50 теоретических ступеней разделения 1281: 1, 2, 3 — см. рис. 255.
341
Рис. 257.
Автоматическая лабораторная установка для полумикроректификации, разработанная Фишером.
Установка содержит системы регулирования мощности электронагревателя масляной банк по температуре масла или перепаду давления потока пара в колонне, мощности электронагревателя кожуха колонны, флегмового числа путем деления потока пара, стабилизации вакуума, объема отбираемых фракцйй дистиллята. Предусмотрена регистрация всех параметров процесса. Установка снабжена предохранительными устройствами против превышения заданной температуры н выхода из строя системы подачи охлаждающей воды.
а — схема участка трубчатой щелевой колонны:
1 — наружная труба со спиралевидной канавкой на внутренней стенке; 2 — внутренняя труба со спиралевидной канавкой иа наружной стенке; <3 — кольцевой канал для прохода паров; *
б — общий вид установки:
1 — масляная баня на подъемной платформе; 2, 8, 10 — термометры сопротивления масляной бани, обогревающего кожуха и головки колонны соответственно; 3 — куб; 4 — контрольный термометр куба; 5 — электромотор мешалки; 6 — трубчатая щелевая колонна; 7 — кожух для компенсационного обогрева колонны; 9 — головка колонны с дефлегматором; 11 — сердечник клапана; 12 — магнитная катушка; 13 — холодильник дистиллята; 14 — вакуумированный сосуд; 15 — сборник фракций; 16 — устройство для установки пробирок; 17 — магнитная муфта; 18 — автоматическое приводное устройство; 19 — холодильник прибора, измеряющего перепад давлений; 20 — шкаф управления; 21 — шесть компенсационных самописцев; 22 — прибор «Миннтрон 5» для регулирования мощности электронагревателей куба и кожуха колонны, а также для установки электронного реле головки колонны; 23 — регулятор перепада давления в колонне и подачи охлаждающей воды; 24 — стабилизатор вакуума; 25 — регулятор сборника фракций; 26 — шкаф для запасных частей н принадлежностей.
Изготовитель: фирма Фишера «Лабораторная и исследовательская техника», Бонн—курорт Годесберг.
жидкости 0,1 — 1,5 мл. Ректификационная установка с этой колонной, оснащенная большим числом автоматических приборов и регуляторов, предназначена для работы при атмосферном давлении и под вакуумом (остаточное давление до 10~3 мм рт. ст.), причем давление в кубе не превышает 0,1 мм рт. ст.
Высокая эффективность массопереноса достигается в колонне Фишера благодаря гидродинамически оптимальному конструктивному выполнению стенок кольцевого канала (рис. 257я). Оптимальную скорость потока пара в колонне шипт можно рассчитать по формуле:
гиопт = 2£> 1^2/а (189)
где D —коэффициент диффузии низкокипящего компонента в паре при рабочих условиях; а — ширина щели.
На рис. 2576 схематически показана автоматическая лабораторная установка Фишера для полумикроректификации. Эта установка может снабжаться семью различными колоннами с числом теоретических ступеней разделения от 10 до 80; объем загружаемой исходной смеси составляет от 0,5 до 4000 мл. Масляная баня с регулируемым подводом тепла позволяет поддерживать постоянную температуру куба с точностью около 0,1 °C. Сведения о других элементах и узлах установки приведены в разд. 8.1.2.
343
7.3.2. НАСАДОЧНЫЕ КОЛОННЫ
Насадочная колонна представляет собой трубу, в которой размещены слои из нерегулярно уложенных элементов насадки (см. разд. 4.10.2). Насадку укладывают на решетки различной формы (рис. 258) или на перфорированные воронки (рис. 259), применяемые например, в ректификационных колоннах Штаге для установок «Лабодест» и в колоннах по нормалям «Дестинорм», разработанных автором. Свободное сечение решеток не должно быть меньше свободного сечения уложенных на них слоев насадки. Обычно сначала засыпают слой более крупной насадки, на которой затем укладывают основную насадку, размеры которой определяют из следующего соотношения:
Внутренний диаметр колонны Ю
Эквивалентный гидравлический диаметр насадки
Чтобы предотвратить растекание жидкости к стенкам (см. разд. 4.2), необходимо неоднократно собирать флегму по высоте колонны и снова распределять ее, возвращая к центральной части колонны. Гроссе-Етрингхауз [32] пытался достичь этого, исполь-зуя колонну, в корпусе которой были кольцевые вмятины глубиной 2 мм на расстоянии 30 мм друг от друга по высоте колонны (рис. 260). С этой же целью колонну можно разделить на отдельные участки и перераспределять флегму в конце каждого участка (см. разд. 4.10.2), а также использовать специальные вставки из металлической сетки с коническими углублениями. Их преимущество состоит в том, что они практически не имеют мертвых объемов [33].
Рис. 258.
Решетки для размещения насадки: спиральная (а); плоская (б);
1 — стеклянный стержень; 2 — носок для стекания капель.
Рис. 259.
Перфорированная воронка для поддержания насадки.
Рис. 260.
Колонна Гроссе—Етрннгхауза с кольцевыми вмятинами в корпусе.
344
Выбор диаметра насадочной колонны определяется требуемой пропускной способностью (см. разд. 4.11). В лабораторных ректификационных установках используют обычно колонны диаметром 10—50 мм. Колонны диаметром 50—200 мм относятся к полупромышленным. Пилотные установки снабжаются ректификационными колоннами диаметром от 150 до 400 мм.
Рабочую высоту слоев насадки обычно выбирают в интервале от 500 до 1000 мм, чтобы легко было сравнивать результаты испытаний насадок различных типов. В зависимости от цели перегонки и необходимого числа теоретических ступеней разделения применяют колонны, составленные из отдельных царг, что особенно выгодно при аналитической дистилляции, так как можно без особых трудностей собрать колонну требуемой высоты. Кроме того, подобные колонны удобны при сравнительной ректификации, моделирующей промышленный процесс, когда рабочая высота насадки точно определена по числу теоретических ступеней разделения, а также по параметрам гидродинамического режима процесса. В этом случае может оказаться, что необходимая высота колонны не совпадает с высотами стандартизованных колонн.
7.3.3. ТАРЕЛЬЧАТЫЕ КОЛОННЫ
Тарельчатые колонны применяют в лабораторных и промышленных установках для специальных задач разделения, например для получения дистиллята с высокой степенью чистоты методом непрерывной ректификации. Эти колонны используют также в лаборатории для проведения сравнительной ректификации, моделирующей промышленный процесс. Колонны с ситчатыми тарелками пригодны для аналитических разгонок при атмосферном давлении. Тарельчатые колонны используют в промышленных ректификационных установках непрерывного действия [34].
Новые типы лабораторных и промышленных тарельчатых колонн подробно рассмотрены Штаге [35]. В этой работе обсуждены также вопросы испытаний эффективности подобных колонн. Преимущества колонн с колпачковыми тарелками для непрерывных процессов перегонки разобраны в разд. 5.2.2.3 (см. также рис. 167). Коэффициент полезного действия колпачковых тарелок при циклическом способе ведения процесса превышает 150%.
По конструктивному выполнению различают фонтанирующие, колпачковые и ситчатые тарелки *. Ректификацию в тарельчатых колоннах проводят в основном при атмосферном давлении, вслед-ствии их высокого гидравлического сопротивления потоку паров, которое существенно превышает гидравлическое сопротивление колонн других типов.
* Как эти, так и ряд других тарелок принципиально сходной конструкции (провальные, клапанные и др.) в отечественной технической литературе принято называть барботажными. — Прим. ред.
345
Рис. 261.
Колонна Кеезома с фонтанирующими тарелками:
1 — ванны; 2 — трубы.
Рис. 262.
Колонна Брууна с колпачковыми тарелками:
1, 3 — трубы для подвода и отвода жидкости; 2 — колпачок; 4 — штуцер для прохода пара.
Принцип действия тарельчатых колонн заключается в барботаже восходящего потока пара через перемещающийся по тарелке слой жидкости определенной высоты. Флегма перетекает с вышележащей тарелки на нижележащую по переливным трубам, расположенным как внутри, так и снаружи колонны (см. разд. 4.2 и 4.7).
Колонна с фонтанирующими тарелками Кеезома [36] показана на рис. 261. Жидкость находится в ваннах 1, а флегма стекает по трубам 2. Во всех колоннах с фонтанирующими и колпачковыми тарелками поток пара при входе в слой жидкости на тарелке меняет направление движения и барботирует через жидкость, двигаясь в виде отдельных пузырьков радиально вверх. Колонны с фонтанирующими тарелками Кеезома применяются сравнительно редко из-за низкого коэффициента полезного действия тарелки, не превышающего 50%. Колонны с колпачковыми тарелками различных конструкций находят широкое применение прежде всего для определения необходимых параметров масштабного перехода.
Штаге [35 ] предложил для непрерывных процессов разделения лабораторные и промышленные колонны с колпачковыми тарелками, предназначенные для работы в широком интервале нагрузки по жидкости. Эффективность этих колонн не зависит от нагрузки по жидкости и соотношения нагрузок паровой и жидкой фаз, а также от кратковременных нарушений технологического режима процесса. В лабораторных установках широко применяют тарельчатые колонны Брууна [37] (рис. 262). По конструктивному выполнению они приближаются к промышленным колоннам с колпачковыми тарелками. Их недостаток заключается в том, что переточ-ные трубы для флегмы размещены снаружи колонны, и это может приводить к значительному переохлаждению жидкости. Степень охлаждения можно уменьшить, поместив колонну в заполненный воздухом или вакуумированный кожух (рис. 263). Трубы для подвода 1 и отвода 3 жидкости (см. рис. 262) расположены таким об-346
разом, чтобы на тарелках образовывались слои жидкости толщиной около 10 мм. На штуцер для прохода паров 4 свободно насажен колпачок 2, имеющий в нижней части прорези для дробления потока паров на пузырьки возможно меньших размеров.
Колонна Креля с колпачковыми тарелками (см. рис. 139) и внутренними переточными трубами для флегмы выполнена подобно промышленным колоннам. Диаметр колонны 50—70 мм; она хорошо зарекомендовала себя при сравнительных разгонках, моделирующих промышленные процессы. Эту колонну можно снабдить пробоотборниками с кранами и шлифами для термометров. Адиабатичность процесса обеспечивается с помощью разъемного обогревающего кожуха.
Многокамерная колонна Клейна, Штаге и Шульце также имеет внутренние переточные трубы для подачи флегмы [381. Распределение пара на тарелках обеспечивается с помощью отверстий, расположенных по окружностям. Через эти отверстия пар выходит в виде небольших пузырьков, интенсивно перемешивая
Рис. 263.
Колонна Брууна с колпачковыми тарелками:
а — колонна с 40 реальными тарелками и съемным изоляционным кожухом, заполненным воздухом; б — то же с 20 реальными тарелками и вакуумированным кожухом.
Изготовитель: народное предприятие «Комбинат технического стекла», Ильменау.
Рис. 264.
Колонна с колпачковыми тарелками, снабженными отражательными перегородками для пара:
а — схема движения потока пара на тарелке; б — схема колонны с тарелками.
347
Рис. 265.
Колонна с колпачковыми тарелкам» Штаге, снабженными защищенными переточными трубами.
слой жидкости, находящийся на тарелке. Штаге разработал колонну с колпачковыми тарелками диаметром от 40 до 130 мм, снабженными отражательными перегородками для пара. На каждой тарелке поток пара через кольцевую щель поступает в перемещающийся по окружности слой жидкости и затем движется радиально от периферии к центру тарелки, где отражается от пластины, установленной напротив кольцевой щели (рис. 264 а). Возникающее при этом вихревое движение фаз существенно ин
тенсифицирует процесс массообмена. Значительная длина пути вынужденного движения жидкости по тарелке от подводящей до выводящей трубы также способствует повышению эффективности разделения (см. рис. 2646 и 167). Коэффициент полезного действия тарелки в интервале применяемых рабочих нагрузок практически не изменяется и лежит в пределах от 80 до 90% [35, 39]. Колпачковая тарелка Штаге с защищенными переточными трубами (рис. 265) обладает высокой пропускной способностью по жидкости.
На рис. 266 представлены сравнительные данные по гидравлическому сопротивлению и к. п. д. тарельчатых колонн различных типов.
В табл. 50 приведены геометрические параметры этих колонн, а на рис. 267 показаны их некоторые конструктивные особенности.
Ситчатые тарелки обычно выполняют в виде перфорированных пластин. Невысокий слой жидкости на этих тарелках поддерживается благодаря динамическому напору восходящего потока паров. Очевидно, что такие тарелки характеризуются минимально допустимой нагрузкой по пару, ниже которой жидкость стекает через отверстия в тарелках.
Колонны с ситчатыми тарелками наиболее пригодны для аналитической ректификации низкокипящих углеводородов. Ситчатые тарелки Олдершоу—Гроля [41 ] выполнены в виде пластин с вертикально просверленными отверстиями диаметром 0,75— 1 мм, расположенными по окружностям (рис. 268). Флегма поступает на нижележащую тарелку по переточной трубе, расположенной в центральной части тарелки (о гидравлическом сопротивлении колонн этого типа см. разд. 4.11, табл. 30). У колонны с ситчатыми тарелками Зигварта переточные трубы установлены 348
снаружи, а тарелки выполнены в виде перфорированных цилиндрических обечаек с глухими днищами (рис. 269).
Коэффициент полезного действия колонн с ситчатыми тарелками зависит от различных факторов. Ван-Вийк и Тиссен [43] исследовали работу колонны с восемью ситчатыми тарелками внутренним диаметром 38 мм при высоте уровня жидкости на тарелке 120 мм. При ректификации бинарной смеси «-гептан — метилциклогексан было установлено, что к. п. д. тарелок значительно снижается, если концентрация любого компонента в смеси ниже 0,1% (мол.) В интервале скоростей потока пара от 10 до 27 см/с к. п. д. изменялся в пределах от 86 до 75%. При постоянной скорости пара 17 см/с и концентрации низкокипящего компонента в кубовой жидкости хв = 50% (мол.) к. п. д. увеличивался от 65 до 85% с ростом флегмового числа от 0,54 до 1,0.
Умхольц и Ван-Винкле [44] изучали работу колонны с ситчатыми тарелками диаметром 25 мм при высоте уровня жидкости на тарелке 50 мм. Свободное сечение тарелки составляло 16,2%. Оказалось, что наибольшей эффективностью обладают тарелки с диаметром отверстий 1,9 мм. При ректификации смеси о-кси-лол —м-кс и лол было установлено значительное снижение к. п. д.
Рис. 266.
Зависимости коэффициента полезного действия и гидравлического сопротивления от нагрузки по флегме для тарельчатых колонн (размеры см. в табл. 50):
1,2 — колонны типа «Лабодест» с колпачковыми тарелками, снабженными отражательными перегородками для пара; 3 — колонны типа «Лабодест» с колпачковыми тарелками, оборудованными защищенными переточными трубами; 4 — колонна с колпачковыми тарелками Шмюклера —Фрица; 5 — колонна с колпачковыми тарелками Бранда; 6 — колонна с колпачковыми тарелками фирмы Нормшлиф.
349
Таблица 50.
Площади прохода пара и флегмы для шести типов колонн с колпачковыми тарелками, испытанных Штаге [35]
(Описание конструкций колонн см. в подписи к рис. 266)
Порядковый номер колонны
Геометрический параметр колонны 1 2 3 4 5 6
Наружный диаметр, мм 55 55 55 55 62 58
Внутренний диаметр, мм 51,5 51,5 51,5 51,5 57,5 54
Площадь поперечного сечения колонны, см2 21 21 21 21 26 23
Площадь поперечного сечения штуцера для прохода паров, рассчитанная по наружному диаметру, см2 3,51 7,8 2,58 3,31 7,9 3,29
Отношение к площади сечения колонны, % 16,7 37 12,3 15,7 30,3 14,4
Площадь поперечного сечения штуцера для прохода паров, рассчитанная по внутреннему диаметру, см2 2,44 5,2 2,12 1,13 3,5 1,76
Отношение к площади сечения колонны, % 11,6 24,7 10,1 5,4 13,7 7,7
Площадь прорезей в колпачке, см2 5,58 3,1 3,1 2,25 1,82 2,1
Отношение к площади сечения колонны, % 26,65 14,7 14,7 10,7 7,0 9,2
Площадь поперечного сечения колпачка, рассчитанная по внутреннему диаметру, см2 5,58 1,83 1,83 0,85 4,33 1,0
Отношение к площади сечения колонны, % 26,65 8,7 8,7 4,05 16,6 4,36
Общая площадь для прохода пара, см2 11,16 4,93 4,93 3,10 6,15 3,10
Отношение к площади сечения колонны, % 53,3 23,4 23,4 14,8 23,6 13,6
Площадь кольцевой щели для входа пара, см2 9,7 4,15 13,45 10,0 8,83 16,3
Отношение к площади сечения колонны, % 46,3 20,0 64,0 47,5 33,9 72,5
Площадь поперечного сечения отводной трубы, см2 9,74 7,17 6,38 6,6 . 7,22 6,4
Отношение к площади сечения колонны, % 46,2 34,1 30,4 31,5 27,8 28,0
Площадь сечения труб для перетока флегмы, см2 1,04 0,67 0,29 0,6 0,39 0,5
Отношение к площади сечения колонны, % 4,93 3,15 1,38 2,38 1,5 2,19
350
тарелки с 90 до 65% при уменьшении абсолютного давления в колонне с 750 до 50 мм рт. ст. С ростом нагрузки коэффициент полезного действия сначала увеличивается, затем в интервале нагрузки от 200 до 250 г/(см2-ч) остается практически постоянным. В работе [45] указывается, что эффективность колонны зависит также от толщины перфорированной пластины тарелки и физико-
Рис. 267.
Схемы новых типов колонн с колпачковыми тарелками:
а — колонна Шмюклера — Фрица [401; б — колонна Бранда [35], нзго товнтель: фирма Р. Бранд, Вертхайм; каталог фирмы Бранда 68; в — ко лонна фирмы Нормшлнф, изготовитель: фирма Фридрнхса—Мачке «Сте клянные приборы Нормшлнф», Вертхайм.
Рис. 268.
Колонна с ситчатыми тарелками Олдершоу—Гроля:
а — колонна в вакуумированном кожухе, снабженном линзовыми ком пенсаторами; б — вид тарелок в работе.
Рис. 269.
Колонна с ситчатыми тарелками ЗиГварта.
Изготовитель: фирма «Йенский завод стекла Шотт», Майнц.
Рис. 270.
Колонна с ситчатыми тарелками модели «Пер-фо-дрип» [46 ].
351
химических свойств разделяемой смеси (поверхностного натяжения жидкости, плотностей паровой и жидкой фаз).
Колонна с ситчатыми тарелками модели «Перфо-дрип» [46] состоит из калиброванной трубы, внутри которой на стержне укреплены перфорированные пластины из коррозионностойкой стали (рис. 270). Расстояние между тарелками можно изменять. Тарелки могут быть выполнены из тефлона. Эта колонна обладает более высокой разделяющей способностью, чем колонна Олдер-шоу—Гроля, и позволяет обеспечивать высокую степень очистки разделяемых компонентов.
При пульсирующей подаче пара в колонну Олдершоу диаметром 32 мм (каждая тарелка имела 82 отверстия диаметром 0,85 мм, высота уровня жидкости на тарелке была равна 30 мм) Мак-Гуре и Маддокс [47] достигли существенного повышения эффективности разделения, которое зависит от амплитуды и частоты пульсаций скорости потока пара.
Основное требование к конструктивному оформлению всех тарельчатых колонн состоит в том, что выбранное межтарельчатое расстояние должно практически исключать унос жидкости с тарелки на тарелку. При размещении тарелок на небольшом расстоянии друг от друга поток пара уносит частицы жидкости на вышележащую тарелку, что значительно снижает к. п. д. тарелок.
Вагнер с сотр. [48] определял с помощью радиоактивных индикаторов степень загрязнения дистиллята высококипящими соединениями при вакуумной ректификации. Невитт с сотр. [49] проводил теоретическое и экспериментальное исследование механизма каплеобразования и распределения капель по размерам. -
Исследованию гидродинамических характеристик колонн с колпачковыми и ситчатыми тарелками посвящена работа Меликяна [50].
Некоторые параметры описанных тарельчатых колонн приведены в табл. 28, составленной Штаге [35, 38].
Значения к. п. д. тарелок различных колонн, приведенные в табл. 28, дают лишь приближенное представление об эффективности данных колонн, так как они были определены в несопоставимых рабочих условиях.
7.3.4. КОЛОННЫ С НЕПОДВИЖНЫМИ КОНТАКТНЫМИ УСТРОЙСТВАМИ
В отличие от насадочных колонн, заполненных беспорядочно уложенной насадкой, и колонн с тарелками, жестко крепящимися к корпусу, к колоннам с неподвижными контактными устройствами, относят аппараты со свободно вставленными элементами, выполненными из непроницаемого или перфорированного материала. В промышленности такие контактные устройства называют насадками *. Жидкость стекает по элементам насадки в виде
* В отечественной литературе такие колонны называют колоннами с регулярной насадкой. — Прим. ред.
352
пленки, причем степень смачиваемости насадки, выполненной из перфорированного материала, выше (см. разд. 4.2). Поскольку эффективность разделения в подобных колоннах зависит от степени смачиваемости материала насадки, их называют «колоннами со смоченными стенками».
Различают следующие виды колонн с неподвижными контактными устройствами: со стеклянной спиралью; со спиралью из металлической сетки; с проволочной спиралью; с насадкой Стедмана и с наклонными орошаемыми пластинами.
Прототипом колонны со стеклянной спиралью Видмера [51 ] (рис. 271) является колонна Голодца [52], снабженная сифонными гидрозатвором и концентрическими трубами. Основной недостаток таких колонн состоит в том, что в них не выдерживается принцип противотока фаз [38], поэтому они не могут быть рекомендованы для использования. Колонну Дафтона со спиралью переменного шага, уменьшающегося по ходу потока паров (рис. 272), целесообразно применять в процессах разделения, при которых не требуется высокой эффективности и необходимо небольшое время пребывания фаз в колонне. ВЭТС у подобных колонн обычно составляет около 10 см.
Значительно более эффективной является колонна Леки и Эвела с перфорированным контактным устройством, выполнен-
Рис. 271.
Колонна Видмера с концентрическими трубами и стеклянной спиралью.
Рис. 272.
Колонна Дафтона со спиралью переменного шага.
Рис. 273.
Колонна Леки—Эвела со спиралью из металлической сетки.
Рис. 274.
Спиральное контактное устройство Бауэра—Кука, выполненное из металлической сетки.
12 Э. Крель
353
Таблица 51.
Характеристика колонны Бауэра и Кука со спиральным контактным устройством из металлической сетки
Диаметр колонны 5 мм; высота колонны 430 мм.
Исследованная смесь: н-гептан —метилцнклогексан
Нагрузка, мл/ч ВЭТС, см Удерживающая способность по жидкости, приходящаяся на 1 теор. ступень разделения, мл
38,5 1.88 0,045
63,0 2,28
84,0 2,54
110,0 2,79 0,066
ным в виде спирали из металлической сетки [53]. В этой колонне полоса металлической сетки намотана винтообразно на стеклянный стержень (рис. 273). ВЭТС у данной колонны лежит в интервале от 1 до 5 см, а удерживающая способность примерно составляет 0,5 мл на одну ступень разделения. Сравнительно сложный способ изготовления контактного устройства Леки и Эвела описан в работе Сталкупа с сотр. [54]. Более просто изготовляется специальная спиральная насадка из металлической сетки, предложенная Бауэром и Куком [55]. Этой насадкой обычно снабжают колонны, диаметр которых не превышает 5 мм. Проволочную сетку 50 меш из монель-металла сгибают под углом 90° таким образом, что она образует расположенные друг за другом вертикальные плоские пластины, между которыми располагаются горизонтально пластины, образующие два открытых сегмента (рис. 274). Такую спираль удобно вставлять в колонну, выполненную из калиброванной трубы, втягивая ее внутрь с помощью медной проволоки, прикрепленной к одному из концов спирали. Стенки колонны предварительно смачивают маслом, которое удаляют после установки насадки с помощью растворителя. Медную проволоку растворяют в концентрированной азотной кислоте. Характеристика колонны этого типа приведена в табл. 51.
Следует особо отметить, что подобные колонны обладают чрезвычайно малой удерживающей способностью по жидкости и сравнительно низким гидравлическим сопротивлением. Эти преимущества позволяют применять данные колонны для вакуумной ректификации при остаточных давлениях до 1 мм рт. ст. и загрузке исходной смеси от 6 до 15 мл.
Сложной в изготовлении, но более эффективной является колонна Подбильняка с проволочной спиралью [56]. Проволочную спираль укладывают по винтовой линии вокруг центрального стержня. Эта спираль, известная как насадка «Хэли-грид», должна плотно прилегать к стенкам колонны, чтобы жидкость не стекала 354.
Таблица 52.
Характеристика колонн Подбильняка с насадкой «Хэли-грид»
Диаметр колонны, мм Нагрузка, мл/ч ВЭТС, см Удерживающая способность по жидкости, приходящаяся на 1 теор. ступень разделения, мл
11,0 200 0,9 0,07
245 1,14
315 1,4
375 1,64 Предел захлебывания
25,0 500 1,0 0,33
1000 1,25 0,53
1500 1,58 0,76
2000 1,90 1,07
по ним (рис. 275). Насадку «Хэли-грид» изготовляют диаметром 8—30 мм из проволоки толщиной 0,25 мм, оставляя зазор между соседними витками до 0,25 мм. Характеристика этих колонн приведена в табл. 52.
Модификация насадки Подбильняка, предложенная Брезина, выполнена в виде трубы из стеклоткани, спирально намотанной на центральный стержень, также обернутый стеклотканью. ВЭТС для данной насадки лежит в интервале от 1,6 до 2,37 см. Высокая эффективность этих колонн обусловлена образованием тонкой пленки жидкости между отдельными витками за счет капиллярных сил [57 ].
Чрезвычайно малая ВЭТС даже при сравнительно больших нагрузках позволяет рекомендовать колонну Подбильняка для аналитической ректификации. Подобные колонны наряду с много-
Рис. 275.
Колонна Подбильняка с проволочной спиралью (насадка «Хэли-грид»).
Рис. 276.
Насадка Стедмана из металлической сетки (а) и модифицированная насадка Коха—Ваи-Рэя (б).
12*
355
Таблица 53.
Характеристика колонн с насадкой Стедмана и с насадкой, модифицированной Кохом и Ван-Рэем
Диаметр колонны 25 мм, высота насадки 1 м
Тип насадки Удельная нагрузка, мл/(см2 • ч) Флегмовое число ВЭТС, см
Насадка Стедмана 30 ОО 1,25
100 1,96
200 2,42
220 2,54
Модифицированная насадка 25 40 1,43
50 100 1,59
120 100 2,35
трубчатыми и трубчатыми колоннами с кольцевой щелью являются наиболее эффективными из всех известных колонн.
Насадка Стедмана [58], выполненная в виде конусов из металлической сетки (рис. 276а), обладает высокой эффективностью, малой удерживающей способностью по жидкости и значительной пропускной способностью. Однако при сборке такой насадки трудно обеспечить правильное размещение конусов, чтобы избежать растекания жидкости к стенкам колонны. Отверстия для прохода пара А в смежных конусах смещены относительно друг друга. Эту насадку можно использовать в колонне, выполненной только из калиброванной трубы.
Кох и Ван-Рэй [59 ] предложили более простую модификацию насадки Стедмана, которая состоит из сферических элементов, придающих насадке большую эластичность (рис. 2766). Отбортованная пружинящая кромка элемента плотно прижимается к стенкам колонны, поэтому можно использовать трубы с отклонением размера диаметра от 0,5 до 1 мм. Значения ВЭТС у оригинальной насадки Стедмана и ее модификации практически равны между собой, что было установлено при исследовании процесса ректификации смеси «-гептан — метилциклогексан (табл. 53). Удерживающая способность этих колонн по жидкости при нагрузках 190— 500 мл/(см2-ч) составляет 0,2—0,75 мл на одну теоретическую ступень разделения.
В соответствии с работой Брэгга [60] число теоретических ступеней разделения для насадки Стедмана диаметром 10— 3300 мм можно определить по следующей формуле
пул = 9,2 + 31/(0,277?) + 5,04/(0,97?) (190)
где Луд — удельное число теоретических ступеней разделения, равное числу теоретических ступеней разделения, приходящихся на 1 м высоты слоя насадки; 7? — расход флегмы при рабочей температуре, л/ч.
Еще более простой в изготовлении является колонна с насадкой из наклонных орошаемых пластин [61 ]. Элементы насадки 356
Таблица 54.
Характеристика колонны с насадкой из наклонных орошаемых пластин
Диаметр, мм Число наклонных пластин, шт. Расход флегмы, мл/ч ВЭТС, см Удерживающая способность по жидкости, приходящаяся на 1 теор. ступень разделения, мл
15 230 250 1,82 0,4
30 115 400 2,40 0,83
в виде овальных пластин располагают в колонне зигзагообразно друг за другом. Материал насадки — проволочная сетка из нержавеющей стали V4A. Отверстия для прохода пара на смежных элементах насадки смещены относительно друг друга (рис. 277). Характеристики колонн с такой насадкой диаметром 15 и 30 мм приведены в табл. 54. •
Благодаря сравнительно высокой эффективности и малого гидравлического сопротивления колонны с регулярными насадками находят все более широкое применение в пилотных и полупромышленных установках для вакуумной ректификации при остаточных давлениях до 1 мм рт. ст. Некоторые насадки обеспечивают практически неизменяющиеся газо- и гидродинамические условия движения потоков фаз (см. разд. 4.2), вследствие чего разделяющая способность насадки остается постоянной при широком интервале диаметра колонны.
На основе насадки Стедмана был разработан ряд насадок, изготовляемых из различных тканей. Сведения об этих насадках можно найти в работе Эллиса и Варяванди [62]. Юхайм [63] испытывал ректификационную установку с колонной, снабженной насадкой «Мультифил». Пакет насадки представляет собой металлическую сетчатую ленту (сталь V4A), свернутую в архимедову спираль. Такое выполнение насадки позволяет равномерно распределить жидкость по ней за счет капиллярных сил и обеспечивает значительное свободное значение для прохода газа (рис. 278). Стандартизованные пакеты насадки
Рис. 277.
Колонна с насадкой из наклонных орошаемых пластин.
Изготовитель: фирма Эрнст Хааге, Мюльхайм (Рур).
357
имеют длину 100 мм и диаметр от 25 до 87 мм. Пакеты насадки устанавливают в колонне друг на друга до достижения слоя необходимой высоты. При электрическом компенсационном обогреве стбнок колонны слой насадки высотой 1 м имел эффективность, равную 60 теоретическим ступеням разделения, что соответствует ВЭТС 1,66 см (разделяли эталонную смесь о-кси-лол—лг-ксилол).
В промышленных установках обычно применяют насадку типа «Гиперфил» с высотой пакетов 100 мм и диаметром от 100 до 375 мм (максимально 900 мм). Эта насадка хорошо распределяет жидкость при диаметре колонны до 300 мм. В колоннах большего диаметра необходимо устанавливать специальные распределители жидкости (рис. 279). Хубэр с сотр. [64] разработал преимущественно для промышленных колонн пакетную насадку типа «Зульцер», которая изготавливается из металлических или из стекловолокнистых сеток и имеет систему регулярно распределенных проточных каналов (разд. 4.2), наклоненных к оси колонны.
Тимофеев и Аэров [65] провели систематическое исследование колонн с сетчатыми насадками диаметром 28 и 150 мм для скорости
Рис. 278.
Насадка из металлической сетки типа «Мультнфил» .
Изготовитель: фирма Юхайм, Золинген.
Рис.. 279.
Колонна с насадкой «Гиперфил» .
Изготовитель; фирма Юхайм. Золинген.
Рис. 280.
Схема распределителя жидкости лабораторной пленочной колонны.
358
Рис. 281.
Пакеты пленочной насадки ACV диаметром 200 мм.
потока пара от 0,07 до 0,47 м/с. В своих исследованиях они изменяли угол наклона гофров, их глубину, а также удельную объемную поверхность насадки.
Весьма близки к рассмотренным сетчатым насадкам некоторые другие пленочные насадки (часто очень дорогостоящие). Необходимым условием их успешного применения является обеспечение равномерного распределения жидкости по стенкам насадки. Насадки выполняют из металла, стекла или керамики. По данным Штаге, сотовые насадки для лабораторных и промышленных колонн изготавливают в виде пакетов высотой 200 и диаметром 25—150 мм с размерами каналов 6 X 6 и 10 X 10 мм; стандартная высота слоя насадки составляет 1000 мм. На рис. 280 представлена схема распределителя жидкости лабораторной пленочной колонны, а на рис. 281 показан пакет пленочной насадки ACV диаметром 200 мм.
Квасняк [65а] предложил насадку, состоящую из металлических пластин, в которых выштамповано большое число уступов, расположенных зигзагообразно.
Колонны, работающие при прямотоке паровой и жидкой фаз, разработаны Жаворонковым и Малюсовым [66]. Они предложили следующие контактные устройства: насадки с вертикальными каналами круглого или прямоугольного сечения, в которых потоки жидкости и пара движутся снизу вверх, и насадки с каналами, снабженными направляющими устройствами, обеспечивающими перекрестное движение потоков фаз за счет спиралеобразного течения и диспергирования потока жидкости. В работе этих авторов приведены экспериментальные данные и методы расчета колонн с такими контактными устройствами. Указано также, что наибольшая трудность при эксплуатации данных колонн заключается в необходимости предотвращения уноса жидкости с одной ступени разделения на другую.
7.3.5. КОЛОННЫ С ВРАЩАЮЩИМИСЯ КОНТАКТНЫМИ УСТРОЙСТВАМИ
Колонны с вращающимися контактными устройствами, или так называемые роторные колонны, известны с 1925 г. В работе Иоста с сотр. [73] рассматривается вопрос о перспективах раз-
359
Рис. 282.
Установка Абегга с колонной, снабженной вращающейся лентой.
а ~ принципиальная схема колонны; б — общий вид автоматизированной лабораторной установки.
Изготовитель: фирма В. Бюхи, Флавиль, Швейцария.
работки роторных колонн и о преимуществах колонн этого типа по сравнению с тарельчатыми, насадочными и колоннами других типов.
Разработка роторных колонн была вызвана необходимостью повышения эффективности разделения без увеличения удерживающей способности по жидкости и гидравлического сопротивления проходу потока паров. Такие высокоэффективные колонны особенно необходимы при микроперегонках с загрузкой смеси 1—5 г.
По конструктивному выполнению роторные колонны можно подразделить на следующие группы (см. разд. 5.1.1): а) колонны с вращающейся металлической лентой (рис. 282), которая может иметь плоскую, спиральную или крестовидную форму; б) колонны с вращающимся цилиндром (ширина щели между цилиндрическим корпусом колонны и вращающимся цилиндром составляет 1—2 мм [см. рис. 2861); в) колонны с цилиндрическим конденсатором, вращающимся внутри обогреваемого цилиндрического корпуса колонны (рис. 283); г) колонны с вращающимися и не
360
подвижными пластинами (или конусами), соединенными соответственно с валом и корпусом колонны (рис. 284); д) колонны с вращающейся спиралью * (рис. 285).
Первая лабораторная роторная колонна с вращающимися и неподвижными пластинами разработана Мейерсом и Джонсом. Урей и Хафмэн предложили для проведения процесса обогащения изотопа кислорода 18О роторную колонну диаметром 15 см и высотой 10,7 м, основанную на том же принципе. Эффективность колонны Урея и Хафмэна составила 500 теоретических ступеней
* Модификацией колонны этого типа является пленочная роторно-спиральная колонна, в которой на вертикальном валу установлены одно- или многоза-ходные рулоны отбортованных по краям лент. Пар проходит между витками рулона перпендикулярно отбортованным лентам (см. Авт. свидетельство СССР № 203621, 1967 г., патент США № 4038353, 1977 г. или патент ГДР № 126495, 1977 г.). В СССР колонны этого типа диаметрами от 240 до 2000 мм применяются для вакуумной ректификации термически нестойких веществ. — Прим. ред.
Рис. 283.
Колонна Байрона, Баумана н Коула с вращающимся конденсатором:
1 — электропривод конденсатора; 2 — холодильник конденсатора; з — охлаждаемая ловушка; 4 — обогреватель стенок колонны; 5 —вращающийся конденсатор, охлаждаемый водой; 6 — куб; 7 — обогреватель куба.
Рис. 284.
Колонна Мейерса—Джонсона с вращающимися и неподвижными пластинами.
361
Рис. 285.
Суперректнфнкатор Подбильняка со спиральным ротором центрифужного типа.
разделения. Лезесне и Лохте, разместив в трубчатой лабораторной колонне вращающуюся металлическую ленту, создали первую модель роторных колонн этого типа, которую затем неоднократно модифицировали различные исследователи.
На рис. 282а показана принципиальная схема колонны Абегга с вращающейся лентой. В этой колонне ВЭТС равна 2,5—3 см, что достигается благодаря диспергированию жидкости на мелкие капли и обеспечению противотока паровой и жидкой фаз. Лента вращается с частотой 1000—3500 об/мин. Удерживающая способность по жидкости этой колонны с внутренним диаметром 6 мм и высотой 375 мм составляет лишь 0,2 мл, а перепад давления потока пара, приходящийся на 1 м высоты колонны, равен 0,5 мм рт. ст. Благодаря таким характеристикам колонны этого типа удобно применять при ректификации очень высококипящих веществ для предотвращения их разложения. Технологические и геометрические параметры колонн различных типов с вращающейся лентой приведены в табл. 33, 55—57. Необходимо отметить, что производительность подобных колонн при работе под вакуумом значительно уменьшается, например при снижении абсолютного рабочего давления от 760 до 10 мм рт. ст. она уменьшается примерно на 70% [67].
Автоматизированная лабораторная ректификационная установка Абегга (см. рис. 2826) имеет колонну диаметром 6,5 мм и высотой 550 мм, снабженную спиралевидной лентой из нержавеющей пружинной стали или тефлона, частота вращения которой равна 1400 об/мин. Регулирование подачи флегмы в этой колонне 362
Таблица 55
Характеристика колонн с вращающимися контактными устройствами (см. также табл. 33)
[73] [25] [71] [69] о [25] [741
Примечания ’ Ширина кольцевого зазо- pa 1 мм Ширина кольцевого за- : зора 1—2 мм То же, 1 мм Ширина кольцевого за- i эора 1,09 мм Ротор высотой 1 м и диа- метром 25 мм. Соотноше- ние количеств тепла, под- веденных к стенкам ко- лонны и к кубу, 20 : 1 621 вращающихся эле- ментов 1 125 вращающихся и 124 неподвижных элементов 175 пар конических сег- ментов
Удерживающая способность по жидкости. Гидравлическое сопротивление 0,17 мл на 1 теор. ступень раз- деления. 0,5 мм рт. ст. на 43 теор. I ступени разделения 17,5 мл 1 2,0 мм рт. ст. Гидравлическое 1 сопротивление 0,2—0,5 мм рт. ст.
ним/ро ‘edoiod KHHatneda BlOlOBh 2600 800 6000 4000 250/1500 700/1700 320
(по 'ме он) 'о
О1ГЭИЬ аоаонаап’ф 8 8 ю О «О фле! 280 •е
h/irw 'хкин -В11Ч11ЭИ HdLI BMeAdjen 069 001 О о о о со О СО 210
wo *эхея ем 0,9 0,935 4,45 2,56 СМ 1,28 0,93
KHHairatfeed ВэиэиЛхэ •doax oiroHh СМ □О 38,9 500
‘1ЧИНО1ГОЯ diawuHl/ см Fw. OS ю 200— 240
WO ‘1ЧННО1Г -ом eiooiqg 58,4 О 00 001 1070 451
О fe О
я я
Тип ротора Цилиндр CU =1 Я 5 Я Д’ Си Я Я я д »я я £ 2 я я CU CQ си о я я <и =1 я о я Вращающиш CU о я я о КС я о я я я 8 я S я о 2 я <и я я <и я я я о 2 я <и я я 8 я я я о н я <и
Таблица 56.
Оптимальная частота вращения ротора в колоннах с вращающимися контактными устройствами
Тип колонны Тип ротора Нагрузка, мл/ч Рабочий интервал частоты вращения ротора, об/мии Оптимальная частота вращения ротора, об/мин ^Литера» тура
С вращающим- Цилиндр со щет-
ся цилиндриче- ками
ским ротором 36 690 400—2600 2000 [73] *
36 1330 400—2600 2000
4с 690 800—3200 2600
4с 950 400—3000 2600
4с 1170 400—2400
4с 1330 400—1600
Гладкий цилиндр; 1550 0—4000 2600 [71] **
ширина кольце- 2000 0—4000 2600
вого зазора 3000 0—4000 2600
1,09 мм 4000 0—4000 2600
С вращающей- Звездообразный 150 0—2000 1400 [75] ***
ся лентой
* Эффективность разделения при превышении оптимального числа оборотов ротора резко снижается. ♦♦ В интервале 1400—2000 об/мин эффективность разделения остается постоянной. *** При превышении 2600 об/мин эффективность разделения не уменьшается. В интервале 2200—2600 об/мин кривые эффективности поднимаются очень полого, превращаясь затем в прямые линии. Теоретически при нагрузке 3000 мл/ч кривая эффективности при превышении 2600 об/мии должна подниматься более полого.
Таблица 57.
Перепад давления в роторных колоннах
Тип ротора колонны Высота колонны, см Диаметр колонны, мм Рабочее давление, мм рт. ст. Перепад давления, Др-103 мм рт. ст. Литература
иа 1 см высоты колонны иа 1 теор. ступень разделения
Ленточный 545 6,7 1 1,3 [75]
82 20,0 1 2,0 34,4— 710
145 15,0 0,5 2,0 4,3
Цилиндр 11,5
Конденсатор 80 0,005— 1,5 4,4 20,0 [69]
осуществляется с помощью качающейся капельницы. Эффективность колонны составляет 30 теоретических ступеней разделения при нагрузке 70 мл/ч, удерживающая способность по жидкости равна 1,2 мл.
Автоматизированная установка, выпускаемая фирмой Эрнст Хааге (Мюльхайм/Рур) предназначена для микроперегонки в вакууме; она имеет колонну с вращающейся лентой. Колонна рассчитана на загрузку разделяемой смеси в объеме от 2 до 100 мл. Она может быть выполнена из кварца, высота ее составляет400 мм (^25 теоретических ступеней разделения) или 1000 мм («^50 теоретических ступеней разделения).
Суперректификатор Подбильняка центрифужного типа со спиральным ротором [68 ] имеет более высокую производительность по сравнению с другими роторными колоннами (см. рис. 285).
Во всех описанных выше колоннах жидкость за счет центробежных сил, создаваемых при вращении ротора, отбрасывается на стенки корпуса колонны в виде аэрозоля. Мелкодисперсный аэрозоль жидкости имеет большую поверхность, что обусловливает интенсивный массо- и теплообмен между жидкостью и восходящим потоком паров. Очевидно, что в таких колоннах несколько возрастает гидравлическое сопротивление проходу потока паров *.
В отличие от рассмотренных колонн Беннер с сотр. [69] предложил другое конструктивное решение. Он поместил вращающийся металлический конденсатор в колонну с обогреваемыми стенками. Эффект разделения в этой колонне возникает как за счет противоточного взаимодействия фаз в поле центробежных сил, так и за счет ряда последовательно протекающих и взаимосвязанных процессов парциальной конденсации и испарения. Байрон с сотр. [70] описал колонну аналогичной конструкции, он разработал лежащую в основе этого принципа разделения теорию «термической ректификации» (см. рис. 283 и разд. 5.4.3).
Прототипом колонн с вращающимся цилиндром являются трубчатые колонны с кольцевой щелью (см. разд. 7.3.1). В этих колоннах также имеется канал кольцевого сечения шириной 1— 2 мм с той, однако, разницей, что внутренний цилиндр вращается для обеспечения интенсивного перемешивания потока пара. Колонны данного типа были предложены Виллингэмом с сотр. [71 ]. Позднее разработкой подобных колонн занимался, главным образом, Пост [72]. На рис. 286 показана схема предложенной им колонны. Пост создал теорию о том, что эффект разделения при перегонке в меньшей степени зависит от степени турбулизации потока пара и обусловлен в основном равномерным распределе-
* Предлагаемый автором механизм интенсификации массообмена не является исчерпывающим, так как не во всех роторных колоннах образуется аэрозоль (капли) жидкости. Кроме того, автором не учитывается значительная турбулизация паровой фазы, что в подавляющем большинстве случаев имеет решающее значение. — Прим. ред.
365
Рис. 286.
Колонна Иоста с вращающимся цилиндром:
/ — куб; 2 — шарикоподшипники; 3 — цилиндрический ротор; 4 — штуцер для отбора дистиллята; 5 — вал привода;
6 — охлаждаемый кожух; 7 — карман для термопары; 8 — кольцевой канал; 9 — фланцевое уплотнение; 10 — дифференциальная термопара; // — компенсационный электронагреватель; 12 — слой изоляции.
нием пленки жидкости по стенкам колонны. На основе этой теории Иост предложил [73 ] снабжать цилиндрические роторы щетками (см. рис. 201), которые обеспечивают более равномерное распределение жидкости *. Из работ Куна [24 ] и Иоста [73 ] следует, что наибольший эффект разделения в роторных колоннах можно получить при равномерном
распределении жидкости по стенкам колонны и небольшой ширине кольцевого зазора, обеспечивающей снижение сопротивления массо- и теплопереносу в паровой фазе.
Необходимым условием эксплуатации роторных колонн всех типов является адиабатичность процесса и незначительное колебание частоты вращения ротора**. Имеются различные точки зрения относительно оптимального числа оборотов ротора и зависимости эффективности от частоты вращения ***. Из данных табл. 55 видно, что частота вращения ротора изменяется в широких пределах от 250 до 6000 об/мин. Согласно работе Иоста [73] число теоретических ступеней разделения зависит как от производительности роторной колонны, так и от частоты вращения и формы ротора. В табл. 56 приведены оптимальные числа оборотов ротора при различной производительности колонн. Соответствующие зависимости представлены на рис. 287—289.
Иост показал, что число теоретических ступеней разделения резко уменьшается при превышении оптимального числа оборотов цилиндрического ротора (см. рис. 288 и 289). Виллингэм с сотр. [71 ], напротив, установил, что число теоретических ступеней разделения непрерывно возрастает при увеличении частоты вращения ротора выше оптимальной (см. рис. 287а). В США
* Необходимо отметить, что работы по созданию роторных колонн с использованием принципа одновременной турбулизации жидкой и паровой фаз с обеспечением равномерного смачивания стенок и обновлении поверхности жидкости были впервые проведены в СССР в Государственном научно-исследовательском и проектном институте азотной промышленности и продуктов органического синтеза и Физико-химическом институте им. Л. Я. Карпова в период 1948— 1952 г. — Прим. ред.
** Это противоречит описанному выше (с. 365) принципу термической ректификации. — Прим. ред.
*** Правильнее связывать эффективность с окружной скоростью вращешя ротора, так как при этом будет учтен диаметр колонны. — Прим. ред.
366
Рис. 287.
Зависимости эффективности роторных колонн от нагрузки по жидкости и частоты вращения ротора:
а — для колонны с вращающимся цилиндром [71]; б — Для колонны со звездообразным ротором [75];
/, 2, 3, 4 — опытные кривые, полученные при различных нагрузках; 5 — теоретическая кривая
а), об/мин
Рис. 288.
Зависимость числа теоретических ступеней разделения от нагрузки и частоты вращения цилиндрического ротора (тип ЗЬ), полученная Иостом.
Рис. 289.
Зависимость числа теоретических ступеней разделения от нагрузки и частоты вращения цилиндрического ротора (тип 4с), полученная Иостом.
должна быть создана колонна с вращающимся цилиндром, которая при частоте вращения ротора 8000 об/мин и пропускной способности 4 л/ч будет иметь эффективность в 500 теоретических ступеней разделения. Оптимальное число оборотов ротора зависит также от конструктивных особенностей колонны и, особенно, ротора (см. табл. 56). Гельперин и Хаценко [74] установили, что в колонне с вращающимися и неподвижными конусами увеличение частоты вращения ротора выше 320 об/мин не влияло на эффективность разделения. Эта скорость вращения является оптимальной для колонн подобного типа; Гафмэн и Юри также нашли, что в интервале 250—1500 об/мин частота вращения ротора не оказывает заметного влияния на эффективность колонны *
Для колонн с вращающейся лентой оптимальная частота вращения ротора по различным данным лежит в интервале от 1000 до 35Q0 об/мин, причем на нее оказывает значительное влияние конструктивное выполнение ротора. Так, Мюррей [75], показал, что для колонн со звездообразным ротором оптимальная частота вращения равна 1400 об/мин при нагрузке 150 мл/ч. При дальнейшем увеличении числа оборотов ротора эффективность разделения остается постоянной (см. рис. 2876).
Следует отметить, что время установления стационарного режима работы колонны составляет для колонн с вращающейся лентой 0,5—1 ч, для колонн с цилиндрическим ротором не превышает, поданным Иоста [73], 1 ч. ИрЛин и Брунс [76] установили, что для колонны с вращающимся цилиндром при ВЭТС, равной 1 см, время выхода на стационарный режим составляет всего 6 мин **.
Как уже указывалось выше, гидравлическое сопротивление проходу паров для насадочных колонн зависит от типа и размеров насадки, нагрузки и давления и лежит в интервале 0,03— 1,3 мм рт. ст. на одну теоретическую ступень разделения, что соответствует 0,005—0,2 мм рт. ст. на 1 см высоты колонны. Для удобства сравнения в табл. 57 приведены данные по гидравлическому сопротивлению различных роторных колонн. Приведенные данные достаточно отчетливо подтверждают преимущество роторных колонн, гидравлическое сопротивление которых, по сравнению с насадочными и, особенно, тарельчатыми колоннами на несколько порядков ниже.
Другие характеристики роторных колонн примерно совпадают с соответствующими параметрами трубчатых колонн с кольцевой щелью, многотрубчатых колонн, а также колонн с проволочной сетчатой и спиральной насадками (см. разд. 7.3.1 и 7.3.4).
Из сравнения различных роторных колонн однозначно следует, что наиболее пригодной для лабораторных установок яв
* См. третье примечание па с. 366. — Прим. ред. 5
** Указанное время является, по-видимому, заниженным.—Прим. ред.
368
ляется колонна с цилиндрическим ротором (см. рис. 287а) *. При использовании этих колонн обычно необходимо обеспечивать относительно большую частоту вращения ротора. При работе в вакууме возникает затруднение, связанное с необходимостью обеспечения уплотнения быстро вращающегося вала. Этого затруднения можно избежать, если использовать электромагнитный привод. Для колонны с вращающейся спиралью Хёхер [77 ] исследовал зависимости числа теоретических ступеней разделения и удерживающей способности по жидкости от частоты и направления вращения спирали.
Нейман [78] анализировал пропускную способность промышленной роторной колонны диаметром 500 мм, состоящей из восьми контактных ступеней. Исследования промышленных роторных колонн, предназначенных для работы в вакууме, провел Мамин с сотр. [78а].
7.4. ХОЛОДИЛЬНИКИ И ДЕФЛЕГМАТОРЫ
В лабораторных и пилотных ректификационных установках холодильники применяют для следующих целей: полной конденсации паров (конденсаторы), частичной конденсации паров (дефлегматоры), охлаждения потоков жидкостей (холодильники).
Во всех этих случаях хладоагент, в качестве которого часто используют воду, служит для приема и отвода тепла. Таким образом, холодильник является теплообменником, который при необходимости может использоваться и для нагревания потока жидкости. Как и в промышленных установках, подобные теплообменники используют для подогревания исходной смеси. Если температура затвердевания дистиллята выше температуры охлаждающей воды из водопровода, то в качестве хладоагента следует использовать воду из термостата, температура которой должна быть выбрана таким образом, чтобы исключалось выпадение твердых частиц дистиллята в холодильнике. Дефлегматором называют такой холодильник, в котором путем регулирования расхода охлаждающей воды конденсируют лишь часть потока пара. Образующийся конденсат подают в качестве флегмы в колонну, а не-сконденсировавшийся остаток паров полностью конденсируют в конденсаторе и отбирают в качестве дистиллята (см. разд. 5.2.3).
Важнейшие типы лабораторных холодильников, стандартизованные по конструктивному выполнению и размерам, приведены в табл. 58.
* На основании долголетнего опыта мы считаем, что наиболее простыми в изготовлении и надежными в работе являются лабораторные роторные колонны с бессальниковым экранированным электромотором и ротором в виде гибкого «ерша». Они позволяют плавно регулировать число оборотов и не нуждаются в подшипниках благодаря свойству ротора самоцентрироваться (см. Голубев И. Ф., Мещеряков Н. В., Олевский В. М., Труды ГИАП, вып. V, 1956, с. 316—329;. — Прим. ред.
369
Таблица 58.
Стандартные холодильники для ректификационных установок
Стандарт Наименование
TGL 40-344 группа 7 Холодильники, интенсифицированные холодильники
8339 группа 1 Холодильники, технические условия изготовления
группа 2 Холодильники Либиха
группа 3 Шариковые холодильники
группа 4 Холодильники Димрота (Все указанные холодильники выполняются со стандартными шлифами или без них)
DIN 12575/53 Холодильники Либиха с кожухом, припаянным к охлаждаемой трубе
12576/56 Холодильники Либиха со стандартными шлифами
12580/53 Шариковые холодильники
12581/56 Шариковые холодильники со стандартными шлифами
12585/57 Змеевиковые холодильники
12586/55 Змеевиковые холодильники со стандартными шлифами
12591/54 Холодильники Димрота со стандартными шлифами
В основном различают холодильники двух типов. Это холодильники с наклонной охлаждаемой трубой (см. рис. 238, 5) и холодильники с вертикальной охлаждаемой трубой (см. рис. 238, 11), причем пары или охлаждающую жидкость можно вводить в холодильник как сверху, так и снизу. При вводе паров в холодильник сверху происходит наиболее интенсивное охлаждение, поскольку при этом пары проходят вдоль всей охлаждающей поверхности. Однако в этом случае возникает опасность, что конденсат переохладится. Этот вариант ввода паров применяют преимущественно при обезвоживании органических жидкостей, так как в отличие от варианта с нижним вводом сконденсировавшиеся капли воды не будут зависать в холодильнике, а будут смываться последующими порциями конденсата. В свою очередь вертикальные холодильники с нижним вводом паров обладают теми преимуществами, что в них конденсат не переохлаждается и дистиллят не смешивается с восходящим потоком несконденсировавшихся паров. Охлаждающую воду и пары обычно подают в холодильник противотоком.
В зависимости от формы выполнения охлаждающей поверхности различают следующие типы холодильников.
1) Холодильники с гладкой трубой: а) воздушные холодильники (см. рис. 238, 3 и 9) для жидкостей с высокими температурами кипения и затвердевания; б) холодильники Либиха с рубашкой 370
сравнительно большого диаметра (см. рис. 238, 5); в) холодильники Веста с рубашкой малого диаметра (рис. 290).
Интенсифицированный воздушный холодильник (рис. 291), используемый при перегонке для охлаждения жидкостей с температурой кипения по меньшей мере на 25 °C выше температуры окружающей среды, описал Тёльдте [79]. Пары поднимаются по трубе 1 и конденсируются на поверхностях кожуха 2 и трубы 4. В результате разогревания в трубе 4 возникает вертикальный ток воздуха, обеспечивающий непрерывный подвод более холодного воздуха из окружающего пространства. Гидрозатвор 5 заполняют на V5 высоты высококипящей жидкостью. Давление выравнивается путем подсасывания воздуха через штуцер 3. В холодильнике, состоящем из трубы и соосного охлаждающего кожуха, целесообразно, чтобы между трубой и кожухом был возможно меньший зазор, как в холодильнике Веста. Данные холодильники изготавливают также с охлаждаемыми шлифами (см. рис. 290).
Рис. 290.
Холодильник Веста с вмятинами на внутренней трубе небольшого диаметра и охлаждаемыми шлифами.
Рис. 291.
Воздушный холодильник Тёльдте:
/ — труба для подвода паров дистиллята; 2 — кожух; 3 — штуцер для подвода воздуха; 4 — внутренняя труба; 5 — гидрозатвор.
Рис. 292.
Холодильник Шотта с волнистой поверхностью охлаждаемой стенки.
Изготовитель: народное предприятие «Йенский завод стекла Шотт», Иена.
371
Рис. 293.
Холодильник Штаеделера с кожухом для твердых хладоагентов.
Рис. 294.
Интенсифицированный холодильник, составленный из холодильников Либиха и Димрота.
Рис. 295.
Холодильник и охлаждаемая приставка к кубу в виде колбы, разработанные Хенкелем.
Рис. 296.
Дефлегматор с изменяющейся охлаждаемой поверхностью [80 1.
2) Холодильники с развитой охлаждаемой поверхностью: а) холодильник Веста с вмятинами на охлаждаемой стенке (см. рис. 290); б) холодильник Шотта с волнистой поверхностью охлаждаемой стенки (рис. 292); в) шариковый холодильник Аллина (см. рис. 238, 8); г) холодильник Фридрихса с винтовой поверхностью охлаждаемой стенки (см. рис. 238, 7).
Большую охлаждающую поверхность на сравнительно небольшой длине можно получить, если применять охлаждаемую трубу с вмятинами или выполнять поверхность охлаждаемой стенки волнистой, шаровой или винтообразной.
3) Змеевиковые холодильники: а) змеевиковый холодильник с подачей хладоагента в кожух (см. рис. 238, 10)', б) холодильник Димрота с подачей хладоагента в змеевик (см. рис. 238, 11)', в) холодильник Штаеделера с кожухом для твердых хладоагентов (рис. 293); г) интенсифицированный холодильник с комбинированными охлаждаемыми поверхностями.
Змеевиковые холодильники наиболее пригодны для охлаждения потоков жидкости или пара при атмосферном давлении. При вакуумной ректификации вследствие значительного увеличения скорости потока паров легко возникают очаги подвисания капель жидкости. Поэтому при вакуумной перегонке лучше использовать холодильники Димрота, обладающие высоким коэффициентом теп-372
лопередачи. Холодильники Штаеделера преимущественно применяют для конденсации низкокипящих веществ, поскольку они позволяют использовать в качестве хладоагента лед или твердый диоксид углерода. Для этой же цели предназначены интенсифицированные холодильники с комбинированной охлаждаемой поверхностью, в которых паровой поток охлаждается как изнутри, так и снаружи. Такие холодильники изготавливают из трубы с волнистыми стенками, которую вставляют в гладкостенную трубу (см. рис. 292), или из комбинации холодильников Либиха и Димрота (рис. 294).
К холодильникам с развитой охлаждаемой поверхностью относится также холодильник Хенкеля [81 ], который представляет собой холодильник Либиха или Веста, свернутый в многовитко-вую спираль (рис. 295). Этот интенсифицированный проточный холодильник особенно пригоден для работы под вакуумом. При высоте 20 см он обладает охлаждающей поверхностью 500 см2 (см. для сравнения характеристики других холодильников в табл. 59).
Проточные холодильники обычно применяют в качестве дефлегматоров, однако при использовании этих холодильников возникают трудности в точной установке требуемого флегмового числа. Охлаждаемую поверхность дефлегматора, показанного на рис. 296, можно изменять, перемещая поршень путем вращения гайки с накаткой [80].
Для стеклянных холодильников минимальное значение коэффициента теплопередачи равно 0,5 кал/(см2-мин-°С). Необходимая охлаждаемая поверхность определяется по формуле:
f = Q/(Wcp) (191)
где Q — передаваемое количество тепла, кал/мин; k — коэффициент теплопередачи, кал/(см2-мин-°C); А/Ср — средняя разность температур между паром и хладоагентом, °C.
Среднюю разность температур Д/ср рассчитывают по соотношению:
(192)
где t — температура насыщенного пара, °C; и /2 — соответственно температура охлаждающей воды на входе в холодильник и выходе из него, °C.
Обширные исследования эффективности холодильников проведены Фридрихсом [83], Фридрихсом и Крузкой [84], а также Махом и Германом [85]. По данным Эйхгорна [86], коэффициенты теплопередачи для стеклянных холодильников длиной 200— 1000 мм изменяются в интервале от 1,5 до 0,5 кал/(см2-мин-°С). Известно, что коэффициент теплопередачи уменьшается с увеличением длины холодильника, поэтому целесообразно вместо одного длинного холодильника применять два холодильника меньших размеров. При этом воду в каждый холодильник необходимо подавать раздельно, поскольку коэффициент теплопередачи
373
Таблица 59.
Характеристики различных холодильников по данным Эйхгорна [861
(Э0-НИН)/1ГВЯ ‘вминчииИ1 -О1ГОХ КЭЧ-ООЕПЭХ OSOlS — ’frf CO 04 r-io io eo —, —, СЧ —• 04 04 — — 04 Tf 04 —. —« CO 04 04
(Эо • HHW • zWO)/LfBM ‘HhBtfadauoiruai хнаи'пиффео’Я 00 04 1Л LOLO LO CO 00ООЮШО4О4^Ь- 04 05 h- Г-- LO CO О О О Ф О О Г-- 04 CO Ю Tf О О 00 lO O4~ lO -Г о o d -" —" cd —- о —1 — о о о о — о
ww ‘MpAdx yowgBtfJKBirxo имнахо BHHtnLfoX 05 05 04 04 04 04 04^ Ф СЛ СО СО О СЭ О О4~
ни ‘rnpAdi HowgBtfJKBirxo dxaw -виЯ UHHHadiAHH 00 LO LO LO Ю lO СО cocococo"coo4cob>h^t^-b-ir^oo о Ь-О4Ь- со
ww ‘вхЛжоя dxawBHtf UHHJKAdBH LO 04 04 СО СО СО СО М" СО СО СО СО О О О 05 Tf 04
ww ‘вхКжоя oxaTn -енв^жв^хо вншг'П' LO О О О О О Ф о О О О О О О—1 LO LO OOOO — OJOOO’tlHlOO 04 LO Tf rf —СОГ'-004СОМ"04С004С00404 —
гиэ ‘qxooHXdaa -он UBWae^Birxo ю ООЮЮ сооооою о о о ф о 00 Ю 00 04 04 04 00 04 О оо —05 04 О— — oq Tf — CW-« 04 —< СО 04 04 04 ’Ф —* 04
WW ‘ВНИ1Г1/ 200 400 700 1000 200 300 400 200 300 200 300 200 1137 205 435
Mid BidBtfuBio dawoH L0 о Ю —' о ь- 00 ОО СП СП L0 L0 Ю LO ю 04 04 04 04 04
Тип холодильника Холодильник Либиха Шариковый холодильник Змеевиковый холодильник Холодильник Димрота Холодильник Штаеделера Интенсифицированный холодильник Шариковый холодильник Сокслета Йенский холодильник Шотта с волнистой поверхностью охлаждаемой стенки Холодильник Фридрихса с винтовой поверхностью охлаждаемой стенки Спиральный холодильник Бахмана
в длинном холодильнике уменьшается вследствие повышенного нагревания охлаждающей воды в нем и снижения вследствие этого средней разности температур между паром и водой *. Наиболее эффективными являются холодильник Димрота и холодильник Фридрихса с винтовой поверхностью, имеющие коэффициенты теплопередачи от 1,5 до 1,25 кал/(см2-мин-°С). Высокий теплосъем холодильника Шотта с волнистой поверхностью обусловлен его весьма большой охлаждаемой поверхностью, равней 1137 см2, в то время как коэффициент теплопередачи этого холодильника очень низок и составляет 0,575 кал/(см2-мин-°С).
Брезина [82] исследовал влияние конструктивных особенностей холодильника Либиха на их эффективность. Он предложил новые математические соотношения для определения эффективности таких холодильников и разработал аппаратуру для их испытаний.
Пользуясь значениями коэффициентов теплопередачи, приведенными в табл. 59 и формулой (191), можно рассчитывать геометрические характеристики холодильников всех типов. Следует иметь в виду, что при выборе холодильников по величине охлаждаемой поверхности необходимо, чтобы она превышала расчетное значение примерно на 50%. Геометрические характеристики лабораторных холодильников, выпускаемых промышленностью, изменяются в широком интервале.
Для правильного выбора охлаждаемой поверхности теплообменники полупромышленных и пилотных установок следует рассчитывать по возможности более точно. При расчете и изготовлении стеклянных элементов подобных холодильников можно пользоваться значениями коэффициентов теплопередачи, приведенными в табл. 60.
Для удобства сборки холодильников с любой охлаждаемой поверхностью стекольная промышленность выпускает охлаждающие элементы в виде пакетов змеевиков диаметром от 40 до 450 мм.
Таблица 60.
Коэффициенты теплопередачи для стеклянных холодильников
Вид теплообмена Коэффициент теплопередачи, ккал/(м2 ч °C)
Конденсация паров в холодильнике 250—300
Охлаждение жидкостей 150
Охлаждение газов 50
* Неточная формулировка, так как при снижении средней разности температур уменьшается не коэффициент теплопередачи, а теплосъем, т. е. количество тепла, отводимого в единицу времени. При увеличении высоты конденсатора снижение коэффициента теплопередачи может происходить из-за возникновения на нижней части поверхности теплообмена относительно толстой пленки стекающего конденсата. — Прим. ред.
375
7.5. ФОРШТОСЫ, ПРИСТАВКИ
ДЛЯ ДИСТИЛЛЯЦИОННЫХ АППАРАТОВ и головки КОЛОНН
Для отвода паров, образующихся в кубе при дистилляции, их конденсации и измерения температуры к кубу присоединяют фор-щтосы или приставки, использующиеся в качестве связующих элементов. При ректификации в колонну возвращают определенное количество флегмы и измеряют при этом флегмовое число. Для подобных измерений колонну снабжают специальной головкой.
При проведении непрерывного процесса ректификации требуются специальные патрубки для ввода газов и жидкостей в колонну. Конструкции форштосов, приставок и головок колонн весьма многочисленны, поэтому ниже рассмотрены лишь те, которые обладают принципиальными отличиями.
7.5.1. ФОРШТОСЫ
Форштосы являются связующим звеном между холодильником и сборником дистиллята (TGL9970—9972). Обычно они снабжаются штуцерами для присоединения к вакуумной линии (см. рис. 238, 6 и 12).
Особенно удобным является вакуумный форштос Аншюца— Тиле (см. рис. 238, 13). Этот форштос предпочтителен также при работах в условиях атмосферного давления, поскольку позволяет отбирать произвольное число фракций (см. разд. 7.2).
7.5.2. ПРИСТАВКИ для дистилляционных
АППАРАТОВ
При дистилляции приставки служат связующим звеном между кубом (в виде колбы) и конденсатором. Приставки обычно снабжаются термометрическими карманами. Приставки простейших видов показаны на рис. 238, 2 и 4. Наиболее предпочтительны приставки со стандартным шлифом NS14,5 для термометра. Они выполнены таким образом, что ртутный шар термометра при измерениях оказывается постоянно в одном и том же месте пароотводной трубы. Это особенно важно при сравнительной перегонке. Некоторые приставки стандартизованы. В табл. 61 приведены стандартные приставки. Некоторые из них показаны на рис. 297.
Приставка Кляйзена (TGL9979) снабжена двумя штуцерами со шлифами NS14.5. Правый штуцер предназначен для установки термометра со стандартным шлифом, а левый может использоваться для установки капилляра (TGL9978, DIN12404/63), мешалки или воронки для ввода жидкости (см. рис. 236). Эта приставка хорошо зарекомендовала себя при дистилляции под вакуумом и при атмосферном давлении. Правый штуцер можно удли-376
Таблица 61.
Стандартные приставки для дистилляционных аппаратов
Стандарт
Наименование
TGL 9974 Приставка с одним боковым штуцером
9975 Приставка с одним параллельным штуцером
9977 Перемычка со шлифом для термометра
9979 Приставка Кляйзена с боковым шлифом
9980 Приставка с припаянным холодильником Либиха
9981 Приставка с припаянным холодильником Димрота
13840 Приставка Праля
40—353 Приставка, применяемая при перегонке с водяным паром
DIN 12594/54 Приставки со стандартными шлифами
пять, выполняя его в виде короткой колонны с насадкой или спиралями Видмера, а также елочного дефлегматора (рис. 298). В этом случае колонна служит в качестве брызгоотделителя, как приставка Райтмайра (см. рис. 239, а) или сферическая приставка с кольцами Рашига (рис. 299). Для сильно вспенивающихся жидкостей разработаны специальные приставки (см. рис. 239,6). При перегонке с водяным паром применяют приставку Праля с трубой для ввода пара (рис. 300). Изготовляют также ряд при-
Приставки для дистилляционных аппаратов с одним боковым штуцером, TGL9974 (а), с одним параллельным штуцером, TGL 9975 (б) и перемычка со шлифом для термометра, TGL 9977 (в).
1 — муфта шлифа В по TGL 9967; 2 — конус шлифа В по TGL 9967; 3 — муфта шлифа A NS 14,5/23 по TGL 9967; 4 — конус шлнфа В NS 29/32 по TGL 9967.
Рис. 298.
Приставка Кляйзена с коротким елочным дефлегматором.
377
Рис. 299.
Сферическая приставка с кольцами Раш ига.
Рис. 300.
Приставка Праля с трубой для ввода пара.
Рис. 301.
Приставка с припаянным холодильником Либиха со штуцером для присоединения к вакуумному насосу и со шлифом для присоединения сборника дистиллята.
ставок с припаянными вертикальными (см. рис. 295) или наклонным холодильниками (рис. 301). Аппараты с наклонными холодильниками занимают сравнительно много места, поэтому чаще используют вертикальные холодильники. При использовании в качестве приставки перемычки (см. рис. 238, 4 и рис. 297в) в сочетании с винтовым холодильником или холодильником Дим-рота дистилляционный аппарат значительно компактнее.
Очень компактна приставка Хенкеля (см. рис. 296), изготавливаемая в различных модификациях [81 ]. Приставки других типов описаны Геммекером и Штаге [87].
Для полупромышленных и пилотных установок приставки в основном собираются из отдельных деталей, применяемых для монтажа трубопроводов (см. разд. 7.2). Для ввода газов и жидкостей в колонну применяют вставки с входным штуцером или кольцевым разбрызгивателем (см. рис. 138 и 142).
7.5.3. ГОЛОВКИ КОЛОНН
Головки колонн являются связующим звеном между колонной и конденсатором или дефлегматором. Поскольку их применяют в основном при ректификации, они снабжаются дополнительными устройствами для регулирования и измерения флегмового числа. В зависимости от способа получения флегмы различают головки частичной и полной конденсации [88].
Головка колонны частичной конденсации подробно описана выше (см. рис. 171). При низкотемпературной ректификации обычно используют дефлегматоры, поэтому дистиллят часто получают в виде пара. В этом случае применимы различные головки
378
частичной конденсации потока паров. Как указывалось в разд. 5.2.3, полная конденсация паров дистиллята при ректификации более предпочтительна. В лабораторной и промышленной практике в основном проводят перегонки с полной конденсацией, особенно в тех случаях, когда необходимо обеспечить высокие флегмовые числа, которые практически не удается регулировать посредством парциальной конденсации в дефлегматоре.
Торман [19], Геммекер и Штаге [87], а также Шнайдер и Шмид [89] показали, что головка колонны должна удовлетворять следующим требованиям: обеспечивать легкость регулировки и измерения флегмового числа; обладать минимальной удерживающей способностью по жидкости; иметь простую и механически прочную конструкцию, применимую как для работы при атмосферном давлении, так и под вакуумом; обеспечивать герметичность аппаратуры при распределении флегмы; предотвращать «подви-сание» жидкости; обеспечивать точность измерения температуры паров и подачу флегмы в колонну при температуре кипения или с небольшим переохлаждением. Кроме того, головка должна позволять регулировать и измерять нагрузку и флегмовое число в любой момент времени. Подобные измерения необходимо проводить в тех случаях, когда нагрузка колонны превышает 500 мл/ч, при которой визуальным путем уже нельзя подсчитать число образующихся капель. Особенно важно беспрепятственно измерять температуру паров. При этом необходимо следить за тем, чтобы на термометрический карман не попадали капли переохлажденной жидкости и давление в точках измерения температуры и давления было одинаковым.
Методы распределения небольших количеств жидкости в массообменных колоннах разобраны Клоссом [89а].
Для точного измерения и регулирования флегмового числа головки колонн снабжаются следующими устройствами и приборами: капельницами; трубками с косым срезом и капиллярами, служащими для подсчета падающих капель флегмы и дистиллята; двумя параллельно соединенными и независимо охлаждаемыми конденсаторами флегмы и дистиллята; регуляторами расхода флегмы и дистиллята в виде капилляров различной длины и диаметра, выбираемыми в соответствии с заданным флегмовым числом; механическими или электронными реле времени, обеспечивающими автоматическое регулирование расхода флегмы путем деления потока пара или жидкости (электронное реле времени обычно выполняется в комплекте с электромагнитным регулятором).
Метод подсчета капель в головках колонн не потерял своего значения до настоящего времени как способ контроля за расходом флегмы и отбираемого дистиллята, особенно при микроперегонках.
Шнайдер и Шмид [89] систематически исследовали работу различных капельниц (рис. 302). Данные, полученные ими при использовании бензина, приведены на рис. 303. Из рис. 303 отчет-
379
Рис. 302.
Капельницы:
а — формы выполнения выходного отвер стия капельниц для подсчета капель по Шнайдеру— Шмиду.
б — наконечник с несколькими капельницами, применяемый при нагрузках по жидкости, превышающих 500 мл/ч.
№ 8234, аналог—патент ФРГ №
ливо видно, что размер капель для различных капельниц существенно зависит от нагрузки по жидкости.
В головке колонны, запатентованной автором (патент ГДР 1011177) и описанной в нормалях
«Дестинорм», в качестве счетчика капель используются капилляры. Эта головка позволяет дополнительно измерять объемные расходы флегмы и отбираемого дистиллята. Плоскошлифованные и полированные капилляры обеспечивают хорошее формирование капель и вследствие небольшой их высоты не вызывают значитель-
ного подъема жидкости в них (см. рис. 128, 162).
Для большинства использующихся в лабораторной и промышленной практике органических жидкостей даже при температурах, приближающихся к температуре кипения, поверхностное натяжение на границе с воздухом составляет от 20 до 40 дин/см. Поэтому капилляры, описанные выше, будут образовывать капли объемом (4—5)-10-2 мл, т. е. объем 20—25 капель будет примерно составлять 1 мл. При повышении поверхностного натяжения объем образующихся капель увеличивается, исходя из этого можно
оценивать значение поверхностного натяжения жидкости.
Зависимости числа капель от нагрузки по, жидкости — бензину, полученные для различных капельниц. Обозначения кривых см. на рис. 302а.
•//--l. число капель, суммарный объем которых равен 1 мл.
380
Другими примерами конструкций головок ректификационных колонн, основанных на методе подсчета капель, являются йенская головка и головка «Нормаг» [91 ]. У йенской головки (рис. 304) штуцер для ввода пара расположен на высоте нижней трети конденсатора, который может быть дополнительно соединен с холодильником. Кран для отбора дистиллята имеет круговые шкалы на корпусе и на тефлоновом сердечнике, с помощью которых можно точно устанавливать воспроизводимые от опыта к опыту флегмовые числа.
Головка «Нормаг», разработанная Реном и Тайлигом (рис. 305), имеет надежно работающий «воротник» для сбора дистиллята. Конструктивной особенностью этой головки является размещение двух кранов на линии отбора дистиллята. Левый кран является регулирующим и служит для установки заданного флегмового числа, а правый кран — запорный. Регулирующие краны обычно являются кранами точной регулировки. Для этой цели они снабжаются установочными коническими углублениями или несколькими каналами. Примером подобного регулирующего крана является многоходовой кран Шульца [92]. Для повыше-
Рис. 304.
Головка лабораторной ректификационной колонны, предназначенная для работы при атмосферном давлении и под вакуумом.
Изготовитель: народное предприятие «Йенский завод стекла Шотт», Иена.
Рис. 305.
Головка ректификационной колонны Рена—Тайлига:
Рис. 306.
Головка ректификационной колонны Шнайдера—Шмида со стеклянным игольчатым клапаном.
381
Рис. 307.
Головка ректификационной колонны Хюбнера, имеющая соединенные конденсатор и холодильник дистиллята, а также клапан с коническим сердечником.
ния точности регулирования флегмового числа Шнайдер и Шмид [89] предложили головку колонны со шлифованным игольчатым клапаном, изготовленным из стекла (рис. 306).
Головку ректификационной колонны, изображенной на рис. 305, также изготавливают со стеклянным игольчатым клапаном, конструкция которого подробно описана Крамером [90]. В головках других конструкций используются клапаны со шпинделем из тефлона (см. рис. 247) или клапаны с сильфонами (см. рис. 248). Для измерения флегмового числа применяют трубки, нижний торец которых срезан под углом 60° к вертикали. Такая форма среза выбрана на основе многочисленных экспериментов (см. рис. 302 и 303). Головка колонны, предложенная Хюбнером [93], имеет соединенные друг с другом конденсатор и холодильник дистиллята. Охлаждаемый конический сердечник клапана (рис. 307) в этой головке служит для регулировки расхода отбираемого дистиллята. Благодаря установке холодильника под некоторым углом к горизонтали головка имеет небольшую высоту и в ней практически отсутствуют «мертвые» зоны. Устройство для замены сборников дистиллята, имеющееся в данной головке, обладает существенными преимуществами по сравнению со сборником Аншюца—Тиле.
Головка ректификационной колонны типа «Корад-хид», предназначенная для работы с постоянным флегмовым числом, имеет независимые конденсаторы флегмы и дистиллята [94]. Принцип 382
работы головки состоит в том, что вращающийся на шлифе конденсатор имеет зубцы 1 различного размера, с которых по каплям стекает конденсат (рис. 308). С помощью шлифа конденсатор можно установить таким образом, чтобы выбранный зубец, с которого стекает определенное количество жидкости, оказался над воронкой 2, служащей для приема дистиллята. Правильно выбрав зубец, можно получить необходимое флегмовое число. Для прекращения отбора дистиллята конденсатор устанавливают таким образом, чтобы конденсат капал с зубцов мимо воронки 2.
Автоматические головки ректификационных колонн обычно работают на принципе регулирования объема отбираемой фракции по времени отбора. В этих головках с помощью механического или электронного реле времени (см. разд. 8.4) устанавливают необходимое отношение промежутка времени включения реле (подача флегмы в колонну) к промежутку времени его выключения (отбор дистиллята), соответствующее заданному флегмовому числу. При этом необходимо, чтобы скорость выкипания жидкости в кубе поддерживалась постоянной, например, с помощью специальных устройств, описанных в разд. 8.4. Подобные головки могут работать по двум методам. По первому из них паровой поток разделяется в определенном соотношении и полученные потоки направляются в раздельно работающие конденсаторы для флегмы и дистиллята. Второй метод заключается в полной конденсации паров с последующим делением образовавшегося конденсата в определенном соотношении.
Автоматические головки колонн обладают тем преимуществом, что они не имеют кранов, вследствие чего исключается возможность загрязнения дистиллята и флегмы жировой смазкой шлифов. Кроме того, они позволяют быстро и надежно устанавливать любое флегмовое число. Удерживающая способность по жидкости у этих головок очень мала. При использовании подобных головок можно работать с флегмовыми числами от 30 до 100, в то время как при применении головок с самыми точными регулировочными н кранами обеспечение флегмового числа бо-
лее 30 представляет значительные труд-fE'l ности.
На рис. 309 (см. также рис. 2576) показана автоматическая головка Коллинза и
I Лэнца [95], основанная на делении паро-
7 вого потока. Восходящий поток паров об-
текает термометр 1 со стандартным шлифом
Щ ПТ Рис. 308.
\JI Головка Ллойда— Горнбахера типа «Корад-хид» с незави-
симыми конденсаторами для флегмы н дистиллята:
J 1 — зубцы для стекания капель дистиллята; 2 — воронка
_______________________ для отбора дистиллята.
383
Рис. 309.
Автоматическая головка Коллинза—Ленца с делением парового потока: 1 — термометр со стандартным шлифом; 2 — соленоид; 3 — дефлегматор; 4 — трубчатый стержень клапана; 5 — клапан; 6 — труба для отвода паров дистиллята.
Рис. 310.
Головка Штаге [97], работающая по методу деления парового потока, снабженная двумя магнитными клапанами и выпускным сифоном.
и поступает в дефлегматор 3. Соленоид 2, соединенный с временным регулятором, поднимает через определенные промежутки времени трубчатый стержень 4 с клапаном 5, размещенном на небольшом сферическом шлифе. При поднятом клапане 5 определенное количество паров в соответствии с заданным флегмовым числом поступает по трубе 6 в холодильник дистиллята.
Описанная головка колонны особенно предпочтительна, если дистиллят является расслаивающимся гетероазеотропом, так как благодаря делению гомогенного потока паров удается сохранить соотношение компонентов в азеотропе. На принципе деления потока паров работают также обе головки типа «Нормаг» (см. рис. 232, а, б). Коллинз и Ленц [95] опытным путем установили, что при одинаковых соотношениях промежутков времени между включением и выключением регулирующего прибора метод деления потока паров обеспечивает меньшее флегмовое число, чем метод деления потока конденсата. Кроме того, при работе по методу деления потока паров требуется надежная термоизоляция головки колонны с помощью вакуумированного кожуха для предотвращения образования «дикой» флегмы. В связи с этим перед проведением ректификации необходимо определить поправки, зависящие от конструктивных особенностей головки колонны и свойств разделяемой смеси.
384
Штаге (97] предложил головку ректификационной колонны с двумя магнитными клапанами, работающую по методу деления парового потока, для которой флегмовое число точно соответствует отношению промежутков времени включения и выключения регулирующего прибора (рис. 310). Промежутки времени, в течение которых клапаны находятся в открытом и закрытом состояниях у обоих клапанов различны.
В головках колонн, работающих по методу деления потока конденсата, широко применяются сферические или конические клапаны, а также качающиеся стержни или воронки с электромагнитным приводом. На рис. 311 показана принципиальная схема головки с клапанами [96]. Восходящий поток паров конденсируется в конденсаторе 2. При закрытом коническом клапане 4
Рис. 311.
Головка Кизельбаха, работающая по методу деления потока конденсата: 1 — соленоид; 2 — конденсатор; 3 — стержень конического клапана; 4 — конический клапан; 5 — запорные клапаны.
Рис. 312.
Автоматическая головка Креля с качающейся воронкой по нормалям «Де-стинорм»:
1,5 — шлифы; 2 — корпус; 3 — регулирующее устройство; 4 — железный сердечник приводного механизма; 6 — кожух; 7 — воронка для приема дистиллята.
Изготовитель: народное предприятие «Комбинат технического стекла», Ильменау.
13 Э. Крел:
385
конденсат полностью стекает обратно в колонну. С помощью соленоида 1 стержень 3 клапана 4 поднимается через определенные промежутки времени, соответствующие заданному флегмовому числу. При этом часть конденсата через клапан 4 проходит в сборник дистиллята. Оба клапана 5 служат для прекращения подачи жидкости (вместо кранов) и могут использоваться при рабочих давлениях до 10 мм рт. ст. Штаге [97] применял простые и двойные клапаны, а также скользящие шайбы.
Головка с качающимся стержнем использовалась в колонне Абегга с вращающейся лентой (см. рис. 282). Конденсат, образующийся на поверхности конденсатора, сначала стекает на пол движный стержень, снабженный железным сердечником, а затем поступает в виде флегмы в колонну. При включении электромагнита стержень отклоняется и конденсат начинает поступать в сборник дистиллята.
При высоких нагрузках более целесообразно применять головку с качающейся воронкой. Подобная автоматическая головка, разработанная автором, показана на рис. 312. В этой головке поток паров проходит мимо делительного устройства. Конденсатор расположен таким образом, чтобы пары не проникали в пространство корпуса 2 и не создавали помех работе регулирующего устройства 3. Качающаяся воронка и приводной механизм с железным сердечником 4, крепящиеся на шлифах 1 и 5 соответственно, могут быть легко демонтированы. Это обеспечивает возможность легко устранять возникающие неполадки. Кожух 6, установленный за приемной воронкой дистиллята 7, служит для охлаждения низкокипящих веществ или для нагревания жидкостей с высокой температурой плавления. Данная головка позволяет регулировать флегмовое число с точностью ±2%.
В головках с механическим регулированием, снабженных медленно вращающимся стержнем, стекающий конденсат, попадая на вращающийся стержень, поступает далее либо в колонну, либо в сборник дистиллята. Благодаря периодическому экранированию высоконапорной струи воздуха направление вращения стержня через определенные промежутки времени меняется.
Головки ректификационных колонн для промышленных установок собирают из отдельных частей конденсаторов и делительных устройств. В этих головках преимущественно применяют клапаны с электромагнитным регулированием. Регулировка клапанов осуществляется также, как и в головках лабораторных колонн (см. рис. 306, 247). С помощью электромагнитов, электромоторов или пневматического привода можно автоматически регулировать работу клапанов (см. рис. 248). Для автоматического деления потока конденсата в основном применяются головки с качающейся воронкой (см. рис. 142). В этих головках колеблющийся сердечник движется с помощью электромагнитов, размещенных на наружной стенке колонны. Отбор дистиллята через боковой штуцер проводят при включенных магнитах.
386
7.6. КУБЫ, ПРИЕМНИКИ И СБОРНИКИ ФРАКЦИЙ ДИСТИЛЛЯТА
Кубы и приемники или сборники фракций дистиллята являются начальными и конечными элементами ректификационной установки. В куб загружают разделяемую смесь, а в приемнике или сборнике дистиллята получают очищенный или разделенный на фракции дистиллят.
7.6.1. КУБЫ
При лабораторной перегонке в качестве кубов применяют обычные круглодонные колбы с коротким или длинным горлом емкостью до 500 мл, снабженные стандартными шлифами. Для стандартизованных методов дистилляции используют колбы специальных конструкций, например колбы Энглера (см. рис. 235) и колбы Сэйболта (TGL 0-12363). Стандартизованы также дистилляционные колбы (рис. 313) емкостью от 25 до 1000 мл (TGL 0-12364).
Стандартные длинногорлые колбы (см. рис. 313) применяют в качестве кубов преимущественно при дистилляции. Благодаря длинному горлу предотвращается унос брызг. Плоскодонные колбы не следует использовать при вакуумной дистилляции, так как они недостаточно прочны. В качестве кубов ректификационных колонн наиболее подходящими являются круглодонные трехгор лые колбы. Два боковых штуцера со стандартными шлифами NS14,5 используют для установки термометра, загрузки и выгрузки веществ, а также для отбора проб, подачи газа или пара через подводящую трубу и ввода капилляра, предотвращающего пульсации давления при кипении. Боковые штуцеры должны составлять с вертикальной осью колбы угол 20°, чтобы оставалось достаточно места для размещения необходимых деталей при установке обогревающего кожуха. Целесообразно снабжать колбы крючками для стягивания пружинами шлифовых соединений. Это необходимо потому, что при ректификации в кубе возникает избыточное давление, которое может при отсутствии стягивающих устройств вытолкнуть детали, вставленные в куб на шлифах. Если в ходе ректификации необходимо часто менять термометры или желательно использовать термометры без шлифов, применяют колбы с термометрическими карманами, в которые для улучшения теплопередачи заливают немного масла (рис. 314). Для ректификации при атмосферном давлении куб заполняют разделяемой смесью на 2/3 объема, а для ректификации под вакуумом — на х/2 объема.
Для микро- и полумикродистилляции как правило необходимы небольшие колбы, которые в основном изготавливают с длинным горлом (см. рис. 127), что позволяет проводить отгонку с минимальным кубовым остатком.
Для проведения перегонки удобны колбы с плоским дном (рис. 315). Их преимущество состоит в том, что поверхность испа-
13*
387
Рис. 313.
Стандартные дистилляционные колбы (TGL 0-12364).
Рис. 314.
Трехгорлая колба с пробоотборником и термометрическим карманом.
Рис. 315.
Плоскодонная колба со сферическим шлифом.
рения в них практически не изменяется до конца отгонки, в то время как у круглодонных колб поверхность испарения постепенно уменьшается с понижением уровня жидкости. При непрерывной ректификации применяют колбы с сифонной трубкой, благодаря которой поддерживается постоянный уровень жидкости и обеспечивается непрерывный отбор кубового продукта.
При низкотемпературной ректификации используют в основном кубы цилиндрической формы, которые удобнее помещать в сосуды Дьюара (см. рис. 173, 175, 176, 180). Другие формы выполнения кубов обуславливаются конструкцией обогревателя (см. разд. 7.7.2).
Для промышленных ректификационных установок, выполненных из стекла, применяют круглодонные колбы емкостью до 4 л с коротким горлом и стандартными шлифами и трехгорлые круглодонные колбы емкостью до 10 л (TGL10102). Стекольная промышленность освоила выпуск стеклянных кубов круглой формы емкостью до 200 л и цилиндрических сосудов объемом до 375 л со штуцерами, расположенными сверху, сбоку и снизу сосуда. Цилиндрические сосуды снабжаются также крышками со штуцерами.
На рис. 316 показаны подвесные сосуды емкостью от 16 до 150 л [1,5]. Переход к сферическому или коническому шлифу может быть осуществлен с помощью промежуточной вставки. Такие большие сосуды можно нагревать с помощью водяного пара, теплоносителей или погружного электрического кипятильника. При перегонке легковоспламеняющихся и взрывоопасных веществ применяют кубы из нержавеющей стали V2A. Штаге [99] разработал металлический куб емкостью от 10 до 200 л (рис. 317) с двойным металлическим кожухом для обогрева куба при помощи масляной бани. Переход от плоских металлических уплотнительных поверх-388
ностей фланцев штуцеров к стеклянным сферическим шлифам в этом случае также можно осуществить с помощью промежуточных вставок. Наиболее безопасным стеклянным кубом является стеклянная колба с защитным металлическим кожухом и баней из расплавленного металла (рис. 318). Такие колбы изготавливают емкостью от 1 до 20 л [99]. Между колбой и металлическим кожухом имеется свободное пространство, которое заполняют расплавленным висмутом или каким-либо металлическим сплавом.*
* Например, сплавом Вуда. — Прим. ред.
Рис. 316.
Подвесной сосуд емкостью 16 — 150 л, используемый в качестве куба ректификационной установки.
Изготовитель: народное предприятие «Йенский завод стекла Шотт», Иена.
Рис. 317.
Металлический куб Штаге с приваренным кожухом для масла [98 ]:
1 — штуцер для присоединения ректификационной колонны; 2, 3, 13 — штуцеры для установки датчиков температуры; 4 — штуцер для установки датчика давления; 5 — штуцер для слива кубовой жидкости; 6 — штуцер для присоединения бака с маслом; 7, 10 — штуцеры для ввода и вывода охлаждающей воды; 8,9 — штуцеры для присоединения к циркуляционному насосу; 11, 14 — штуцеры для вывода и ввода масла; 12 — штуцер для присоединения нагревателя.
389
Рис. 318.
Куб в виде стеклянной колбы с защитным металлическим кожухом и баией из расплавленного металла (по патенту Германии).
Изготовитель: фирма Эрнст Хааге, Мюльхайм (РУР).
Кожух с расплавленным металлом обеспечивает равномерный подвод тепла к колбе. Для наблюдения
за процессом кипения и уровнем жидкости в колбе в металличе-
ском кожухе имеется два смотровых окна, размещенные напротив.
7.6.2. ПРИЕМНИКИ И СБОРНИКИ ФРАКЦИЙ ДИСТИЛЛЯТА
В качестве приемников дистиллята можно использовать практически любые колбы (см. рис. 238). Очень удобны цилиндрические градуированные приемники (см. рис. 238, 15), которые позволяют непрерывно следить за объемом отбираемого дистиллята. Если получаемый дистиллят имеет высокую температуру плавления, то его собирают в приемник с обогревающим термостатирующим кожухом, который охватывает и сливной кран (рис. 319). При аналитических разгонках необходимо также устанавливать холодильник дистиллята и термостатировать приемник, чтобы можно было измерять объем дистиллята при постоянной температуре. При обычной перегонке приемники должны сообщаться с окружающей атмосферой, а при перегонке под вакуумом их соединяют с вакуумной линией. В приемном сосуде, изображенном на рис. 320, предусмотрено охлаждение штуцера, присоединяемого к вакуумной линии [100].
Для отбора четырех фракций дистиллята без нарушения процесса вакуумной дистилляции применяют вращающийся на шлифе приемник Бредта (см. рис. 238, 14 и рис. 321), а для отбора семи фракций — приемник Брюля (см. рис. 238, 16) с семью сборниками размером 120 X 30 мм (см. рис. 127, 130). Любое число фракций можно отбирать с помощью форштоса Аншюца—Тиле (см. рис. 238, 13) со сменными приемниками (см. разд. 7.5.1). На рис. 322 показан вакуумный приемник Штаге с переключаемыми сборниками, а на рис. 137 и 142 — приемники для сбора больших количеств дистиллята, снабженные штуцерами для продувки и присоединения к вакуумной линии, с помощью которых можно работать без нарушения процесса перегонки. Приемник Рока и Янца [101 ] выполнен в виде «коровьего вымени». Он (рис. 323) снабжен магнитным регулирующим устройством. Магнит 1, укрепленный на кольце 2, притягивает к соответствующему сборнику штифт 4, подвешенный на крючке 3. Преимущество данного 390
Рис. 319.
Приемник дистиллята с термостатирующим кожухом.
Рис. 320.
Приемник дистиллята с термостатирующим кожухом и охлаждаемым штуцером А для присоединения к вакуумному насосу.
Рис. 321.
Приемник Бредта с градуированными сборниками.
Рис. 322.
Приемник Штаге с переключаемыми сборниками.
приемника заключается в том, что дистиллят не соприкасается с жировой смазкой шлифов.
Для отбора газообразного дистиллята при низкотемпературной ректификации необходимы специальные устройства.
В лабораторных ректификационных установках, работающих при атмосферном давлении, предпочтительно использовать автоматические сборники фракций, аналогичные применяемым при хроматографических работах. Эти сборники обычно имеют вра-
щающиеся диски, на которых по окружности или по спирали
Архимеда установлены пробирки. Регулирование объема отбирае-
мой пробы дистиллята осуществляется в этих сборниках следующими способами: подсчетом капель, применением сифона или фотоэлемента,
Рис. 323.
Приемник Рока—Янца с магнитным регулированием:
1 --- магнит; 2 — кольцо для крепления магнита; 3 — крючок; _____________ 4 - - штифт,
391
Рис. 324.
Сифонный приемник Креля с точной регулировкой объема отбираемых фракций дистиллята методом погружения груза.
1 — мерный сосуд с погружным грузом; 2 — фотоэлемент; 3 источник света; 4 — измерительная бюретка.
Рис. 325.
Фотоэлектрический регулятор сборника фракций Грассмана—Дефнера:
1 — источник света; 2 — мениск жидко сти; 3 — фотоэлемент.
заменой пробирок через определенные промежутки времени. Подобные принципы регулирования используют также и в специальных устройствах для вакуумной перегонки.
Сборники фракций, в которых осуществляется подсчет капель, работают таким образом, что падающие капли дистиллята приводят в действие контактное устройство или фотоэлемент; получаемые при этом импульсы электрического тока передаются через реле на счетчик капель. На шкале счетчика можно устанавливать любое заданное число капель, по достижении которого приводное устройство поворачивает вращающийся стол на определенный угол, подставляя следующую пробирку к месту приема дистиллята. При этом счетчик капель возвращается в нулевое положение.
При аналитических разгонках обычно отбирают фракции одинакового объема, фиксируя температуру в момент отбора. Объем фракции определяется загрузкой куба и составляет обычно 1 — 20 мл. Для отбора небольших фракций объемом 1—5 мл целесообразно применять метод подсчета капель, а для отбора фракций объемом свыше 5 мл лучше использовать метод регулирования, основанный на принципе сифонирования (рис. 324). Мерный сосуд 1 снабжен изогнутой трубкой, через которую осуществляется сифонирование жидкости. Поток жидкости проходит при этом мимо фотоэлемента 2 с источником света 3, дающего импульс на 392
реле. Реле срабатывает с запаздыванием на несколько секунд, при этом включается поворотное устройство стола с пробирками. Мерный сосуд с сифонной трубкой снабжен подвижным погружным грузом. Меняя глубину погружения этого груза, можно устанавливать требуемый объем отбираемой фракции с точностью =±=0,1 мл, чем устраняется необходимость замены сосудов с сифоном при изменении объема фракций.
Процесс отбора дистиллята в полностью автоматизированном сборнике фракций Грассмана и Дефнера [102] регулируется с помощью фотоэлемента. Благодаря тому, что в данном сборнике объем дистиллята непосредственно измеряется в пробирке, можно устанавливать любой объем отбираемых фракций от 0,5 до 0,8 мл с точностью до одной капли (около 0,03 мл). Регулирование объема осуществляют, поднимая или опуская источник света 1 (рис. 325). Фотоэлектрическое устройство приводит в действие поворотный механизм в тот момент, когда мениск жидкости 2 в пробирке пересекает световой луч, при этом световой пучок, преломляется в вертикальном направлении. Преломленный световой пучок воспринимается фотоэлементом 3, расположенным под приемной пробиркой. Необходимо отметить, что вогнутый мениск жидкости значительную часть светового луча отражает вниз, т. е. в глубь жидкости. В результате повторного отражения от стеклянных стенок пробирки световой пучок достигает ее выпуклого дна, которое служит своеобразной собирательной линзой, обуславливающей формирование направленного луча света.
Для регулирования отбора малых объемов около 1 мл в качестве импульса, который приводит в действие механизм поворотного стола, используют ток, возникающий при контакте между двумя электродами [103]. При этом работают с очень слабыми токами около 0,1 мА, что позволяет применять такой метод для перегонки всех обычных растворителей за исключением углеводородов. Погрешность регулирования не превышает 1 %.
При регулировании объема пробы дистиллята по времени вращающийся стол с пробирками поворачивается при помощи электрических контактных часов через заданные промежутки времени. Для отбора фракций одинакового объема по этому методу необходимо обеспечивать строгое постоянство нагрузки ректификационной колонны. Данный метод применяется в основном при микроректификации (см. рис. 2576).
7.7. НАГРЕВАТЕЛЬНЫЕ ПРИБОРЫ
И ТЕПЛОИЗОЛЯЦИЯ
Для проведения процессов перегонки необходим непрерывный подвод тепла. Это тепло в основном используется для нагревания и испарения перегоняемого вещества. Остальная часть подводимого тепла идет на термоизоляцию аппаратуры, которая должна
393
работать по возможности в адиабатических условиях. В тех случаях, когда перегонку ведут при температуре до 100 °C для предотвращения самопроизвольной конденсации паров зачастую бывает достаточно хорошей термоизоляции, в то время как при более высоких рабочих температурах в слой изоляции необходимо помещать нагревательные приборы.
Нагревательные системы в установках для перегонки предназначены для обогрева куба, для нагревания исходной смеси и кубовой жидкости и для термоизоляции.
Нагревательные приборы различных конструкций, предназначенные для нагревания и испарения перегоняемых веществ, подробно рассмотрены в работе Штаге и Геммекера [104].
7.7.1. ОБОГРЕВ КУБОВ
Дистилляционные кубы обогревают с помощью газовых горелок, электронагревателей или теплообменников. Горелку Бунзена применяют в основном только для обогрева небольших кубов, например при дистилляции по Энглеру (см. рис. 235) и при микродистилляции. Этот метод обогрева особенно удобен при перегонке сильно вспенивающихся жидкостей, поскольку уменьшением пламени горелки можно предотвратить чрезмерное вспенивание жидкости в кубе. Кубы больших размеров редко обогревают непосредственно газовым пламенем, так как это связано с опасностью перегревания и затрудняет точное регулирование температуры. Для предотвращения перегрева при работе с газовыми горелками куб помещают на металлической сетке с асбестом или применяют воздушную баню [105]. В последнем случае куб обогревается в мягких условиях более равномерно нагретыми отходящими газами. Выполнение нагревателя в виде дымовой трубы позволяет эффективно использовать тепло пламени (рис. 326).
Более надежное регулирование обогревания кубов достигается с помощью электрических нагревательных приборов. Различают электронагреватели следующих типов: открытые, закрытые, фасонные, инфракрасные, погружные кипятильники.
Плоские нагревательные элементы с открытой спиралью позволяют легко регулировать нагрузку ректификационной колонны по перепаду давления потока пара в ней (см. разд. 8.4), в то время как плоские нагревательные элементы с закрытой спиралью обладают слишком большой тепловой инерцией, что приводит в отдельных случаях к захлебыванию колонны при выходе ее на заданный температурный режим. Рекомендуется между кубом и плоским нагревательным элементом оставлять воздушный зазор и обеспечивать термоизоляцию куба для уменьшения теплоизлучения (рис. 327).
Кипятильники с зеркальными отражателями, заменяющие горелки Бунзена, являются модификацией нагревателей, работающих по принципу обогревания куба горячим потоком воздуха, 394
Действие подобных кипятильников конструкции Клейна [106] основано на концентрировании теплового излучения, испускаемого нагревательным элементом, в одной точке над выходным окном нагревателя с помощью отражательных зеркал. Наличие регулирующего устройства (электропереключателя) позволяет поддерживать требуемую температуру. Электрогорелка Хофмана [107] обеспечивает нагрев куба до 800 °C.
К фасонным нагревателям относятся нагревательные элементы с электроспиралью, которым придают форму обогреваемой поверхности (см. рис. 149, 184). При необходимости перемешивать разделяемую смесь с помощью электромагнитных мешалок можно рекомендовать куб с нагревателем, показанный на рис. 328. Для обеспечения вращения мешалки нижний тубус куба имеет плоское дно и обогревается с помощью коаксиального цилиндрического подогревателя с ленточным элементом. Безопасность при перегонке легковоспламеняющихся и взрывоопасных веществ достигается при использовании эластичных обогревающих кожухов, пригодных для колб'различных размеров. С помощью этих нагревателей, в которых спирали вплетены в стекловолокно, можно получить температуру до 400 °C. Система пружинных колец обеспечивает плотное прилегание стекловолокна к стенкам колбы (рис. 329). Наличие круглых отверстий в днище нагревателя предотвращает образование воздушных «мешков» и позволяет применять данный прибор для обогревания колб с нижним отводящим штуцером и воронок [108]. Расположение нагревательных эле-
Рис. 326.
Обогревательное устройство Хюбнера, работающее по принципу воздушной бани.
Рис. 327.
Колбонагреватель по нормалям «Дестинорм» .
Изготовитель: народное предприятие «Комбинат технического стекла», Ильменау.
395
Рис. 328.
Перегонная колба с тубусом, обогреваемым ленточным электронагревательным элементом.
Рис. 329.
Универсальный обогревающий кожух для обеспечения температур до 450 °C.
Рис. 330.
Колба с наружным кипятильником.
Рис. 331.
Колба с внутренним погружным кипятильником Юиге.
ментов непосредственно у стенок колбы обеспечивает высокую эффективность теплопередачи и уменьшает возможность перегрева при кипении, особенно при перегонке вязких жидких смесей.
Значительная эффективность теплообмена достигается при использовании погружных кипятильников (см. разд. 7.6.1). В приборах этого типа нагревательный элемент помещается в трубу. На рис. 147 показан аппарат для перегонки воды с нагревательным элементом, изготовленным из кварца. В некоторых случаях применяют колбы с карманом в днище, в который вставляют нагревательный элемент соответствующей формы (рис. 330). В подогревателе «Интус»», предложенном Юнге (рис. 331), погружной кипятильник вводят в колбу сверху и вставляют в карман на дне колбы, предназначенный для обеспечения более полной отгонки. Следует иметь в виду, что применение термоизоляционного кожуха мешает наблюдать за процессом кипения и уровнем жидкости в кубе. Непрерывное перемешивание кубовой жидкости достигается в колбе с циркуляционным обогревом (рис. 332). На тубус с вставленным нагревательным элементом наплавлен стеклянный порошок, что обеспечивает равномерное кипение кубовой жидкости. Для полупромышленных и пилотных ректификационных установок можно использовать погружные нагревательные 396
элементы типа THQ 6 мощностью б кВт (см. разд. 5.1.3.3), выпускаемые «Йенским заводом стекла Шотт» (ГДР).
Наряду с нагревателями, работающими по принципу передачи тепла теплопроводностью, в последнее время все более широкое применение находят подогреватели, передающие тепло излучением. К подогревателям такого типа, обеспечивающим равномерный подвод тепла в мягких условиях, относится инфракрасный излучатель. Различают светящиеся теплоизлучатели, в качестве которых используют лампы накаливания мощностью 150, 250 и 500 Вт, и теплоизлучатели ИК-лучей мощностью до 1000 Вт, выполняемые из металлических труб. Применяя несколько светящихся теплоизлучателей, можно получать температуры до 300 °C. ИК-теплоизлучатель можно сравнительно легко выполнить в форме обогреваемой поверхности. Эти излучатели особенно пригодны для микродистилляции. Преимущество применения инфракрасных нагревателей заключается в том, что они обеспечивают очень малую разность температур между наружной и внутренней стенками обогреваемого стеклянного аппарата. Методы использования в лаборатории высокочастотных нагревателей описаны в работе Клиса [ПО].
Обогрев кубов с помощью теплоносителей применяют в тех случаях, когда непосредственный обогрев аппарата невозможен и требуется особенно мягкий и равномерный режим нагревания. Например, применение теплоносителей необходимо при отгонке низкокипящего компонента от основной высококипящей фракции, поскольку при непосредственном обогреве в этом случае после отгонки низкокипящего компонента температура резко повышается.
Рис. 332.
Колба с циркуляционным обогревом.
Рис. 333.
Куб с нагревательным змеевиком по нормалям «Дестииорм» .
Рис. 334.
Куб с обогревающим кожухом по нормалям «Дестннорм> .
397
Рис. 335.
Погружные теплообменники для пилот* иых и промышленных аппаратов без мешалок (а) и с мешалками (б).
Изготовитель: фирма «Стеклянная
техника», Висбаден — Шнрштайн.
зуют перегретый пар (см. разд, тели — парафиновые масла,
Обычно теплоносители пропускают через открытые жидкостные бани (см. рис. 203), змеевики (рис. 333) или кожухи (рис. 334), которыми снабжается куб колонны. В тех случаях когда для получения температур выше 100 °C нельзя применить пар высокого давления, исполь-6.1). Жидкие теплоноси-
ицерин или триэтиленгликоль — нагревают в замкнутом контуре с помощью обогревающего змеевика (см. рис. 317) или термостата. Для обогрева пилотных и промышленных стеклянных аппаратов в качестве теплоносителей в основном используют водяной пар и нагретое масло. На рис. 335 показаны погружные теплообменники для пилотных и промышленных аппаратов с мешалками и без них. В качестве открытых жидкостных бань используют водяные бани для температур до 80 °C, масляные бани для температур до 330 °C (см. табл. 39), бани из расплава солей для температур 150— 550 °C (см. табл. 39); песчаные бани для любых температур, бани с расплавленным металлическим сплавом для температур выше 70 °C (см. рис. 318).
Следует иметь в виду, что при применении песчаных бань трудно регулировать температуру, а при использовании бань из расплава солей или металлов стеклянный куб необходимо вынимать из бани до начала затвердевания расплава. В противном случае куб можно разбить. Наиболее пригодными металлическими сплавами для бань являются: сплав Вуда с температурой плавления 71 °C, состоящий из 1—2 ч. кадмия, 2 ч. цинка и 7—8 ч. висмута, и сплав Розе с температурой плавления 95 ° С, состоящий из 2 ч. висмута, 1 ч. свинца и 1 ч. цинка. Применять ртуть и сплавы с более высоким содержанием свинца не рекомендуется вследствие токсичности их паров.
7.7.2. НАГРЕВАНИЕ ИСХОДНОЙ СМЕСИ И КУБОВОЙ ЖИДКОСТИ
ПРИ НЕПРЕРЫВНОЙ РЕКТИФИКАЦИИ
При проведении непрерывных процессов ректификации необходимо, чтобы исходная смесь, поступающая в колонну, была нагрета до температуры, соответствующей температуре жидкости внутри
398
колонны в месте ввода исходной смеси (см. рис. 142, 167). Для нагревания исходной смеси до температур, не превышающих 100 °C, обычно достаточно змеевикового теплообменника (см. разд. 7.4), в котором в качестве теплоносителя используется термостатированная жидкость. Через змеевик, погруженный в масляную баню, можно также пропускать кипящую жидкость. При этом температура масляной бани регулируется с помощью контактного термометра.
В полупромышленных и пилотных ректификационных установках преимущественно применяют стеклянные змеевиковые пакеты с предохранительным устройством против выталкивающего действия жидкости (рис. 336). В качестве теплоносителей в этих случаях используют водяной пар (избыточное давление 3 кгс/см2 и температура 147 °C) или горячее масло (максимальная температура 220 °C). Пар подают в верхний штуцер, а конденсат выводят из нижнего штуцера, расположенного под углом к горизонтали. Левый верхний штуцер предназначен для установки термометра.
На рис. 138 показан нагреватель, выполненный в виде наружной нагревательной обмотки, а на рис. 337 изображен нагреватель типа «Лабодест VD2», снабженный погружным нагревательным элементом, крепящимся с помощью фланца. Мощность этих нагревателей можно регулировать с помощью контактных термометров и реле (см. разд. 8.2.2).
При непрерывной перегонке кубовую жидкость можно нагревать с помощью устройств, показанных на рис. 328—335. При использовании циркуляционных испарителей количество жидко-
Рис. 336.
Пакет обогревающих змеевиков с предохранительным устройством против выталкивающего действия жидкости. NW — диаметр условного прохода. Изготовитель: фирма «Стеклянная техника», Висбаден — Ширштайн.
Рис. 337.
Нагреватель для исходной смеси типа «Лабодест VD2» с погружным электронагревательным элементом.
Изготовитель: фирма Фишера «Лабораторная и исследовательская техника», Бонн —курорт Годесберг,
399
Рис. 338.
Циркуляционные испарители системы «Нормшлнф» (а), системы «Нор-* маг» (5) и системы «Шмидт» (в) [1041.
Рис. 339.
Проточный куб типа «Лабодест» с раздельным вводом пара и флегмы, разработанный Штаге.
Изготовитель: фирма Фишера «Лабораторная и исследовательская техника», Бонн—курорт Годесберг.
ста, находящееся в кубе колонны, и, прежде всего, среднее время пребывания ее в кубе могут быть значительно уменьшены [104].
На рис. 338 показаны некоторые циркуляционные испарители с вертикальными и наклонными погружными нагревательными элементами. Горизонтальный циркуляционный испаритель типа «Лабодест» (рис. 339), предназначенный для подогрева высоко-кипящих жидкостей под вакуумом, имеет раздельный подвод греющего пара и кубовой жидкости. Аппараты, работающие с этим испарителем, обычно применяют в крупных установках. Данный испаритель целесообразно использовать также в пилотных аппаратах, показанных на рис. 141 и 142.
400
7.7.3. ТЕПЛОИЗОЛЯЦИЯ
Пары, выходящие из куба, обычно должны достигать конденсатора в адиабатических условиях, т. е. без подвода и отвода тепла. При температурах до 100 °C эти условия могут быть обеспечены с помощью соответствующих теплоизолирующих веществ. При температурах, превышающих 100 °C, снаружи изоляционного слоя необходимо размещать дополнительные подогревающие устройства. Потери тепла в окружающую среду, вызванные конвекцией и излучением, должны быть компенсированы путем подвода соответствующего количества тепла. Обычно теплоизолируют куб и все части колонны, размещенные над кубом. Достаточная термоизоляция обеспечивает не только более хорошее и воспроизводимое разделение, но и лучшее использование подводимого тепла (см. разд. 4.12).
Для термоизоляции лабораторных, пилотных и промышленных аппаратов применяют теплоизоляционные материалы с малой теплопроводностью, вакуумированные кожухи, обогревательные системы с циркулирующим теплоносителем [111 ], электронагревательные элементы, размещенные в слое изоляции.
В обычных ректификационных установках, не требующих точного регулирования флегмового числа; в качестве теплоизоляционного материала широко используют асбестовый шнур. Следует отметить, что в большинстве случаев применяют слой изоляции недостаточной толщины, в то время как этот слой должен составлять 50—60 мм. Очень удобна термоизоляция в виде полуцилиндров из стекловолокна, которые легко накладываются на колонну любой длины (рис. 340), затем эти полуцилиндры дополнительно обматывают снаружи лентой из стеклоткани. Если в качестве теплоизоляционных материалов используют магнезию или минеральное волокно, то их помещают в кожух, изготовленный из тонкого листового металла. Эффективно также дополнительно обматывать стенки колонны алюминиевой фольгой. Описанные виды термоизоляции, а также изоляция с помощью кожуха, заполненного воздухом, применимы при температурах стенок аппаратов не превышающих 60—80 °C.
Вакуумированный кожух обеспечивает надежную термоизоляцию аппаратов, работающих при температуре до 250 °C, если остаточное давление в кожухе составляет от 10“4 до 10-6 мм рт. ст. [112]. Решающее значение при этом имеет способ покрытия серебром стенок кожуха.
При необходимости обязательного наблюдения за ходом процесса перегонки часто используют колонны с прозрачными вакуумированными кожухами. В этих случаях необходимо непрерывно вакуумировать кожухи с помощью диффузионных насосов, поскольку в выпускаемых промышленностью колоннах указанные выше значения остаточного давления соблюдаются очень редко. Для уменьшения теплопотерь излучением вакуумированные ко-
401
100
100 , 500 WOO 2000 3000
Нагрузка, мл/ч
Рис. 340.
Термоизоляционный кожух колонны в виде соединенных полуцилиндров нз стекловолокна.
Рис. 341.
Зависимость числа теоретических ступеней разделения п от нагрузки колонны по жидкости [141].
Колонна диаметром 25 мм н высотой 1200 мм с посеребренным вакуумированным кожухом.
Насадка колонны: 1 — сетчатая насадка из широкотканной сетки марки DNW1 с малым гидравлическим сопротивлением; 2 — сетчатая насадка из узкотканной сетки марки DNW2; 3 — то же с использованием дополнительного компенсационного электрообогревателя кожуха.
жухи колонн обычно серебрят изнутри. В слое серебра можно оставлять окно шириной 5—10 мм для наблюдения за процессом, протекающим в колонне. Однако наличие этого окна существенно снижает эффективность теплоизоляции. Теплопотери за счет излучения можно уменьшить также путем установки внутри вакуумированного кожуха цилиндров из тонколистового металла или алюминиевой фольги. Кожух с перфорированной фольгой обладает таким же изолирующим действием, как и полностью посеребренный кожух, при этом, в отличие от последнего, позволяет наблюдать за ходом процесса в различных частях колонны [113]. Различие тепловых расширений внутренней и наружной обечаек кожуха компенсируют, снабжая наружную обечайку кожуха линзовыми компенсаторами (см. рис. 257, б, 282) или выполняя внутреннюю обечайку, кожуха со спиральным участком. Колонны диаметром менее 10 мм и высотой более 500 мм, работающие при температуре выше 150 °C, рекомендуется снабжать теплоизолированной электронагревательной обмоткой, наматываемой на вакуумированный кожух. Качество теплоизоляции можно приближенно оценивать по температуре наружной стенки аппарата, которая не должна значительно превышать комнатную температуру-
Фишер и Вейанд [27 ] показали (см. гл. 8), что дополнительный электронагреватель, размещенный на вакуумированном кожухе 402
колонн, значительно повышает эффективность разделения (рис. 341). Не рекомендуется выполнять термоизоляцию в виде кожуха с проточным теплоносителем — паром или термостатированной жидкостью (см. разд. 7.7.1), поскольку в этом случае вследствие непосредственного контакта теплоносителя с колонной даже при небольшой разности температур имеет место значительный подвод или отвод тепла от стенок колонны [112]. В результате будет образовываться «дикая» флегма или может быть вызвано дополнительное испарение жидкости по высоте колонны. Во избежание этого Коллинг и Трамм [114] предложили помещать слой термоизоляции между стенкой колонны и кожухом с циркулирующим маслом, а также снаружи кожуха.
Надежную термоизоляцию лабораторных и пилотных аппаратов удается обеспечить только при использовании электрообогре-ваемого кожуха со слоем термоизоляции необходимой толщины. Такая термоизоляция особенно необходима при перегонке под вакуумом. На рис. 342 показаны нагревательные элементы со стекловолокном, которые были использованы для термоизоляции куба и колонны в установке молекулярной дистилляции [115]. Вебер [112] показал, что тепловой поток через стенку колонны не зависит от того, на каком расстоянии от стенки в слое теплоизоляции расположена электронагревательная обмотка. Намотка спирали непосредственно на корпус колонны не вызывает каких-либо нарушений технологического режима процесса. *
Очевидно, что вследствие наличия температурного градиента по высоте колонны, необходимо выполнять обогревающий кожух из отдельных секций, мощность которых целесообразно регулировать с помощью контактных термометров (см. разд. 8.2.2). Не рекомендуется регулировать температуру обогревающего кожуха по температуре кипения жидкости в головке колонны за исключением тех случаев, когда разность температур между головкой и кубом не превышает 1 —2 °C.
* Без особой необходимости наматывать спираль непосредственно на корпус колонны не рекомендуется во избежание местных перегревов и возможных поломок вследствие термических напряжений. — Прим. ред.
Рис. 342.
Нагревательные элементы со стекловолокном, устанавливаемые на аппарате для молекулярной дистилляции.
403
Рис. 343а.
Зависимость температуры первого нагревательного контура от длины колонны, полученные Кортюмом н Биттелем.
I — расстояние от верхнего конца колонны; N — число витков нагревательной спирали, приходящееся иа 10 см ее длины.
Рис. 3436.
Схема обогревающего кожуха с термопарами.
Кортюм и Биттель [116] предложили применять для обогрева колонны два нагревательных элемента, которые позволяют получать любую постоянную разность температур по длине обогревающего кожуха. Для этой цели они использовали два нагревательных контура, в первом из которых нагревательная спираль наматывалась таким образом, что расстояние между витками увеличивалось от нижней к верхней части колонны. Соответствующее понижение температуры регистрировали термопарами, установленными через каждые 10 см высоты колонны (см. рис. 343, а). Оказалось, что при любой силе тока в контуре обеспечивается линейное изменение температуры по высоте колонны.
Нагревательная спираль второго контура, изолированная бусами, наматывалась на первый контур, причем расстояние между витками во втором контуре было одинаковым и соответствовало расстоянию между витками в средней части первого контура. Снаружи спирали изолировали толстым асбестовым шнуром и кожухом, заполненным магнезией или кизельгуром. Два контактных термометра устанавливали таким образом, чтобы их сосуды для ртути соприкасались со стенкой колонны в местах, удаленных на 10 см от верхнего и нижнего ее концов. Верхний контактный термометр регулировал с помощью реле мощность второго контура, а нижний —мощность первого контура. При этом в каждом контуре регулировалась только часть общей мощности.
404
На рис. 343, б показана принципиальная схема обогревающего кожуха, регулируемого с помощью термопар [117]. Мощность обогрева 85 Вт (220 В; 0,39 А) достаточна для получения температур до 200 °C; для достижения более высоких температур необходима мощность обогрева 100 Вт (220 В; 0,46 А). При калибровке мощности подобного кожуха в него помещают колонну соответствующей длины с термометрами, введенными на шлифах в местах, удаленных от концов колонны на расстояниях, равных 1/3 её длины. При различных мощностях обогрева измеряют температуры, установившиеся внутри колонны, и берут среднее значение из показаний обоих термометров. Строят график зависимости температуры внутри колонны от мощности обогрева.
Втабл. 62 приведены опытные данные Вебера [112], полученные при калибровке обогревающих кожухов. Мощность обогрева соответствует теплопотерям, которые имели бы место при работе колонны без внешнего обогрева. Подводимая теплота отнесена к 1 м высоты колонны, а эквивалентное этому теплу количество конденсирующихся паров рассчитано на основе правила Трутона по скрытой теплоте парообразования при атмосферном давлении.
Используя график зависимости мощности обогрева от температуры внутри колонны можно регулировать реостатом мощность обогрева кожуха по показаниям ваттметра или амперметра. Более точное автоматическое регулирование можно обеспечить, применяя термопары или газонаполненные термометры (см. разд. 8.2.2).
Необходимо изолировать также и различные детали между кубом и колонной, колонной и конденсатором, и участки колонны между отдельными секциями обогревающего кожуха. Если эти участки не изолировать, то они начинают играть роль парциальных дефлегматоров, образующих дополнительное количество флегмы. При перегонке высококипящих веществ часто бывает затруднительно выводить пары из колонны. В этих случаях применяют паровые трубы с вакуумированным кожухом (см. рис. 309, 310), которые обеспечивают надежную термоизоляцию при температуре паров до 150°С. Для получения более высоких температур предпочтительно использовать дополнительный нагревательный элемент
Таблица 62.
Результаты калибровки обогревающего кожухв колонны
Температура, °C Мощность обогрева, Вт Подводимая теплота, кал/(м-ч) Количество конденсирующихся паров ректифицируемого вещества, эквивалентное подводимой теплоте, моль/(м«ч)
75 14 9 300 1,27
157 42 28 700 3,2
219 77 51 000 5,0
297 114 76 000 6,3
405
Рис. 344.
Нагревательный элемент в виде электропроводящей ленты типа «Хемотерм».
Изготовитель: народное предприятие «Электрическое тепло Бельциг», Бельциг.
в виде электропроводящей ленты (рис. 344). Такие ленты изготавливают длиной 600—2500 мм и шириной 6—90 мм [118]. Их потребляемая мощность составляет 36—850 Вт, что при удельной мощности 0,4—1,25 Вт/см2 позволяет получать температуры выше 400 °C. Для достижения более высоких температур используют гибкие проволочные спирали из нержавеющей жаропрочной стали. Диаметр проволоки равен 6 мм. Применяя спирали различной длины, получают максимальные температуры 450 и 800 °C. Удельная мощность спирали составляет 4,65 Вт/см2. Ленточный нагревательный элемент фирмы «Электротермаль энджиниринг» (Лондон) выпускается в виде ленты, наматываемой на барабан. По заданной мощности электрообогрева с помощью калибровочного графика определяют необходимую длину электронагревательной ленты, которую можно разрезать на участки любой протяженности. К концам отрезанного участка ленты прикрепляют вручную с помощью небольшого простого приспособления соединительные клеммы.
При использовании ленточных нагревательных элементов необходимо следить за тем, чтобы пары не перегревались, в противном случае возможно искажение температуры в головке колонны. Для предотвращения этого следует предварительно откалибровать нагревательный элемент тем же способом, что применяется для калибровки обогревающего кожуха колонны. При перегонке веществ с высокой температурой плавления легко поддерживать необходимую температуру соединительных коммуникаций перед приемником дистиллята и самого приемника с помощью ленточных нагревателей.
Для термоизоляции стеклянных трубопроводов применяют трубчатые кожухи из меди и свинца, плотно прилегающие к стенкам стеклянных труб. Эти кожухи обогревают снаружи паром и теплоизолируют. Эластичный профилированный шланг «Калорекс» [119] из синтетического каучука также можно использовать для термоизоляции стеклянных коммуникаций. Выпускают шланги с диаметрами условного прохода 8, 15 и 30 мм. Коэффициент теплопередачи от таких шлангов к стеклу толщиной 1,5 мм лежит в пределах от 165 до 260 ккал/(ч-м2 град).
406
7.8. НАСАДКИ
Нерегулярно уложенные насадки (см. разд. 4.10.2) применяют для получения в ректификационных колоннах возможно большей поверхности, по которой жидкость распределяется в виде тонкой пленки (см. разд. 4.2). * В некоторых случаях подобного эффекта достигают, используя устройство, симметрично размещенное в свободном пространстве колонны [118а]. Часто поверхность насадки, на которой происходит лассо- и теплообмен, называют активной поверхностью. Чем меньше по размеру элементы насадки, тем больше их суммарная поверхность, приходящаяся на единицу объема колонны. Однако при этом соответственно возрастает удерживающая способность насадки по жидкости, что снижает разделяющую способность колонны (см. разд. 4.10.5). Таким образом, приходится выбирать для конкретного процесса перегонки оптимальные форму, размер и материал насадки с учетом всех необходимых факторов. В работе Лева [120] приведены обширные сведения о характеристиках и методах расчета различных полупромышленных и промышленных насадочных колонн.
7.8.1. ФОРМЫ ВЫПОЛНЕНИЯ ЭЛЕМЕНТОВ НАСАДОК
Разработаны и испытаны насадки с элементами разнообразной формы, однако наиболее широко применяют насадки с цилиндрическими элементами. Из насадок с элементами нецилиндрической формы наиболее применимы в лаборатории шаровая и седловидная насадки. Исследованы насадки следующих видов: участки стеклянных спиралей, стекловолокно, цепная насадка, скобы, гвозди, полусферы из листового металла. Характеристики насадок различных видов приведены в табл. 28.
В систематической работе Керени [121] рассмотрены методы выбора высокоэффективных насадок для ректификационных установок. В данной работе на многочисленных диаграммах представлены зависимости эффективности разделения от формы и размера элементов насадок, а также приведены рабочие параметры массообменных процессов. Кроме того, в статье [121 ] предложено характеризовать эффективность разделения с помощью фактора G, определяемого из соотношений:
С, = nlVH~Ap ~AL~ri (193)
IgG =lg (п/1/'7ГДр)= IgA— В lg L (194)
. где ii — число теоретических ступеней разделения; Н — удерживающая способ-, ность колонны по жидкости; Др — перепад давления в колонне; L — пропускная способность колонны, мл/ч; А, В — константы.
г * Данисе положение не относится к насадочным колоннам, работающим ;В режиме эмульгирования с затопленной насадкой. — Прим. ред.
407
О 200 W ООО 800 1000 1200 1400 1600 1800 2000 Нагрузка, мл/v
Рис. 345.
Сравнительные характеристики различных насадок, полученные Керенн [121].
Результаты получены при разделении смеси н-гептан—метилциклогексаи в колонне с диаметром 24 и высотой разделяющей части 500 мм. Ректификацию проводили с бесконечным флегмовым числом при атмосферном давлении.
УС — удерживающая способность по жидкости, приходящаяся иа одну теоретическую ступень разделения; Др — перепад давления пара, приходящийся иа одну теоретическую ступень разделения.
В логарифмических координатах зависимость фактора эффективности G от нагрузки представляет собой прямую линию, поэтому сравнительно легко можно выбрать наиболее эффективную насадку. На рис. 345 показаны сравнительные характеристики новых насадок Керени (/—4), а также насадок из участков спира
408
лей (5 и 6), навитых с широким и узким шагом соответственно.
Райхельт [121а] провел сравнительные испытания -насадок с цилиндрическими элементами (рис. 346, а—и и м), а также насадок в виде пружин, перфорированных и разрезанных металлических колец. Эти исследования показали, что невозможно получить удовлетворительную сравнительную оценку их массообменных и гидродинамических характеристик, хотя многочисленные типы насадок были разработаны, исходя из различных химико-технологических требований к их конструктивному выполнению.
Насадки с элементами цилиндрической формы, основным типом которых являются кольца Рашига, показаны на рис. 346. Некоторые насадки нецилиндрической формы изображены на рис. 347. К ним относятся также проволочная насадка «Хэли-пак» [122], насадка из проволочной сетки «Окта-пак» (рис. 348, 349), разработанные Подбильняком, и насадка Кдннона из перфорированных полуцилиндров (рис. 350), обладающие высокой эффективностью.
Для лабораторной и пилотной перегонки применяют в основном следующие насадки с элементами цилиндрической формы: кольца Рашига с габаритными размерами 2—10 мм из гладкого или матированного стекла, из керамических материалов и металлов (см. рис. 346, а); кольца «Прим» из металлов —2—10 мм (см. рис. 346, 5); кольца Рашига из проволочной сетки с плоской поперечной перегородкой или без нее—3—10 мм (см. рис. 346, е);
Рис. 346.
Элементы насадок цилиндрической формы (типа колец Рашнга)
а — кольца Рашига гладкие и матированные; б — кольца Рашига с поперечными канавками; в -- кольца Рашига с продольными канавками; г — кольца Рашнга с перемычками; д — кольца «Прим»; е — кольца Рашига из проволочной сетки; ж — кольца «Иитос»; з — участки спиралей; и —-спирали Вильсона; к — шпульки; л — ролики; м — кольца Паля.
409
участки спиралей из стекла и металлов —1,4—10 мм (см. рис. 346, з); спирали Вильсона из стекла —3—6 мм (см. рис. 346, и).
Кольца «Прим» в основном изготавливают из металла; их используют при сравнительной ректификации, моделирующей промышленный процесс. Для этой насадки опубликовано весьма мало данных по измерению числа теоретических ступеней разделения. Согласно работе Тормана [19], ВЭТС для колец «Прим» 2,5 X2,2 мм соответствует ВЭТС для насадки из участков металлических спиралей 4X4 мм. Металлические кольца Рашига, также как и элементы насадки «Перфо-ринг», могут иметь отверстия круглой формы [123].
Все более широкое применение находят модифицированные Диксоном кольца Рашига с плоской поперечной перегородкой или без нее. Их изготавливают путем сворачивания проволочной сетки (примерно 1500—3000 отверстий наем2) в один или два слоя [124, 125]. Сравнительные характеристики различных насадок, выполненных из проволочных сеток, приведены в работе Эллиса и
Рис. 347.
Элементы насадок нецилиндриче-ской формы:
а — шары гладкие и матированные; б — сплошные тела Киршбау-ма; в — бусы гладкие и матированные; г — седла Берля; д — иасадка из сдвоенных элементов; е — звездообразная иасадка.
Рис. 349.
Насадка Подбильняка «Окта-пак» .
«Хэли-пак» .
Рис. 350.
Элемент насадки Каниона из металлических полуцилиндров с мелкой перфорацией.
410
Рис. 351.
Зависимость числа теоретических ступеней разделения от скорости пара в колонне, полученная для насадки из колец Рашига размером 3X3 мм без перегородки, выполненных из однослойной бронзовой сетки (данные Киршбаума).
Диаметр колонны 100 мм, высота слоя насадки 1000 мм. Разделяемая смесь н-гептан —метилциклогексан.
Варяванди [126]. Эти насадки состоят преимущественно из элементов с габаритными размерами, не превышающими 3 мм, и обладают ВЭТС лежащей в пределах 1,5—2,5 см. Существенным преимуществом подобных насадок явля-
; ется незначительное уменьшение эффективности разделения : с увеличением диаметра колонны. По данным Киршбаума [127] ВЭТС для слоя насадки длиной 1 м из сетчатых колец i Рашига 3x3 мм без перегородки даже в колонне диаметром f 100 мм находится в интервале 1,67—2,8 см при скорости i потока пара 0,02—0,27 м/с (рис. 351). Кольца Рашига были изго-товлены из однослойной бронзовой сетки. Опыты проводили 1 с использованием смеси «-гептан—метилциклогексан при бесконечном флегмовом числе.
Насадка Гроссе-Етрингхауза с элементами в виде участков : спиралей из тонкой проволоки, которые благодаря работам Шульце ; и Штаге стали известны под названием «брауншвейгские спирали», ‘ обладает очень высокой разделяющей способностью. Исследования Я автора показали, что эта насадка имеет также сравнительно Я высокую пропускную способность.
Я^^ Недостатком насадки Гроссе-Ётрингхауза является ее относи-тельно высокая стоимость, обусловленная тем, что изготовлять г спирали диаметром и высотой менее 2 мм весьма затруднительно.
Кроме того, вследствие возможной коррозии тонкой проволоки Я| в качестве материала насадки часто необходимо использовать доро-<Я гостоящую нержавеющую сталь V2A. Наибольшей эффективностью Я| разделения обладают насадки с элементами размером 1,3—2 мм. Я: Они имеют ВЭТС от 1 до 1,6 см. Если элементы спиральной насадки Я должны иметь диаметр и высоту 2 мм и менее, то применяют про-Я( волоку толщиной 0,15—0,2 мм. В зависимости от размеров участок ков спиралей для колонны объемом 1 л необходимо следующее Я| число элементов насадки:
Размер элемента, мм .................
Число элементов в 1 л ...............
2 4 20
115 000 12 500 100
| Предложено также изготавливать эти спирали из стекла. Автор I испытал насадку из плотнонавитых стеклянных спиралей разме-
411
Таблица 63.
Результаты испытаний насадки нз стеклянных
спиралей размером 4 и 7 мм
Колоииа диаметром 30 и высотой 1000 мм с длиной разделяющей части 920 мм. Разделяемая смесь: четырех хлор истый углерод — бензол. Концентрация четыреххлористого углерода в кубовой жидкости 40% (мол.); флегмовое число v = со.
Нагрузка, мл/ч ВЭТС (см) для спиралей Нагрузка, мл/ч ВЭТС (см) для спиралей
размер 4 мм размер 7 мм размер 4 мм размер 7 мм
100 5,2 5,4 400 5,6 8,0
200 6,4 800 6,1
ром4х4х0,3 и 7X7X0,5 мм при атмосферном давлении с использованием смеси четыреххлористый углерод —бензол. Результаты этих опытов при различных нагрузках приведены в табл. 63.
Спирали Вильсона являются стеклянной насадкой, элементы которой, в отличие от плотно навитых металлических спиралей, имеют 3—4 витка с шагом около 0,6 мм (см. рис. 346, и). Эта насадка имеет эффективность того же порядка, что и насадка из колец Рашига равного размера, однако она обладает значительно более высокой пропускной способностью.
Насадку с элементами в виде одновитковых проволочных спиралей изготавливают также, как и насадку из участков стеклянных спиралей. Значения ВЭТС, полученные Торманом [19 ] для насадки из одновитковых металлических спиралей, приведены ниже;
Диаметр витка, мм ... . 1,6 2,5 4,0 5,0
ВЭТС, см........... 2,5—3 4 4—8 12
Эффективность насадки из одновитковых проволочных спиралей соответствует эффективности насадок из стеклянных колец Рашига или шаров с элементами равных размеров.
Из группы насадок с элементами нецилиндрической формы в лаборатории и промышленности, в основном, применяют насадки, имеющие следующие элементы: шары из гладкого или матированного стекла размером 3—8 мм (см. рис. 347, а) и седловидные тела из фарфора, специальных материалов и проволочной сетки — 4—10 мм (см. рис. 347, а), а также насадки «Хэли-пак» из проволоки размером элементом 1,8—4,5 мм (см. рис. 348) и «Окта-пак» из проволочной сетки —5,0—7,5 мм (см. рис. 349).
Насадки из гладких и матированных шаров используют прежде всего для препаративных работ. Однако из-за большой массы сплошных шаров высота колонн с шаровой насадкой ограничена.
Насадка из элементов седловидной формы (см. рис. 347, а) обладает наилучшими гидродинамическими характеристиками.
412
Любая часть поверхности седловидного элемента, независимо от его положения, наклонена так, что создает благоприятные условия для контакта паров и жидкости. При центральной подаче потока жидкости угол ее растекания по седловидной насадке составляет 55—60°. Удельная поверхность этой насадки при размере элементов 4—10 мм превышает поверхность насадки из колец Рашига тех же размеров на 30—60%. Эта насадка имеет малое гидравлическое сопротивление потоку паров, которое, например, для насадки с элементами размером 8x8 мм примерно в два раза ниже гидравлического сопротивления насадки с кольцами Рашига того же размера. Хорошие эксплуатационные качества седловидной насадки обусловлены тем, что ее элементы не имеют острых кромок. Седловидную насадку сравнительно легко изготовлять. Следует отметить, что эту насадку выполняют также из латунной сетки 100 меш (насадка Мак-Магона) [128].
Модификацией насадки Мак-Магона является седловидная насадка «Инталокс» [129], которая отличается от обычной седловидной насадки большими поверхностью и свободным объемом, что обуславливает ее меньшее гидравлическое сопротивление (рис. 352) Для лабораторных и промышленных ректификационных колонн такую насадку изготавливают размером 6 и 12,5 мм. Видоизменением этой насадки является насадка «Гидронил-пластик-инталокс» [130], элемент которой имеет два отверстия (рис. 353). Коэффициент полезного действия данной насадки совпадает с коэффициентом полезного действия керамической насадки «Инталокс». При равном гидравлическом сопротивлении насадка «Гидронил-пластик-инталокс» обладает вдвое большей пропускной способностью по сравнению с насадкой из колец Паля. Другой модификацией седловидной насадки из металла является суперседловидная
Рис. 352.
Элемент насадки «Инталокс» .
Рис. 353.
Элемент насадки «Гидроннл-пластнк-ннталокс» .
Изготавливается из различных пластмасс размером 30,48—76,4 мм (1,2 — 3 дюйма).
413
Рис. 354.
Элемент суперседловидной насадки Кнршбаума.
Рис. 355.
Элемент насадки «Интерпак» из металла.
насадка Киршбаума [131], элементы которой имеют выпуклые с зазором седловидные поверхности (рис. 354).
Насадку «Интерпак» (рис. 355) можно изготавливать практически из всех металлов, а также из пластмасс. Эту насадку выпускают с размерами элементов 10x10x0,3; 20x25x0,4 и 30x35x0,8 мм [132]. Она обладает относительно низким гидравлическим сопротивлением.
Таблица 64.
Значения ВЭТС для насадок нз элементов нецилиндрической формы
Колония диаметром 30 мм с высотой разделяющей части 450 мм. Разделяемая смесь четыреххлористый углерод — бензол; флегмовое число v — ©о.
Тип иасадки Размер элемента, мм ВЭТС, см Нагрузка, мл/ч Плотность орошения, мл/(ч«см2) Примечание
Насадка Каннона из пер- 4 1,3—2 [19 к гл. 7]
форированных металлы-
ческих полуцилиндров.
Насадка «Хэли-пак» 1,8 0,5
Седловидная насадка нз 4 4,73 200 28,3 Данные
фарфора 6 6,00 200 28,3 автора
8 7,50 200 28,3
10 9,0 200 28,3
4 5,29 400 56,6
6 6,92 400 56,6
8 8,18 400 56,6
10 10,00 400 56,6
4 6,0 600 84,9
6 8,18 600 84,9
8 9,00 600 84,9
10 11,24 600 84,9
4 6,43 800 113,2
6 9,00 800 113,2
8 11,24 800 113,2
10 12,87 800 113,2
414
Значения ВЭТС для насадок из элементов нецилиндрической формы приведены в табл. 64. Характеристики фарфоровой седловидной насадки получены автором.
7.8.2. ВЛИЯНИЕ МАТЕРИАЛА НАСАДКИ НА ЭФФЕКТИВНОСТЬ РАЗДЕЛЕНИЯ
В разд. 4.2 сообщалось о влиянии химической природы материала насадки на разделяющую способность колонны. Насадки для лабораторных колонн в основном изготавливают из стекла, фарфора, глины, различных металлических сплавов и в последнее время также из пластмасс. Предпочтение обычно отдают стеклу и керамическим материалам благодаря их коррозионной стойкости в среде агрессивных жидкостей. Преимущество фарфора заключается в том, что он после обжига становится твердым и не содержит железа, которое может оказывать каталитическое воздействие на разделяёмые вещества. Проволочные или сетчатые насадки из нержавеющей стали V2A обеспечивают наибольшую эффективность разделения.
Различную эффективность сетчатых насадок, изготовленных из разных сплавов, Фухс [133] объясняет, основываясь на работах Форсайта с сотр. [134 ], неодинаковой смачиваемостью поверхности насадок и возможностью возникновения неодинаковых адсорбционных эффектов в зависимости от химической активности этих сплавов. Вольф и Гюнтер [135] разделяли эталонные смеси с различной полярностью веществ для определения влияния последней на эффективность насадок из различных металлов. В результате опытов было найдено, что в зависимости от материала насадки эффективность разделения изменяется на 10—12%.
Автором показано, что степень смачиваемости и, следовательно, эффективность насадки в значительной степени зависят также и от средней высоты выступов шероховатости поверхности элементов насадки [136]. Кроме того, установлено, что насадки из пластмасс с пористостью около 10% превосходят по степени смачиваемости сетчатые насадки.
Основные преимущества насадок из пластмасс по сравнению с насадками из других материалов заключается в строгой однородности размеров элементов, малой массе, большой доле свободного объема (вследствие малой толщины стенок), высокой механической прочности, относительно высокой коррозионной стойкости, а также в возможности легкой очистки [137].
Тэллер [138] объясняет сравнительно высокую
Рис. 356.
Насадка «Тэллерэттен» из полиэтилена, предложенная Тэллером [138].
415
эффективность насадок, изготовленных из плохо смачиваемых пластмасс, их низкой адгезионной способностью (см. разд. 4.2), которая обуславливает увеличение удерживающей способности жидкости свободным объемом насадки. Это приводит к возрастанию времени пребывания жидкой фазы в колонне. Насадка «Тэллерэт-тен» из полиэтилена (рис. 356), разработанная Тэллером, имеет долю свободного объема, равную 83%. Благодаря тому, что элементы насадки выполнены в форме розеток, газовая и жидкая фазы то вступают в контакт на относительно малых поверхностях элементов, то вновь разделяются на отдельные потоки.
Насадку из колец небольшого размера можно изготавливать как из металла или керамики, так и из полимеров [139]. Егбе-ронгбе [140] рассмотрел высокоэффективные компактные насадки из термопластичных пластмасс различных конструкций.
При использовании полиэтиленовых насадок в ректификационных колоннах из стекла или металла возникает опасность значительного растекания жидкости к стенкам колонны. Это растекание обусловлено разностью в силах адгезии, действующих в системах жидкость—стенки колонны и жидкость—элементы насадки (см. разд. 4.2). Согласно работе Штюрмана [141 ] растекание к стенкам можно уменьшить используя колонны с волнистыми стенками. Автором [137] показано, что путем специальной обработки внутренних стенок колонны можно также получить удовлетворительное распределение жидкости по насадкам, выполненным из пластмасс. Растекание жидкости к стенкам удается значительно уменьшить и с помощью покрытия внутренних стенок колонны полиэтиленовой или тефлоновой фольгой.
8
Автоматические установки, измерительные и регулирующие устройства
Высокую степень разделения и воспроизводимость результатов перегонки в лабораторных и пилотных ректификационных установках можно обеспечить, только применяя точные физические методы измерения.
Наряду с измерением температуры и давления при ректификации необходимо также определять флегмовое число и нагрузки колонны по пару и жидкости. Определение физико-химических свойств полученного дистиллята приобретает важное значение только в тех случаях, когда эти измерения проводят непосредственно в процессе перегонки.
Надежные измерения являются предпосылкой для регулирования и автоматизации процессов ректификации. При лабораторной и пилотной ректификации частично используют методы регулирования, применяющиеся в промышленности. Современное состояние техники регулирования рассматривается в специальных журналах.
8.1. АВТОМАТИЧЕСКИЕ УСТАНОВКИ
Основные стадии процессов ректификации во всех современных дистилляционных установках автоматизированы. Степень обогревания во многих случаях регулируют с помощью контактных термометров, скорость выкипания контролируют по дифференциальным манометрам. Для автоматического регулирования вакуума широко используют стенды с вакуумными насосами.
Современные исследования направлены на создание полностью автоматизированных ректификационных установок с одновременной регистрацией измеряемых величин. При работе на полностью автоматизированной установке вручную только загружают разделяемую смесь и через определенный промежуток времени, требуемый для проведения процесса ректификации, получают продукты разгонки; во время перегонки регистрируют кривые температуры в головке и кубе колонны и физико-химические свойства дистиллята.
Степень оснащения ректификационной установки периодического или непрерывного действия измерительными и регулирующими приборами зависит от конкретной задачи разделения и прежде всего от имеющихся экономических возможностей [12]. Различают полуавтоматизированныеи полностью автоматизирован
14 Э. Кррль
417
ные установки. Под полностью автоматизированной установкой понимают установку, в которой все рабочие процессы, включая пусковой период, протекают самостоятельно. Термин полуавто-матизированный целесообразнее заменить выражением «частично автоматизированный», под которым имеют в виду, что автоматизирована лишь часть рабочих процессов.
По степени оснащенности измерительными и регулирующими приборами различают следующие установки.
А. Частично автоматизированная установка, в которой осуществляется:
а) регулирование обогрева куба с помощью контактного термометра и контактного манометра посредством электронного пропорционального регулятора (см. разд. 8.2.2 и 8.4.2); обогрева кожуха колонны с помощью термопар, контактных или воздухонаполненных термометров (см. разд. 7.7, 8.2.1, 8.2.2); подогрева исходной смеси при непрерывной работе ректификационной установки с помощью контактного термометра (см. разд. 8.2.2); расхода исходной смеси при непрерывной работе с помощью дозирующих устройств (см. разд. 8.6); скорости выкипания и, следовательно, нагрузки колонны по пару, с помощью контактного манометра (см. разд. 8.4.2); вакуума с помощью регулятора давления или стенда с вакуумным насосом (см. разд. 8.3); расхода охлаждающей воды с помощью сосуда с постоянным уровнем и расходомера (см. разд. 8.6); а также предохранительного выключателя, отключающего установку при аварийном отключении системы подачи воды;
б) регистрация температур (см. разд. 8.2.1) верхней части колонны, исходной смеси и куба, а также температуры стенки обогревающего кожуха колонны (см. разд. 7.7.3) с использованием термопар или термометров сопротивления вместо - стеклянных термометров.
Б- Полностью автоматизированная установка, в которой осуществляется:'
а) регулирование тех же параметров, что и в частично автоматизированной установке; кроме того, регулируется расход исходной смеси с помощью небольших дозировочных насосов или посредством автоматического поддержания постоянного уровня в напорном баке (см. разд. 8.6); флегмовое число с помощью автоматической головки колонны, приводимой в действие механическим или электронным управляющим устройством (см. разд. 7.5 и разд. 8.4.1); объем отбираемых фракций дистиллята с помощью автоматического сборника фракций (см. разд. 7.6);
б) регистрация температуры (см. разд. 8.2.1) верхней части колонны в зависимости от количества дистиллята, а также температур исходной смеси, куба и обогревающего кожуха колонны с помощью термографа; флегмового числа косвенным путем посредством измерения разности температур между верхней и средней частями колонны; давления с помощью записывающего прибора.
418
8.1.1. АВТОМАТИЗИРОВАННЫЕ УСТАНОВКИ
ДЛЯ СТАНДАРТНОГО ДИСТИЛЛЯЦИОННОГО АНАЛИЗА
Примером полностью автоматизированной дистилляционной установки является установка D86, работающая по методу ASTM [1 ] и предназначенная для разгонки по Энглеру с автоматической записью диаграмм кипения. При работе на этой установке вручную приходится лишь загружать анализируемую пробу, устанавливать начальную мощность электрообогрева, извлекать зарегистрированную диаграмму разгонки и вынимать колбу с кубовым остатком. Электрическая схема данной установки представлена на рис. 357. Кроме указанных на рис. 357 устройств, в этой установке используют также следующие элементы: специальный электро-
Рис. 357.
Схема полностью автоматизированной дистилляционной установки D86, работающей по методу ASTM:
1 — термопара; 2 — баия с тающим льдом; 3 — нагревательное устройство; 4 — выключатель, служащий для отключения установки в момент отгонки 95% (об.) исходной смеси; 5 — потенциометр счетчика времени;
6 — потенциометр измерителя объема; 7 — индикатор отгонки 95% (об.) исходной смеси; 8 — мотор счетчика времени; 9 — регулятор обогрева; 10 — выключатель, служащий для отключения установки по окончании разгонки; 11 — включатель для пуска установки в работу; 12, 29 — пружины;
13, 14 — источники света для фотоэлемента; 15 — мотор для измерителя объема; 16 — мотор и пускатель для регистратора времени и температуры; 17 — фотоэлектрический усилитель в измерителе объема; 18 — усилитель;
19 — магнитная катушка термографа; 20 — фотоэлемент для регистрации первой капли дистиллята; 21 — фотоэлемент измерителя объема; 22 — электрический усилитель; 23 — мотор барабана самопишущего прибора;
24 — концевой выключатель; 25 — отключатель; 26 — потенциометр с реохордом; 27 — реле; 28 — включатель; 30 — барабан для миллиметровой бумаги.
14
419
Рис. 358.
Ректификатор-автомат для моторных топлив по DIN 51751 (февраль 1964 г.) в соответствии с A STM D86 или D158 с компенсирующим леиточиым самописцем (ширина леиты 210 мм).
Изготовитель: фирма Эрист Хааге, Мюльхайм (Рур).
нагреватель, который реагирует на резкие изменения температуры и работает таким образом, что первая капля дистиллята появляется через 5—10 мин после начала процесса перегонки; устройство, которое автоматически «наносит» отметки времени на диаграмму разгонки через 2 или 10 мин; прибор, автоматически фиксирующий момент отгонки 95% (об.) общего количества исходной смеси и обеспечивающий поддержание в дальнейшем постоянной мощности обогрева куба; приспособление для автоматического возврата всех частей установки в исходное положение после окончания разгонки; устройство, обеспечивающее автоматическое охлаждение приемного сосуда для дистиллята до 15±3 °C.
Точность измерения температуры составляет 0,5 °C, а объема — 0,1 мл. Установка работает при одной из двух предварительно установленных скоростях перегонки 4,5 или 9 мл/мин и рассчитана на круглосуточную эксплуатацию.
Подобная установка «Асда» типа 3 (фирма «Галленкамп» Лондон) может быть использована для проведения следующих операций: ASTM D86-JP 123 группы 1, 2 и 3, а также D1078, DIN 51751 и 51752; ASTM D86-JP 123 группа 4; скоростная перегонка (не стандартизированная).
В одной лаборатории, оснащенной 15 установками «Асда», в течение 8 месяцев было проведено 80000 дистилляционных анализов. Это соответствует производительности установки, равной 666 анализам в месяц. Один исследователь одновременно может обслуживать около 10 таких установок без дополнительных приспособлений. Преимущество данных установок состоит в том, что они 420
полностью исключают субъективные погрешности, возможные при обычной разгонке по Энглеру.
В Советском Союзе для дистилляционного анализа нефти используют автоматические ректификационные установки, предложенные Кантерманом с сотр. [3], с точностью определения температуры начала перегонки —2 °C *. Ректификатор-автомат Зальцера [4] по DIN 51751, предназначенный для разгонки моторных топлив и растворителей, выпускается предприятием АЕГ в Берлине.
На рис. 358 показана установка, описанная Ирманном [5]. Автомат состоит из пульта управления, снабженного компенсирующим линейным самописцем термометра сопротивления, и двух взаимозаменяемых ректификационных агрегатов. Установка предназначена для перегонки в температурных пределах от 0 до -|-285 °C и от -ф-100 до -j-385 °C. Ректификатор-автомат Герцога [6] одновременно с выдачей цифровых данных регистрирует результаты на бумажной ленте. Цаппе с сотр. [7] разработал установку для
* Кроме установок Кантер мана с сотр., в СССР разработаны и выпускаются лабораторные установки других типов, например |полуавтоматизированная установка для полумикроректификации типа ЛМР (СКТБ стеклянных приборов, г. Клин Московской обл.). — Прим. ред.
Рис. 359.
Установка для дистилляционного анализа под вакуумом с автоматическим регулированием температуры, разработанная Цаппе с сотр. [7 ]:
1 — самописец; 2 — термостат; 3 — усилитель; 4 — регулятор давления;
5 — наклонные трубчатые манометры; 6 — фотоэлементы; 7 — сборник с поплавком; 8 — форштос; 9 — стеклянный патрубок; 10 — капилляр для устранения пульсаций давления при кипении; 11 — обогреватель куба с регулятором; 12 — куб; 13 — ректификационная колонна с теплоизолирующим кожухом; 14 — счетчик пузырьков.
421
проведения дистилляционного анализа под вакуумом при стандартизованных условиях. Схема этой установки представлена на рис. 359 [6].
8.1.2. ПОЛНОСТЬЮ АВТОМАТИЗИРОВАННЫЕ
УСТАНОВКИ ДЛЯ ФРАКЦИОННОЙ ПЕРЕГОНКИ
В принципе степень автоматизации ректификационных установок периодического или непрерывного действия можно увеличить настолько, что их можно рассматривать как полностью автоматизированные установки. Полностью автоматизированная установка Улусоя [8] разработана специально для препаративных работ. Для низкотемпературной ректификации предназначена описанная выше автоматическая установка Подбильняка (см. рис. 178), схема включения измерительных и регулирующих приборов которой представлена на рис. 360.
Штаге [9] разработал ряд лабораторных ректификационных установок типа «Лабодест-робот». Примером такой установки является установка с трубчатой щелевой колонной, предназначенная для полумикроректификации (см. рис. 2576). Преимущество данных установок состоит в том, что их измерительную и регулирующую системы можно использовать для оснащения различных ректификационных колонн [10], например для трубчатых щелевых колонн с емкостью куба 5—100 мл, работающих при абсолютном давлении от 0,01 до 760 мм рт. ст. с эффективностью, равной примерно 40 теоретическим ступеням разделения, или для колонн с металлической сетчатой насадкой с емкостью куба 100—1000 мл, работающих при атмосферном давлении или остаточном давлении до 5 мм рт. ст. с эффективностью около 60 теоретических ступеней разделения.
На передней стороне установки для полумикроректификации (см. рис. 2576) расположены специальные вставки, выдвигаемые с помощью телескопических направляющих. Все функциональные элементы размещаются на штеккерной печатной плате, используемой в транзисторной технике. Установка имеет следующие системы регулирования: температуры масляной бани с помощью пропорционального электронного регулятора, работающего по предварительно заданной температуре в пределах от 0 до 300 °C, или переключаемого по предварительно заданной нагрузке, выражаемой числом капель в 1 мин; температуры компенсационного нагревателя кожуха колонны по предварительно заданной температуре от 0 до 300 °C, или с помощью переключения регулятора подстройки по температуре пара в верхней части колонны; флегмового числа путем деления парового потока с периодами включения и выключения реле времени от 0,5 до 200 с; других температур и объема отбираемых фракций; работы сборника фракций путем замены сосудов через заданный промежуток времени (в минутах). Кроме того, установка снабжена системами, обеспечивающими стабилиза
422
цию вакуума, а также предохраняющими от производственных неполадок, таких как прекращение подачи охлаждающей воды, превышение верхних пределов температуры и уровня заполнения сосудов в сборнике дистиллята.
В насадочных колоннах регулирование нагрузки и перепада давления потока пара осуществляется относительно просто, в то время как для трубчатых щелевых колонн, в которых перепад давления составляет порядка 10~3 мм рт. ст., требуется специаль-
ные. 360.
Электрическая схема автоматизированной ’низкотемпературной ректификационной установки Подбильняка «Термокон» (см. также рис. 178):
1 — электронный прибор для контроля количества дистиллята; 2 — трубопровод для паров дистиллята; 3 измерительная ячейка; 4 — измерительный контур для определения теплопроводности; 5 — передача привода устройства для передвижения бумажной ленты; 6 — регистрирующий манометр; 7 — прибор, контролирующий давление и степень охлаждения флегмы; 8 — прибор, регистрирующий теплопроводность; 9 — мотор; 10 — вывод паров; 11 — штуцер для подачи воздуха; 12 — штуцер для продувки; 13 — воздушный термостат; 14 — устройство для автоматического ввода проб газа.
423
| Рис. 361.
i: Полностью автоматизированная ректификационная установка «Аутома-
Ь; тик 62» :
г 1 — расходомер жидкости, стекающей в куб; 2 — капельница; 3 — пере-ходы; 4 — колонна; 5 — обогревающий кожух колонны; 6 — дифмаио-I метры; 7 — воздухонаполнениые термометры; 8 — головка колонны;
9 — термометры; 10 — холодильник; И — вакуумный соединительный V патрубок; 12 — охлаждаемая ловушка; 13 —магнитные клапаны; 14 — погружной груз; 15 — соединительный патрубок; 16 — заглушка; 17 — £ пробоотборник; 18 — дозировочное устройство; 19 — вакуумный прием-ник; 20 — электрические контактные манометры; 21 — трубопроводы; ; 22 — холодильник прибора, измеряющего перепад давления; 23 — рас-
пределитель дистиллята; 24 — подающие трубы; 25 — сосуд для уста-I. иовки сборника фракций; 26 — пробирки; 27 — сборник фракций; 28 — пробка со шлифом; 29 — куб; 30 — пробка; 31 — приемная бюретка; 32 — I вакуумный приемник Аншюца —Тиле; 33 — пульт управления.
к Изготовитель: народное предприятие «Комбинат технического стекла», £ Ильменау.
8 ная регулирующая система. Фишер и Вейанд [10] с помощью электронных приборов измеряли число капель флегмы, подаваемое | в единицу времени. Исходя из результатов этих измерений, регу-[ лировали мощность нагревателя куба по требуемой нагрузке пря-| мопропорционально разности между действительным числом капель [ и предварительно заданным значением. Оказалось возможным I точно и абсолютно воспроизводимо регулировать нагрузку в пре-f делах от 50 до 250 мл/ч при давлении в кубе от 760 до 10~3 мм рт. ст. [ В автоматизированную установку «Аутоматик 62», соответ-I ствующую нормалям «Дестинорм» (рис. 361), разработанную авто-I ром, входят следующие отличительные элементы [11 ]: куб с элек-i тронагревателем «Интус»; устройство для измерения расхода t жидкости, стекающей в куб, и устройство для отбора проб этой I жидкости перед кубом; автоматический электронный регулятор I обогрева кожуха колонны по средней температуре внутри колонны I (см. рис. 368); автоматическая головка колонны с качающейся | воронкой для регулирования флегмового числа с помощью элект-F ровного или механического реле времени (см. рис. 312); автомати-I ческий сборник фракций, содержащий 60 пробирок и управляемый I при помощи сосуда с сифоном и погружным грузом (см. рис. 324); | автоматический электронный регулятор вакуума с точностью I ^tzO, 1 мм рт. ст., работающий с помощью термостатированного ре-I гулятора давления (см. рис. 377); автоматический регулятор к скорости выкипания и, следовательно, нагрузки колонны по пару, |. снабженный контактным манометром (см. рис. 387, 388); оптиче-I ская система для визуального наблюдения за температурой и электронный компенсограф для регистрации температуры.
К Пары дистиллята (см. рис. 361) попадают сначала в термоста-I' тированную зону, где конденсируются низкокипящие компоненты, К при этом легко затвердевающие вещества можно разогревать К в термостате. Расположенный ниже магнитный клапан 13 служит Е для измерения объема жидкости по градуированной трубе. Пробо- отборное устройство 17 с капельницей позволяет отбирать пробы | для испытаний и промежуточных анализов при аналитической
425
ректификации. Можно установить также термометр или приемную воронку для калибровки сосуда с сифоном и погружным грузом. Этот сосуд снабжен вертикально перемещаемым погружным грузом 14, который позволяет регулировать объем фракций с точностью ±0,1 мл. При прекращении подачи дистиллята в сосуд манометр 20, заполненный органической токопроводящей жидкостью, с помощью электронного реле включает специальное устройство, которое устанавливает в сборнике фракций следующую пробирку. Одновременно импульс тока, поступающий от электронного реле, может приводить в действие перо с красителем, встроенное в электронный термограф, которое посредством магнитной катушки отмечает на полосе бумаги число отобранных фракций.
Сборник фракций имеет обычно 60 пробирок емкостью 20 мл каждая. Он помещен в эксикатор, защищенный кожухом из металлической сетки. Сборник фракций применяют при ректификации, протекающей как при атмосферном, так и остаточном (до 1 ммрт.ст.) давлении. После заполнения 30 пробирок следует звуковой сигнал или автоматически подается следующий диск с пробирками. При заполнении всех пробирок по соответствующему сигналу регулятор флегмы отключается и колонна начинает работать с бесконечным флегмовым числом, т. е. без отбора дистиллята.
Соединительная коммуникация между сосудом с сифоном и сборником фракций представляет собой легко разбираемую систему, в которой в значительной мере приняты предосторожности, чтобы дистиллят не соприкасался с жировой смазкой шлифов.
При препаративной ректификации используют сосуд с сифоном или применяют сборники, при достижении в которых определенного уровня жидкости подается звуковой сигнал и ректификационная колонна переключается на режим работы с бесконечным флегмовым числом.
Решая задачу автоматизации процесса ректификации по Мико для качественных испытаний при перегонке вин, Коуколь с сотр. [12а] разработал ректификационный автомат для фракционной перегонки. Эта установка обеспечивает регулирование мощности электронагревателя куба в зависимости от количества дистиллята, получаемого в единицу времени. При постоянной скорости перегонки можно разделить смесь на большое число фракций одинакового объема, получаемого в течение предварительно заданного постоянного промежутка времени, например 15 мин. Объем фракций измеряют с помощью термистора, который отрегулирован на определенную высоту уровня жидкости. Этот термистор обеспечивает получение фракции в объеме 25 мл с точностью ±0,1 мл.
Автоматические установки для непрерывной ректификации разработаны Коллингом и Траммом (см. рис. 166) и Штаге (см. рис. 167).
Рёк с сотр. [13] предложил ряд устройств для автоматизации лабораторных колонн, в частности предохранители, предотвращающие возможность прекращения подачи охлаждающей воды, и 426
Рис. 362.
- Экспериментальная установка непрерывного действия с колоннами, сна* i. бженнымн колпачковыми тарелками с отражательными перегородками для к пара, предназначенная для моделирования процесса перегонки сырой нефти: | 1 — дозировочные иасосы; 2. — пульт управления; 3 — регулятор вакуума; £4 — предохранительное реле.
t Изготовитель: фирма Фишера «Лабораторная и исследовательская тех* = ника», Бонн —курорт Годесберг.
противопожарные предохранители. Подробные обзоры по регулирующей технике для лабораторных колонн представлены в работах Аббега [14], Кесслера и Фоденаля [16], а также Кадлека [17].
Предпосылкой автоматизации непрерывно работающих пилотных ректификационных установок является решение задачи получения достоверных опытных данных, на основе которых можно разрабатывать промышленные установки. На рис. 362 показана экспериментальная установка, предназначенная для моделирования промышленного процесса перегонки сырой нефти. Установка работает непрерывно. Она состоит из одной основной и трех дополнительных колонн, предназначенных для отгонки низкокипящих фракций. Данная установка служит для разгонки многокомпонентных смесей, которые разделяются на четыре фракции. Кубовый продукт отбирается из куба основной колонны. Ректификационные колонны снабжены колпачковыми тарелками с отражательными перегородками для пара. По экспериментальным данным, получаемым при перегонке в этих колоннах, можно непосредственно разрабатывать установки больших размеров. Потоки паровой и жидкой фаз дозируются насосами 1 (см. разд. 8.6). Пульт управления 2 позволяет регулировать скорости выкипания, температуры обогревающих кожухов колонн и флегмовые числа. Регулятор вакуума 3 обеспечивает постоянную степень разрежения, а предохранительное реле 4 отключает установку, как только прекращается подача охлаждающей воды. Температуры на основных стадиях процесса непрерывно регистрируются электронным самописцем [17а].
8.2. ИЗМЕРЕНИЕ И РЕГУЛИРОВАНИЕ
ТЕМПЕРАТУРЫ
8.2.1. ИЗМЕРЕНИЕ ТЕМПЕРАТУРЫ
При ректификации прежде всего необходимо измерять температуру пара в верхней части колонны перед конденсатором и температуру кубовой жидкости. При работе с термолабильными веществами следует обязательно наблюдать за температурой в кубе. При непрерывной ректификации необходимо подогревать исходную смесь до температуры, соответствующей температуре жидкости в колонне в месте ввода в нее исходной смеси. Кроме того, требуется измерять температуру обогревающего кожуха колонны и жидкого теплоносителя (при использовании обогревающей бани) или потока греющего пара.
В лабораторных и пилотных ректификационных установках все еще преимущественно используют стеклянные термометры, однако при небольших интервалах измеряемой температуры предпочтительно применять термопары, полупроводниковые термометры или термометры сопротивления, которые позволяют авто-428
магически регистрировать и регулировать температуру *. О измерении температуры в лаборатории информирует обзорный доклад Фанфора [18].
В работе Лапорта [15] обсуждаются вопросы измерения, регулирования и стабилизации высоких и низких температур. Важно правильно выбрать точки измерения температуры в ректификационном аппарате, так, например, точка измерения температуры в верхней части колонны должна находиться примерно на 10 мм ниже штуцера для вывода пара. Температуру в кубе измеряют в самой нижней его части, чтобы можно было сразу же обнаружить перегрев. Датчики температуры, располагаемые в потоках жидкости или пара, тщательно изолируют и размещают таким образом, чтобы каждый датчик находился в средней части потока.
Стандартные стеклянные термометры, применяемые в ректификационных установках, представлены в табл. 65. Кроме указанных стандартных термометров выпускается большое число стеклянных термометров специального назначения, например угловые термометры со стандартным шлифом для измерения температуры внутри колонны. Термометры с присоединительным фланцем
Таблица 65.
Стандартные стеклянные термометры, применяемые в ректификационных установках
Стандарт Наименование
TGL 40-335 Стеклянный термометр для измерения температуры жидкости
40-336 Термометр для ректификационных установок Стеклянный термометр для измерения температуры жидкостей, термометр для измерения температуры минеральных масел и топлива, термометр типов А и В для измерения температуры кипения
40-339 Лабораторный термометр с коническим шлифом NS14,5/23
D1N 12779/71 12784/55 группа 1 Термометр для ректификационных установок Термометр со стандартным шлифом Термометр для ректификационных установок (эта-
12784/63 группа 2 лонный) Термометр со стандартным шлифом Термометр для измерения температуры в стеклянных
12785/58 группа 1 кубах-колбах (эталонный) Термометр для измерения температуры кипения
12788/63 группа 1 (эталонный) Карман для термометра со стандартным шлифом 14,5/23
* При наличии соответствующих измерительных приборов можно успешно использовать термопары и термометры сопротивления также при широком интервале температур. — Прим. ред.
429
Рис. 363.
1 2 Термометр для ректификационных установок со стандартным шлифом
2} АЛ 14,5 (TGL40-339, DI N12784/55):
ц г-Ц-| / — крепление шкалы по Рихтеру; 2 — крепление шкалы по Штифту;
п 3 — предохранительный расширительный участок; 4 — последнее де-
)JL QU леиие шкалы; /х — длина ножки погружения; /2 — глубина погруже-
ги п иия.
удобны при монтаже; термометры с капиллярами, L ]1 окрашенными с обратной стороны, облегчают визу-fS альное наблюдение за показаниями [19].
1Р В ректификационных установках в основном
тп[ используют термометры, у которых капилляр со шкалой запаян в трубчатый кожух (рис. 363). Тер-z, 1 мометры, предназначенные для измерения темпера-L 1г тур в интервале от —58 до +360 °C, заполняют ' у] ртутью или ее сплавами. Для низкотемпературной ректификации в области температур от —58 до —200 °C применяют термометры, заполненные пентаном или другой подходящей жидкостью. Следует принять во внимание, что кроме термометра со стандартным шлифом, показанного на рис. 363, все термометры градуируют при полном погружении, т. е. при измерениях термометр необходимо погружать в исследуемую среду на всю высоту столбика ртути.
При использовании термометров, устанавливаемых в спе-
циальные карманы, заполненные маслом, в результатах измерения могут возникать заметные погрешности. Пиати и Марти (20 ] показали, что погрешность измерения меньше, если ртутный шар термометра погружен в масло, служащее для передачи тепла. На практике термометр без стандартного шлифа помещают на всю его
Таблица 66.
Характеристики термометров для ректификационных установок со стандартным шлифом по TGL 40-339 DIN 12784/55
Форма выполнения Термометра Интервал шкалы, °C Цена деления, °C Интервал измеряемой температуры, °C Погрешность калибровки, °C
От 0 до +110 0,5 ±0,5
От 0 до +250 1,0 0—50 ±0,7
50—100 ±1,0
А или В 100—200 ±1,5
200—250 ±2,0
От 0 до +360 1,0 0—50 ±0,7
50—100 ±1,0
100—200 ±1,5
200—300 ±2,0
300—360 ±2,5
Длина ножки погружения Ц = 50 110 Глубина погружения /2 = 32 92 200 мм 182 мм
430
длину в поток пара или жидкости, хотя это и затрудняет визуальное наблюдение за показаниями. В других случаях необходимо вводить поправку на выступающий столбик ртути (см. разд. 4.13). Поэтому при ректификации предпочтительны термометры со стандартным шлифом, которые отградуированы при постоянной глубине погружения ножки (от нижнего края шлифа до ртутного шара). Характеристики термометров со стандартным шлифом представлены в табл. 66.
Термометры со стандартными шлифами выпускают с интервалом измерения от 0 до 50 и ценой деления 0,1 °C. Изготавливают также угловые термометры. Стеклянные термометры, с ценой деления 0,1 °C, отградуированные соответствующими метрологическим учреждением, служат в качестве эталонов для термометров со стандартными шлифами и применяются при аналитических разгонках. В паспорте термометра указывается глубина его погружения и средняя температура столбика ртути во время градуировки. Для термометров с ценой деления 0,1 °C погрешность измерения лежит в следующих пределах:
0—50 +0,05 °C 150—200 ±0,2 °C
50—100 ±0,05 °C 200—250 ±0,5 °C
100—150 ±0,2 °C
Поскольку погрешность измерения градуированных термометров изменяется в пределах от ±0,2 до ±6 °C, а стекло, из которого они изготовлены, подвержено процессу старения, необходимо периодически проводить проверку термометров. Для этой
цели на термометры наносят вспомогательные метки, которые позволяют установить’изменение погрешности измерения во времени. Термометры других типов необходимо проверять через определенные промежутки времени с помощью стандартного прибора. В качестве стандартного прибора'может быть использован прибор Юнге-Риделя, работающий по принципу аппарата Тиле, который
служит для измерения температуры плавления. Он пригоден для проверки термометров, предназначенных для измерения температур до 300 °C, которые’градуированы при полном погружении. Показания проверяемых термометров сравнивают с показаниями’подобных термометров, отградуированных метрологическим учреждением.
Для проверки термометров со стандартным шлифом автор применял прибор, показанный
Рис. 364.
Прибор Креля для проверки термометров со стандартным шлифом и термопар.
431
Таблица 67.
Вещества, рекомендуемые для проверки термометров в среде пара
Вещество Температура кипения при 760 мм рт. ст., °C Вещество Температура кипения при 760 мм рт. ст., °C
0—5 0 °C 150— 200 °C
Диэтиловый эфир 34,6 Бромбензол 155,6
Хлористый метилен 40,67 Декан 174,0
Ацетон 56,13 Анилин 184,4
50—100 °C Этиленгликоль 197,4
Метил ацетат 56,96 200—300 °C
Хлороформ 61,0 Бензиловый спирт 205,2
Метанол 64,72 Нитробензол 210,6
Четыреххлористый угле- 76,69 Нафталин 217,96
род Дифенил 254,9
Этилацетат 77,06 Бензофенон 305,9
Циклогексан 80,8
Вода 100
100- 150 °C
Толуол 110,6
н-Бутанол 117,75
н-Бутилацетат 126,2
Хлорбензол 131,69
изо-Амилацетат 142,0
на рис. 364, который также пригоден для калибровки термопар. Проверку термометров с помощью этого прибора проводят в среде пара. В табл. 67 указаны вещества, пригодные для проверки термометров в интервале температур от 34 до 300 °C. Для достижения более высоких температур используют глицерин или другое высококипящее термически стабильное вещество и работают под вакуумом. Прибор позволяет проверять одновременно три термометра со стандартным шлифом. При этом следует учитывать нормированные погрешности измерения четвертого термометра, отградуированного метрологическим учреждением. При проверке термометра следят за тем, чтобы температура столбика ртути этого термометра не сильно отличалась от температуры столбика ртути эталонного термометра, при которой проводили его градуировку. В других случаях вводят соответствующую поправку.
Как уже указывалось, для измерения температуры вместо стеклянных термометров можно использовать термопары или термометры сопротивления, которые имеются в продаже [181.
Электрические методы измерения температуры применяют в тех случаях, когда необходимо непрерывно автоматически регистрировать температуру. Преимущество термопар состоит в том, 432
| что с их помощью можно проводить измерения в очень небольшом | объеме, например на поверхности стеклянных стенок пленочной I ректификационной колонны (см. разд. 5.4.3) или других пленочных I ректификационных аппаратов. Кроме того, термопары и термо-I метры сопротивления характеризуются высокой чувствительностью г и широким интервалом измеряемых температур. Для термопар |- такой интервал лежит примерно в пределах от —200 до Д-1800 °C. | Для фиксации температуры холодного спая термопары целе-[‘ сообразно использовать термостаты, работающие по принципу | Пельтье (термоэлектрическое охлаждение) [211.
I Термометры сопротивления, изготовляемые преимущественно К из платины, предназначены для измерения температур в интервале | от —220 до 750 °C. Принцип действия платинового термометра | сопротивления основан на том, что омическое сопротивление термо-| метра, выполненного в виде пластины, цилиндра, прямолинейно К натянутой или свернутой в спираль проволоки, изменяется при- мерно на 0,4% при изменении температуры на 1 °C. Это означает, К что для обеспечения точности измерения в 0,01 °C требуется В фиксировать изменение омического сопротивления в несколько стотысячных долей от его первоначального значения при 0 °C. В Метод измерения температуры выбирают в зависимости от требуе-Е мой точности. Отметим, что при использовании моста Уитстона
можно измерять и регистрировать также разность температур и, следовательно, регулировать мощность электрообогрева кожуха ; колонны по температуре внутри нее и косвенно регистрировать ; флегмовое число. Применяя напыляемые термометры сопротивле-s ния [22 ], можно точно определить среднюю температуру поверх-( ности испарительных свечей или температуру теплопередающих [ поверхностей.
[.. Для измерения температуры паров и жидкости в пилотных I ректификационных установках часто применяют термометр сопро-| 1тивления, снабженный двумя отдельными измери-г тельными обмотками, служащими для одновре-
| l Т менной регистрации и регулирования измеряемой
д-к величины. Термометр Квикфита в полностью
I I---I стеклянном выполнении (рис. 365) имеет соедини-
I п—<===jw тельные штуцеры с диаметрами условного про-| п хода NW 25 и NW 40.
I — •— Для измерения давления паров и определения
I- характеристик фазовых равновесий применяют ус-
I тройства с полупроводниковыми датчиками, свя-
I занными с компенсографами, которые обеспечивают
Рис. 365.
Термометр сопротивления в полностью стеклянном выполнении.
Интервал измерения от -200 до +300 °C.
Изготовитель: фирма «Стеклянная техника». Висбаден —Шир-штайн.
433
точность регистрации температуры до ±0,001 °C. Таким прибором является термометр «Термофил», который особенно пригоден для измерения температур поверхностей, например в пленочных ректификационных колоннах, аппаратах для молекулярной дистилляции и в обогревающих кожухах колонн. В измерительный зонд этого прибора вмонтирован германиевый полупроводник диаметром 0,25 мм, который отбирает от измеряемого объекта только ничтожное количество тепла. Интервал температур, измеряемых этим прибором, лежит в пределах от —50 до Д45О°С. Измеряемый электрический ток усиливается электронным усилителем и фиксируется на градуированной шкале прибора. Кабельные провода, идущие к усилителю от нескольких датчиков, не искажают результатов измерений и к усилителю можно подключать до 10-ти измерительных зондов. Зонд выполняется из стекла или металла.
Для регистрации температур используют выпускаемые промышленностью термографы, которые снабжены 1, 2, 4, 6 и 12 измерительными зондами.
Лабораторную и пилотную ректификацию часто проводят при температурах примерно до 200 °C. Из этого следует, что для регистрации температуры с точностью, характерной для стеклянных термометров, необходимо пользоваться только теми приборами, которые имеют относительную точность не более 0,1% при абсолютной погрешности до 0,25% от интервала измерения. Этому условию удовлетворяют электронные приборы, записывающие показания, и компенсационные самописцы, которые можно подключать непосредственно к платиновому термометру сопротивления (100 Ом при 0 °C). Самописец можно использовать при непрерывной ректификации для регистрации отклонения температуры от заданного значения.
8.2.2. РЕГУЛИРОВАНИЕ ТЕМПЕРАТУРЫ
Для регулирования степени нагревания неподвижных и движущихся жидкостей, а также мощности электрообогрева различных частей аппаратов, например обогревающего кожуха колонны и нагревательных трубчатых змеевиков, широко используют контактные термометры. Эти термометры также имеют стандартные шлифы NS 14,5 (рис. 366). Они выполняются как с жестко впаянными контактами для любой фиксированной температуры, так и с магнитным регулятором. Термометры с впаянными контактами служат для поддержания требуемой температуры, а термометры с магнитным регулятором применяют в качестве датчиков температуры. Высокую чувствительность имеют контактные термометры со спиральным резервуаром для ртути (рис. 367а). Этот резервуар имеет оптимальную поверхность. Рис. 3676 иллюстрирует чувствительность ртутных термометров со спиральным и цилиндрическим резервуарами [23].
Контактные термометры изготавливают в виде максимальноминимальных Термометров, в которых электроконтакт замыкается 434
при переходе через верхний или через нижний предварительно установленные пределы температуры соответственно. С помощью ролика для точной регулировки можно устанавливать заданную температуру с точностью до 0,1 °C [23]. Температуру можно регулировать также и автоматически с помощью синхронного электромотора в зависимости от показаний задающего термометра.
Удобны контактные термометры с трехполюсным штепсельным разъемом и передвижным стандартным шлифом, благодаря которому можно ступенчато изменять длину погружной трубки [24 ].
Контактный термометр, описанный в работе [25], соединяют через реле с регулируемым потребителем электроэнергии, который включается или выключается при замыкании контактов термометра. В лабораторной практике предпочтительны реле с вертикальными ртутными резервуарами, в которых отсутствуют подверженные естественному износу подвижные детали, такие как рычаги, шарниры и гибкие провода [26].
Подобные транзисторные реле, используемые также и для регулирования мощности потребителей электроэнергии с помощью контактных манометров (см. разд. 8.3.1 и 8.4.2), выпускаются комбинированными в виде реле уровня, совмещенного с реле подстройки. Универсальные лабораторные реле снабжены переключателями для работы в нормальноразомкнутом и нормальнозамкнутом положениях. При замыкании контактов термометра реле, работающее в нормальноразомкнутом положении, включает потребитель электроэнергии, например сигнальные звонок и
Рис. 367.
Термометр со спиральным резервуаром для ртутн:
а — общий вид термометра; б — сравнительные данные о чувствительности термометров со спиральным (/) и цилиндрическим (2) резервуарами для ртути, полученные при одинаковом объеме резервуаров.
Рис. 366.
Контактный термометр с вращающимся магнитным регулятором.
435
лампу, а реле, работающее в нормальнбзамкнутом положении, наоборот выключает потребитель электроэнергии, например нагреватель куба, обмотку обогревающего кожуха колонны, клапан. Эти реле применяют и для фотоэлектрических коммутаторов, например для регулирования уровня жидкости или вакуума (см. разд. 8.6 и разд. 8.3.1.1). При использовании этих реле в качестве предохранительных, они отключают потребитель электроэнергии при выходе из строя контактного термометра.
Универсальные электронные приборы, которые можно использовать для настройки и регулирования ректификационной аппаратуры, описаны в работе Фишера [27]. Как указано в этой работе, наряду с электронным реле для регулирования температуры в интервале от —200 до -|-800 °C можно применять пропорциональные регуляторы. В этом случае в качестве измерительного зонда используют термометр сопротивления со стандартным шлифом или фланцем. Преимуществом этих приборов является возможность их использования для регулирования мощности электронагревателей с малой тепловой поверхностной нагрузкой, что особенно необходимо, если для обогрева применяют электронагреватель, который при замыкании цепи включается сразу же на полную мощность.
Особые сложности возникают при регулировании температуры обогревающего кожуха колонны в зависимости от температуры внутри нее. В разд. 7.7.3 были описаны различные способы тепловой изоляции колонн. Вследствие того, что при непрерывной ректификации, и особенно при ректификации многокомпонентных смесей температура внутри колонны постоянно, а часто и скачкообразно повышается, необходимо соответствующим образом регулировать мощность нагрева электроспиралей. При этом из-за тепловой инерции электроспиралей между температурой внутри колонны и температурой обогревающего ее кожуха может возникать градиент до 30 °C. Обеспечивая автоматическое регулирование мощности электроспиралей, удается существенно уменьшить этот температурный градиент. В этом случае в качестве температурных датчиков применяют воздухонаполненные термометры и термопары, или термометры сопротивления. При регулировании температуры с помощью термопар (см. рис. 343), установленных внутри колонны, а именно в ее верхней части и несколько выше куба, они воздействуют на показывающий прибор, который подает через короткие промежутки времени импульсы на коммутатор. При этом электрический контур, который включает электронагреватель кожуха колонны, замыкается [28]. В качестве температурных датчиков автоматических регуляторов мощности электронагревателей кожуха по температуре внутри колонны используются также и контактные термометры Хутла [29].
Автоматический регулирующий прибор для ректификационных установок «Минитрон 5» (см. рис. 386), разработанный Фишером [27], снабжен двумя электронными пропорциональными регуля
436
торами, которые соединяются с электронагревателями куба и кожуха’колонны. В качестве измерительного зонда служит термометр сопротивления Pt 100, работающий в интервале температур 0—350 °C. Температуру обогревающего кожуха колонны можно регулировать по температуре, предварительно установленной на приборе «Гелипот», или посредством автоматической подстройки по температуре внутри колонны. На термометре сопротивления, расположенном в верхней части колонны вместо прибора «Гелипот», устанавливается заданная температура. Автоматическая регулировка обогревающего кожуха колонны осуществляется по температуре пара внутри нее.
Применяя воздухонаполненные термометры, один баллончик которых размещен внутри колонны, а другой —в обогревающем кожухе, автору [11 ] удалось достигнуть точности регулирования ±0,5 °C. Эта точность была обеспечена при использовании контактного манометра с органической токопроводящей жидкостью, который посредством электронного реле включал и выключал электронагревательную обмотку (рис. 368). С помощью данного
Рис. 368.
Схема системы автоматического регулирования температуры обогревающего кожуха колонны по температуре внутри нее:
/ — колонна; 2 — обогревающая электрообмотка; 3 — воздухонаполненный термометр; 4 -- теплоизоляция нз стекловолокна; 5 — кожух колонны;
6 — регулирующий манометр; 7 — электронное реле; 8, 9 — стандартные шлифы NS29 н NS14.5 соответственно.
437
устройства можно обеспечить до 400 включений в 1 ч. Даже при резком повышении температуры внутри колонны температура обогревающего кожуха изменяется конформно температуре внутри колонны. Преимущество данного метода состоит в возможности регулирования температуры обогревающего кожуха, который целесообразно разделить на отдельные секции длиной 25—50 см, по средней температуре внутри соответствующего участка колонны. Если же регулировать обычно принятым способом, т. е. регулировать температуру верхней секции обогревающего кожуха колонны по температуре верхней части колонны, то при отгоне промежуточной фракции температура кожуха будет значительно ниже истинной температуры внутри колонны. В этом случае верхняя часть колонны будет работать как парциальный конденсатор, образующий дополнительное количество флегмы. Кроме того, при использовании этого метода регулирования можно поддерживать температуру обогревающего кожуха выше или ниже заданной величины по сравнению с температурой внутри колонны.
8.3. ИЗМЕРЕНИЕ И РЕГУЛИРОВАНИЕ
ДАВЛЕНИЯ
Перегонку проводят обычно в определенных интервалах давления. По рабочему давлению можно следующим образом классифицировать процессы перегонки: область давлений выше 760 мм рт. ст. — ректификация поддавлением; область давлений 760 —1 мм рт. ст. — вакуумная дистилляция и ректификация, расширительная перегонка, пленочная ректификация; область давлений 1—1(Г6ммрт. ст. — пленочная перегонка, молекулярная дистилляция. •
Для каждой из указанных областей применяют различные методы измерения и регулирования давления [2 ], которые рассматриваются в следующих разделах [30]. Подробный обзор методов измерения остаточного давления при перегонке под вакуумом представлен Леком [31 ].
8.3.1. ИЗМЕРЕНИЕ И РЕГУЛИРОВАНИЕ ДАВЛЕНИЯ
В ОБЛАСТЯХ ВЫШЕ 760 И ОТ 760
ДО 1 мм рт. ст.
При ректификации под давлением до 2 кгс/см2 (см. разд. 5.4.5) для измерения избыточного давления применяют U-образные манометры * высотой до 1 м (рис. 369). Давление в ректификационной колонне можно рассчитать по формуле:
рк = 6 4-Др (195)
где b — барометрическое давление с учетом необходимых поправок, мм рт. ст.; Др — избыточное давление по показаниям манометра.
* Манометры данного типа обычно заполняют ртутью. — Прим. ред.
438
При необходимости измерения более высоких давлений можно пользоваться выпускаемыми промышленностью мембранными манометрами или манометрами с трубчатой пружиной (Бурдона), которые пригодны также для измерения давления в интервале от 760 до - 0,1 мм рт. ст. Один из методов регулирования избыточного давления в процессе ректификации описан в разд. 5.4.5.
Анероид-регулятор абсолютного давления типа FA 149 (изготовитель: фирма «Баллас Тирнау-хлоратор», Гюнцбург) позволяет регулировать давление в интервале от 20 до 1520 мм рт. ст. с точностью до 0,25 мм рт. ст.
В ходе ректификации, протекающей при атмосферном давлении, необходимо также непрерывно регистрировать давление в колонне, которое в этом случае равно барометрическому давлению. Это давление нужно знать для того, чтобы привести значения температуры к давлению 760 мм рт. ст. (см. разд. 4.13).
Для измерения атмосферного давления с точностью ±0,1 мм рт. ст. следует применять прецизионные ртутные барометры, которые изготавливаются в виде рычажных, чашечных или комбинированных рычажно-чашечных моделей [32]. В лабораторной практике часто пользуются барометрами Гей-Люссака и Шродта-Кифера. Надежный’отсчет показаний с точностью ±0,1 мм рт. ст. обеспечивает нониус, или еще лучше —катетометр. Прибор для измерения давления жидкости, разработанный Никелем [32], учитывает влияние поверхностного натяжения исследуемой жидко
сти и позволяет корректировать смещение начала отсчета, возникающее, например,* при изменении температуры.
Для получения поправок в определенном интервале измерения
давления рекомендуется в течение длительного времени сравнивать показания барометра с' показаниями образцовых стационарных
манометров метеорологических станций. Подобным образом необ-
ходимо проверять также барографы, снабженные анероидами. Точность показаний таких приборов обычно составляет ±0,5 мм рт. ст. Эти приборы имеют устройства для регистрации показаний на бумажной ленте в течение суток или недели. Пользуясь такими лентами с записанными показаниями, можно позднее определить изменения давления в определенные промежутки времени, интересующие исследователя. Погрешность в измерениях
Рис. 369.
Манометр для измерения избыточного давления нлн вакуума в процессе ректификации:
1 — предохранительный сосуд; 2 — штуцер^для присоединения манометра; 3 —^шкала; 4 — сосуд для залива манометрической
жидкости.
439
Рис. 370.
Баровакуумметр
Изготовитель: народное предприятие «Комбинат технического стекла», Ильменау.
Рис. 371.
Укороченный манометр Беннерта для измерения остаточного давления от 0 до 180 мм рт. ст.
давления по анероиду, обусловленную изменением температуры в интервале от —40 до 4-40 °C, обычно устраняют с помощью биметаллического компенсирующего устройства.
При ректификации в условиях пониженного давления также применяют U-образные манометры высотой 1 м с одним открытым концом (см. рис. 369). Хорошо очищенную ртуть заливают в манометр, избегая попадания пузырьков воздуха, через предохранительный сосуд 4. Манометр присоединяют к аппарату с помощью штуцера 2. Сосуд 1 является также предохранительным на случай резкого возрастания вакуума в системе. При помощи шкалы 3, передвигающейся по вертикали, измеряют остаточное давление в системе Др. Абсолютное давление в колонне вычисляют по формуле:
рк=Ь —Др (196)
440
Преимущество этих манометров состоит в том, что они позволяют регистрировать показания во всем интервале давления от 760 до 1 мм рт. ст. с точностью ±0,5 мм рт. ст. Для более точного определения расстояния между менисками двух столбиков ртути необходимо применять вертикальные компараторы или катетометры.
Фаркварсон и Кермицле [331 предложили устройство для автоматической регистрации высоты столбика ртути. Прибор работает при высоте столбика ртути до 80 см с точностью, превышающей ±0,05 мм, и максимальной погрешностью ±0,11 мм.
Снятие показаний существенно облегчается при использовании баровакуумметров (рис. 370), в которых совмещено измерение остаточного и избыточного давлений. Приборы, выпускаемые промышленностью, пригодны для измерения барометрического давления в интервале от 600 до 820 мм рт. ст. и остаточного давления в пределах от 1 до 310 мм рт. ст. Высокая точность измерения обеспечивается благодаря применению калиброванных труб с отклонением внутреннего диаметра от заданного размера в пределах ±0,01 мм и устройства для автоматической поправки на отклонение уровня ртути от нулевого положения [32].
Укороченные манометры Беннерта (рис. 371) с закрытым вакуумированным коленом предназначены для измерения остаточного давления от 0 до 180 мм рт. ст. Точность отсчета показаний у этих приборов составляет ±0,5 мм рт. ст. Однако при измерениях в интервале давлений от 0 до 10 мм рт. ст. могут возникать значительные погрешности вследствие того, что воздух не полностью удаляется из закрытого колена манометра или проникает в него во время эксплуатации. Для предотвращения подобных ошибок измерения необходимо периодически сверять показания данного прибора с показаниями U-образных манометров полной длины (см. рис. 369). В модифицированном укороченном манометре Штрё-лейна на конце левого колена предусмотрен ртутный затвор, позволяющий удалять попавший в манометр воздух.
Наиболее вероятные источники погрешностей измерения при работе с жидкостными манометрами рассмотрены в работе Доша [34]. С помощью прецизионных масштабных линеек и нониусов можно определять высоту столба жидкости в манометре с максимальной погрешностью до 0,2 мм. Точный вакуумметр системы Голланд-Мертена [35 ] в интервале от 200 до 20 мм рт. ст. обеспечивает точность измерения ±0,5 мм рт. ст. Этот прибор позволяет измерять с помощью наклонного колена остаточное давление в пределах от 20 до 0 мм рт. ст. с точностью ±0,1 мм рт. ст. (рис. 372).
Точность измерения давления может быть значительно повышена путем замены ртути на силиконовое масло. Такой наполнитель используется в манометре Лапорта, предназначенном для измерения давления в пределах от 0,2 до 20 мм рт. ст., и в вакуумметре, работающем на принципе вытеснения наполнителя [35 ], с точностью 0,01 мм рт. ст. в интервале от 0 до 10 мм рт. ст. В каче
441
стве наполнителей манометров не следует использовать масло для вакуумных насосов, парафиновое масло и жидкость «амоил S». Повышенная способность к поглощению влаги, характерная для этих жидкостей, приводит к значительным погрешностям измерения. Бахман рекомендует при заполнении манометров водой добавлять к ней вещества, уменьшающие поверхностное натяжение воды. Для измерения давления в интервале 760— 1 мм рт. ст. можно использовать также манометр Мак-Леода. Этот прибор применим в основном для измерения остаточных давлений от 10-1 до 10-6 мм рт. ст., поэтому он описан ниже в соответствующем разделе. Для интервала давления от 0 до 60 мм вод. ст. можно применять указанные выше контактные манометры [36а].
Основное преимущество мембранных манометров заключается в том, что точность измерения с помощью этих приборов не зависит от химической природы исследуемых газов или паров [371. Измерительным устройством в данных приборах является тонкая металлическая мембрана. Мембранный вакуумметр VM-M (фирма Хэраеуза, Ханау) работает в интервале остаточных давлений от 50 до 0,1 мм рт. ст. с точностью ртутного манометра (рис. 373). Данный вакуумметр особенно пригоден для лабораторных и пилотных ректификационных установок. Существенное отличие этого прибора заключается в том, что его чувствительность не зависит от возможного проникновения в него воздуха. Прецизионные приборы для измерения давления, эталонные ртутные дифманометры и анероиды с круглой шкалой выпускает большими партиями фирма «Баллас Тирнау-хлоратор» в Вассербурге. В пилотных
Рис. 372.
Точный вакуумметр системы Голланд-Мертена.
Рис. 373.
Мембранный вакуумметр VM-M
Изготовитель: фирма Хэраеуза, Ханау.
442
Рис. 374.
Схемы систем регулирования давления методом подсоса воздуха (а) и методом частичного вакуумирования (6)'.
1 — вакуумный насос; 2 — буферная емкость; 3 — манометр; 4 — регулятор давления; 5 — электронное реле.
ректификационных установках целесообразно использовать также пружинные манометры различных конструкций [2 1.
При измерении давления с помощью любых приборов важно устанавливать перед манометром охлаждаемую ло-
вушку для задержки конденсирующихся паров. Необходимо также наблюдать за тем, чтобы между участком конденсации паров и точкой измерения давления не возникал градиент давления. С этой целью
до начала разгонки следует проводить сравнительные измерения давления, поочередно присоединяя манометр к шлифу для термометра и к штуцеру, расположенному за охлаждаемой ловушкой.
Для автоматического регулирования остаточного давления в интервале от 760 до 1 мм рт. ст. применяют механические и комбинированные электронномеханические приборы [38]. При
этом различают метод подсоса воздуха и метод частичного вакуумирования. При использовании метода подсоса воздуха вакуумный насос работает непрерывно, и в буферный сосуд с помощью крана тонкой регулировки подают такой объем воздуха, чтобы в системе установилось заданное давление. Основным условием успешного применения данного метода является равномерная работа вакуумного насоса и полная герметичность аппаратуры (рис. 374, а), поэтому этот способ используется только в исключительных случаях.
I 8.3.1.1. Методы частичного вакуумирования
| При регулировании давления методом частичного вакуумирования I вакуумный насос включается только тогда, когда давление в бу-I ферном сосуде превысит предварительно заданное давление, на J которое настроен регулятор. Схема системы регулирования давле-I ния по этому методу представлена на рис. 374, б.
I В интервале от 760 до 10 мм рт. ст. можно применять, например, I регулятор давления с пористой пластиной [39], показанный I на рис. 375. В качестве запорной жидкости в этом регуляторе I применяют ртуть. Количество ртути определяют из условия, что В ее уровень должен быть выше примерно на 3 мм нижнего края I пористой пластины. При открытых кранах 1—3 ректификационную
443
Рис. 375.
Регулятор давления с пористой пластиной;
1 — 3 — краны; 4 — сосуд с пористой пластиной; 5 — сосуд, находящийся под заданным остаточным давлением.
колонну вакуумируют до тех пор, пока давление, показываемое манометром, не будет превышать заданное на 1 —2 мм рт. ст., после
чего кран 3 закрывают. Давление в сосуде 5 начинает уменьшаться,
что вызывает соответствующее снижение уровня ртути в сосуде с пористой пластиной 4. Как только Пористая пластина 4 станет освобождаться от ртути, кран 1 перекрывают. При этом остаточное давление в сосуде 5 окажется меньше, чем давление в сосуде с пористой пластиной 4, и эта разность давлений будет определяться расстоянием между уровнями ртути в сосудах. При возрастании давления в ректификационной установке расстояние между уровнями ртути еще более увеличится и пористая пластина полностью освободится от ртути. В этот момент включается вакуумный насос, который вакуумирует систему до тех пор, пока давление в ректификационной установке не уменьшится до заданного значения и пористая пластина вновь покроется ртутью. При термостатировании данного регулятора давления, удается поддерживать заданный вакуум с точностью ±0,5 мм рт. ст.
Механический регулятор давления Джильмонта [40 ] работает на принципе поплавка с переменной глубиной погружения. Его можно использовать для регулирования остаточного давления не ниже 6 мм рт. ст. На рис. 376 показан этот регулятор, смонтированный на деревянном щите вместе с манометром Штока и обратным клапаном Фридрихса—Антлингера [41 ]. Точность регулирования данного устройства составляет ±0,5 мм рт. ст.
Применяя электронные приборы, можно повысить точность регулирования давления до ±0,1 мм рт. ст. Ртутный манометр можно заменить манометром, заполненным какой-либо высоко-кипящей, электропроводной и дегазированной жидкостью, что обеспечивает повышение чувствительности прибора примерно в 10 раз. При этом разность давлений в 1 мм рт. ст. будет соответствовать разнице уровней жидкости в 10—13 мм. Наименьшее давление, измеряемое с помощью прибора, в этом случае определяется давлением паров заполняющей жидкости. В жидкостном манометре Дубровина, который основан на фотоэлектрическом методе измерения, на фотоэлемент направляют тонкий световой пучок. При увеличении давления в аппаратуре поплавок, всплывая, перекрывает луч света, и неосвещаемый фотоэлемент включает через реле вакуумный насос [42].
444
Другой метод регулирования давления заключается в том, что к манометрической жидкости, например бутилгликолю, добавляют небольшое количество соли, например нитрита натрия, для повышения ее электропроводности. При этом сила тока увеличивается до 10"6 А (напряжение 6—8 В) и с помощью электронного реле (см. разд. 8.2.2) ее можно усилить до 10—15 А при напряжении 220 В. Для регулирования давления в правом колене манометра создают заданное давление, причем это колено термостатируют для исключения влияния изменений температуры (см. рис. 374,6). При равенстве давлений в правом колене и в ректификационной установке уровень манометрической жидкости в обоих коленах манометра одинаков. При повышении давления в установке уровень жидкости в правом колене понижается. В результате повышения уровня жидкости в левом колене электрический контакт замыкается и через электронное реле включается вакуумный насос, который вакуумирует систему до выравнивания давлений.
С помощью автоматического стенда с вакуумным насосом (рис. 377) во время испытаний с чистыми веществами в интервале давлений от 300 до 1 мм рт. ст. была достигнута точность регулирования ±0,1 мм рт. ст. [431. Такая точность для перегонки фенола при давлении 20 мм рт. ст. соответствует разнице температур кипения ±0,1 °C. Если, например, при разделении изомеров ксилола при давлении 70 мм рт. ст. необходимо определять темпера-
Регулятор давления Джильмоита, основанный на принципе поплавка с переменной глубиной погружения.
Рис. 377.
Автоматический стенд с вакуумным насосом, разработанный Крелем. Изготовитель: народное предприятие «Комбинат технического стекла», Ильменау.
445
Рис. 378.
Автоматический регулятор давления Штаге:
1 — поплавок; 2 — масляный манометр; 3 — контакт; 4,6 — штуцеры для присоединения к аппаратуре и вакуумному насосу; 5 — электромагнитный клапан.
Рис. 379.
Прибор для поддержания постоянного вакуума VK1:
1 — «автоматика»; 2 — «установка»; 3 — штуцер для залива запорной жидкости (уровень жидкости должен быть ниже конца контакта примерно на 2 мм).
Изготовитель: фирма Фишера «Лабораторная и исследовательская техника», Бони —курорт Годесберг.
туру кипения с точностью 0,1 °C, то для этого потребуется регулировать давление с точностью, превышающей ±0,15 мм рт. ст. [44 ]. Регулятор давления Штаге (рис. 378) имеет поплавок 1, который размещается во внутреннем колене масляного манометра 2, причем верхний конец контакта 3 лежит непосредственно на весах. Штуцер 4 служит для присоединения регулятора к колонне. При возрастании давления в колонне уровень масла вместе с поплавком
1 в манометре 2 опускается и реле открывает электромагнитный
клапан 5, соединенный с вакуумным насосом с помощью штуцера
6. В приборе Фишера [27], служащем для поддержания в системе
постоянного вакуума, регулирование осуществляется посредством
манометра с наклонной трубкой; манометр заключен в покрытый
слоем серебра вакуумированный кожух (рис. 379).
8.3.2. ИЗМЕРЕНИЕ И РЕГУЛИРОВАНИЕ ДАВЛЕНИЯ
В ИНТЕРВАЛЕ ОТ 1 ДО 10 * 1 * * * * * * 8 мм рт. ст.
Измерение вакуума в области очень низких давлений связано со значительными трудностями, поскольку при очень малых плотностях газа непосредственное измерение его давления затруднено. В этих случаях применяют косвенные методы измерения. Методом, основанным на применении тлеющего газового разряда,
446
можно лишь приближённо определять область измеряемых давлений. Флуоресцентный вакуускоп Бюргера позволяет определять остаточные давления, равные 10'1, 10'3 и 10'5ммрт. ст., а промежуточные значения давлений можно только оценивать. Для обеспече-
ния воспроизводимости результатов при пленочной и молекулярной дистилляции рекомендуется применять методы измерения давления, приведенные в табл. 68.
Методы измерения сверхвысокого вакуума, соответствующего остаточным давлениям от 2-10'10 до 10'12 мм рт. ст., описаны Хохом [46]. Сведения о преимуществах и недостатках манометров, приведенных в табл. 68, можно найти в работе Штекельмахера [47 ].
Альфатрон представляет собой ионизационный манометр, в котором в результате бомбардировки молекул остаточного газа «-частицами образуются ионы [45]. В молекулярном вакуумметре (абсолютном манометре Кнудсена) для измерения вакуума используют эффект радиации. Широкое применение находят молекулярные вакуумметры Геде [48] и вакуумметры Лангмюра с кварцевой нитью [49]. Работа мембранного микроманометра, так же как и
молекулярного вакуумметра, не зависит от химической природы исследуемого газа. Этот прибор позволяет измерять давление в интервале от КГ4 до 1,5-10'1 мм рт. ст. Методика измерения остаточных давлений с помощью диффузионных ртутных насосов описана Мюллером [50].
В настоящей монографии рассмотрены только некоторые модели вакуумметров, выпускаемых промышленностью. Подробное и полное описание этих приборов можно найти в справочнике по
Таблица 68.
Область применения манометров и интервал измеряемых давлений (по Мёнху [45])
Тип манометра или вакуумметра Вид измеряемого давления Зависимость показаний от вида газа-наполиителя Интервал измеряемых давлений, мм рт. ст.
Спиральный и мембран- Общее давление Не зависят 760-Ю-3
ный манометры Газоразрядный манометр Общее давление Зависят 10-2—Ю-»
(вакуумметр Филипса) Демпферный и вязкост- Общее давление Зависят ю-1—10'4
ный манометры Манометр Пирани, ос- Общее давление Зависят ю-1—10'4
нованный на измерении теплопроводности -
Компрессионный мано- Парциальное дав- Не зависят (толь- 760—10"1
метр Мак-Леода ление не конденсирующихся паров ко для газа) Ю-1— ю-s
Ионизационный манометр Общее давление Зависят IO-3— ю-6
Радиометр (молекулярный вакуумметр) Общее давление Не зависят 10"1—IO'8
447
технике высокого вакуума [49]. Для измерения давлений от 10"1 до 10"4 мм рт. ст. используются манометры Пирани, основанные на принципе измерения теплопроводности, и в интервале от 10-3 до 10~6 мм рт. ст. применяются ионизационные манометры. В настоящее время промышленностью выпускаются приборы, являющиеся комбинацией двух последних манометров.
Преимущество электрических вакуумметров состоит в том, что их показывающее устройство непрерывно фиксирует измеряемое давление, благодаря чему появляются возможности для его регулирования и записи показаний. Как вакуумметр, основанный на цринципе измерения теплопроводности, так и ионизационный вакуумметр, измеряют общее давление газов и паров, причем для определения остаточного давления газа их необходимо подключать к системе через охлаждаемую ловушку.
Калибруют эти приборы известным методом с помощью вакуумметра Мак-Леода при использовании предварительно осушенного воздуха. Соответствующие калибровочные кривые представлены в технической документации на прибор. Точность измерения манометра Пирани составляет 5% в интервале давления от 1 до 5-10~3 мм рт. ст., а ионизационного вакуумметра —3% в интервале давления от 1СГ3 до 10'6 мм рт. ст. Для подсоединения этих приборов к вакуумированной ректификационной установке они снабжаются штуцером со стандартным шлифом NS 14,5/23 или небольшим фланцем.
8.3.2.1. Компрессионный манометр Мак-Леода
Компрессионные манометры точно измеряют давление только идеальных газов. Они основаны на принципе сжатия определенного объема газа при измеряемом давлении в капиллярной трубке до давления, которое можно легко измерить. Измеряемое давление с достаточной точностью можно рассчитать по формуле:
p=A/tV2/V1 (197)
где р — измеряемое давление, мм рт. ст.; Д/г — разность уровней ртути в капиллярах, мм; Vi — объем стеклянного шараЗ (рис. 380) с капилляром 5 (считая от места присоединения 2 трубы 7), мл; У2 — объем сжатого газа в капилляре 5.
На рис. 380 показана одна из выпускаемых промышленностью моделей манометра Мак-Леода, в которой уровень ртути поднимается не с помощью груши со ртутью, а под действием атмосферного давления. Сосуд 12 заполняют тщательно очищенной ртутью и к штуцеру 6 подключают аппарат, в котором требуется измерить давление. Штуцер 10 соединяют с водоструйным насосом. При вакуумировании аппарата необходимо одновременно вакуумировать и пространство над ртутью в сосуде 12. С помощью крана 9 регулируют давление таким образом, чтобы не возникало слишком больших перепадов давления между объемом манометра и свободным объемом сосуда 12, так как иначе либо ртуть может подняться 448
очень высоко по манометрической трубке 1, либо воздух по этой трубке проникнет в сосуд с ртутью 12.
После установления в аппарате заданного давления с помощью крана 9 через штуцер 11 в сосуд 12 подают воздух, что приводит к подъему ртути в манометрической трубке 1. Для предотвращения попадания частиц пыли и капелек влаги в сосуд 12 целесообразно снабдить штуцер 11 специальным фильтром. Скорость подачи воздуха регулируют таким образом, чтобы ртуть медленно поднималась до отметки 8, указанной на верхнем конце капилляра 5. Измеряют разницу уровней ртути Ай и по формуле (197) рассчитывают давление р. Значение объема указано на стеклянном шаре 3, а объема V2 —на шкале капилляра 5.
Большинство Манометров данного типа откалиброваны таким образом, что измеряемое давление можно непосредственно регистрировать по шкале. При установке уровня ртути на отметке 4 по шкале, размещенной у трубы 7, определяют давление в интервале до КГ2 мм рт. ст. Перед каждым новым измерением с помощью подключения водоструйного вакуумного насоса нужно опустить уровень ртути в приборе ниже места присоединения 2 трубы 7. Объем стеклянного шара 3 и количество ртути, необходимое для заполнения манометров Мак-Леода, выпускаемых промышленностью, приведены в табл. 69.
Многие модели манометров Мак-Леода рассмотрены в книге Эберта [51 ]. Пневматически регулируемый и самоустанавливаю-щийся компрессионный манометр Мак-Леода, показания которого не зависят от давления окружающего воздуха, подробно описан Пехе [52]. В этой работе приведены также рекомендации по обеспечению воспроизводимости результатов измерений.,
На рис. 381 показана одна из моделей вакуускопа, являющаяся комбинацией укороченного U-образного манометра и компрессионного манометра. Данным прибором можно измерять давление
Рис. 380.
Компрессионный манометр Мак-Леода:
/ — манометрическая трубка; 2 — место присоединения трубы 7; 3 — стеклянный шар; 4, 8 — верхние отметки шара и капилляра; 5 — капилляр; 6, 10 — штуцеры для присоединения аппарата и водоструйного насоса; 9 — край; 11 — штуцер для подачи воздуха в сосуд со ртутью; 12 — сосуд со ртутью.
Рис. 381.
Вакуускоп.
Интервалы измеряемых давлений от 80 до 1 мм рт. ст. (по U-образному манометру) и от 4 до 10“2 мм рт. ст. (по компрессионному манометру).
V215 Э. Крель
449
Таблица 69.
Объем стеклянного шара и количество ртути, необходимое для заполнения манометров Мак-Леода
Интервал измеряемых давлений» мм рт. ст. Объем стеклянного шара, мл Объем ртути, мл
2—10-» 300 450
2—IO"4 100 250
15—IO'3 15 50
в интервале от 80 до 1 мм рт. ст. с помощью U-образного манометра и от 4 до 10"2 мм рт. ст. с помощью компрессионного манометра.
Для лабораторных ректификационных установок очень удобны поворотные манометры Мозера (рис. 382). Они рассчитаны на три интервала измеряемых давлений 700—1, 1—10-2 и 10"2—10"4 мм рт. ст.; для их заполнения требуется всего лишь 6—7 мл ртути. Переход от одного интервала к другому может быть осуществлен поворотом манометра на шлифе против часовой стрелки. Прибор заполняют ртутью настолько, чтобы в положении «г» ртуть, находящаяся при разряжении, только на половину заполняла расширение А. Манометр поворачивают до тех пор, пока столбик ртути не достигнет отметки Мг (интервал измеряемых давлений от 700 до 1 мм рт. ст.) или М2 (интервал от 1 до 10-2 мм рт. ст.). В каждом интервале измерений давление определяют по шкалам пъ п2 и п3. Как и большие манометры Мак-Леода, этот прибор перед каждым новым измерением необходимо возвращать в исходное положение. Манометр Мозера можно проверять и калибровать с помощью большого манометра Мак-Леода.
Компрессионный манометр Каммерера [53] также отличается весьма малым количеством используемой ртути (рис. 383). Его
Рис. 382.
Поворотный манометр Мозера:
а — вид сбоку; б — исходное положение; в — положение при измерении интервале от 700 до 1 мм рт. ст.; г — то же в интервале от 1 до 10“*; д —
то же в интервале от 10“2 до КГ* мм рт. ст.
450
Рис. 383.
Компрессионный манометр Каммерера.
При измерении давлений от 10 до 10‘а мм рт. ст. требуется 16 мл ртути, а в интервале* от 1 до 10“* мм рт. ст. — 17 мл.
преимуществом является также весьма простой метод заполнения ртутью с по-мощью насоса новой модели. Такие приборы выпускаются для интервалов измеряемых давлений от 10 до 10-8 мм рт. ст. и от 1 до 10-4 мм рт. ст.; для измерения остаточных давлений до 80 мм рт. ст. их снабжают U-образными трубчатыми манометрами.
В заключение еще раз отметим, что все компрессионные манометры можно применять для измерения парциального давления газов, неконденсирующихся при за-
данных степени сжатия и температуре. Пары веществ, имеющие при комнатной температуре давление выше 4 мм рт. ст., можно легко обнаружить по расхождению результатов измерений, воз-
никающему при переходе от одного интервала измерений к другому [51 ].
8.3.2.2. Регулирование давления в интервале от 100 до 10-8 мм рт. ст.
Об автоматическом регулировании остаточного давления в областях среднего и высокого вакуума в литературе имеется сравнительно мало сведений. При использовании вакуумметров, основанных на принципе измерения теплопроводности газа, Лапорт [49] рекомендует подключить к мостовой схеме Пирани сигнальное устройство, которое дает звуковой сигнал при увеличении давления выше заданного предела. Нисбет [54 ] описал прибор, позволяющий поддерживать в сосуде, продуваемом воздухом, постоянное давление 10-4 мм рт. ст. Мельпольдер [55] описал регулятор давления, обеспечивающий в интервале от 10-8 до 10-8 мм рт. ст. точность регулирования, равную ;±10'4 мм рт. ст. Схема данного регулятора приведена на рис. 384. Принцип его работы заключается во введении в манометр Мак-Леода четырех впаянных контактов 9—12. С помощью устройства 13 в манометре Мак-Леода каждую минуту поднимают уровень ртути. Регулирование давления осуществляется с помощью контактов 9 и 11. При уменьшении давления в системе ниже заданного контакт 10 замыкается, при этом он через реле 3 и 2 закрывает электромагнитный клапан 5. Этот клапан размещен на штуцере 4, соединяющем систему с вакуумным насосом. Вакуумированный аппарат подсоединяют к шту-
451
4
5
Рис. 384.
Схема регулятора давления Мельпольдера, работающего в интервале от 10"* до 10"® мм рт. ст.:
1 — переключатель; 2,3 — реле; 4,6 — штуцеры для присоединения вакуумного иасоса и аппарата; 5 — электромагнитный клапан; 7 — буферная емкость; 8 — охлаждаемая ловушка; 9—12 — впаянные контакты;
13 — устройство для подъема уровня ртути в манометрической трубке манометра Мак-Леода; 14 — манометр Мак-Леода.
церу 6, буферная емкость 7 служит для уменьшения колебания давления. В ловушке 8, охлаждаемой жидким азотом, задерживаются пары конденсирующихся веществ. При вакуумировании системы в период подготовки регулятора к работе переключатель 1 устанавливают в положение «автоматическое регулирование», как только остаточное давление в системе достигнет 10 мм рт. ст.
Электронный прибор Фишера [27] VKH для поддержания постоянного вакуума, основанный на принципе измерения теплопроводности газов, работает в интервале остаточных давлений от 100 до 10-3 мм рт. ст. (рис. 385). Датчик прибора подключается к вакуумируемой системе с помощью стандартного шлифа. Прибор
имеет переключатель, служащий для переключения регулятора с одного интервала
Рис. 385.
Электронный прибор для поддержания постоянного вакуума VKH
Изготовитель; фирма Фишера, «Лабораторная и исследовательская техника», Бонн— курорт Годесберг.
452
йЗМёрёния на другой. К выходному концу компенсационного усилителя непосредственно подсоединен магнитный клапан, размещенный на трубопроводе, который связывает систему с вакуумным насосом.
8.4. РЕГУЛИРОВАНИЕ ФЛЕГМОВОГО ЧИСЛА
И СКОРОСТИ ВЫКИПАНИЯ
8.4.1. РЕЛЕ ВРЕМЕНИ ДЛЯ РЕГУЛИРОВАНИЯ ФЛЕГМОВОГО ЧИСЛА
Автоматические головки ректификационных колонн, как правило (см. разд. 7.5), работают по принципу регулирования по времени. Флегмовое число при этом определяется отношением промежутка времени выключения реле (подача флегмы в колонну) к промежутку времени его выключения (отбор дистиллята). Устанавливаемые на головках колонн механические или электронные реле времени должны обеспечивать любую продолжительность отбора дистиллята. При малых флегмовых числах от 1 до 5 можно, например, устанавливать продолжительность отбора дистиллята, равной 1 с, что соответствует продолжительности подачи флегмы в колонну от 1 до 5 с. При работе с высокими флегмовыми числами в интервале от 10 до 50 продолжительность включения реле должна быть настолько уменьшена, чтобы скорость отбора дистиллята соответствовала скорости обогащения жидкости, орошающей колонну, высококипящим компонентом. Это означает, что особенно в период отбора промежуточных фракций промежуток времени включения реле (отбор дистиллята) необходимо постепенно сокращать, а промежуток времени выключения реле (подача флегмы в колонну) — соответственно увеличивать.
Эти условия соблюдены в механическом реле времени, разработанном автором [11]. Прибор имеет контактную шайбу, приводимую во вращение синхронным электромотором. Необходимое флегмовое число устанавливается с помощью шибера. Клеммы служат для подключения электронного реле, приводящего в действие электромагнит, установленный в головке колонны. Выходное напряжение электронного реле составляет 220 В. С увеличением флегмового числа одновременно сокращается период включения реле для отбора дистиллята:
Флегмовое число.....................
Период включения реле, с ...........
5 10 20 50
2 1 0,5 0,2
Если электронное реле снабдить переключателем для работы в нормальнозамкнутом и нормальноразомкнутом положениях, то
15 Э. крель 453
можно устанавливать обратные величины флегмовых чисел, т. е. получать флегмовые числа меньше единицы:
При нормальнозамкнутом положении .......................... При нормальноразомкнутом положении ........................
1 2 3 5 10
1 1/2 1/3 1/5 1/10
Длительность и частоту включений реле можно произвольно варьировать путем замены контактных шайб. Этот метод обеспечивает повышенную точность регулирования флегмового числа в интервале от 20 до 100 при аналитической ректификации. При опытно-промышленных и промышленных разгонках период включения реле при отборе дистиллята увеличивают до 30 с.
В настоящее время более предпочтительными являются электронные реле, не имеющие каких-либо механических устройств. Их продолжительность включения и выключения изменяется в интервале от 0,1 с до 20 мин с точностью до 1% 156]. Для задания определенного флегмового числа с помощью этих реле необходимо устанавливать две величины — продолжительность включения и продолжительность выключения. Промышленностью выпускается ряд моделей таких реле, которые обычно снабжены устройством для контроля за температурой кипения. При переключении головки колонны на работу с бесконечным флегмовым числом в верхней части колонны температура повышается до значения, установленного на контактном термометре, который размещен в головке колонны. При понижении температуры в головке колонны ниже установленной прибор автоматически восстанавливает предварительно заданное флегмовое число.
На рис. 386 показан ректификационный регулирующий прибор «Минитрон 5», который одновременно с регулированием продолжительности включения и выключения реле регулирует также температуры куба и обогревающего кожуха ректификационной
Рис. 386.
Регулирующий прибор для ректификационных установок «Мииитрои 5»: 1 — регулятор температуры куба; 2 — стабилизатор температуры обогревающего кожуха; 3 — регулятор температуры обогревающего кожуха; 4 — переключатель периода времени подачи флегмы; 5 — переключатель периода времени отбора дистиллята^ 6 — амперметр; 7 — выключатель контактного термометра; 8 — розетка для подключения к электросети; 9 — функциональный переключатель.
Изготовитель: фирма Фишера «Лабораторная и исследовательская техника», Бонн —курорт Годесберг.
454
колонны (см. разд. 8.2.2). Реле времени имеет 11 ступеней для установки продолжительности отбора дистиллята в интервале от 0,5 до 10 с и 11 ступеней для установки продолжительности подачи флегмы в колонну в интервале от 1 до 100 с. При этом для каждого выбранного периода времени включения реле можно устанавливать любой из одиннадцати промежутков времени подачи флегмы, что позволяет изменять флегмовое число в интервале от 1 до 200. Преимуществом прибора является также возможность подключения к транзисторному реле устройства, обеспечивающего прекращение отбора дистиллята или общее отключение ректификационной установки, что сопровождается звуковым и оптическим сигналами.
Техника регулирования подачи флегмы с помощью автоматических головок ректификационных колонн и электронных реле времени подробно рассмотрена в обзорной работе Рёка с сотр. [13 ]. Геммекер и Штаге [57] показали, что постоянный расход флегмы, а также его воспроизводимость и независимость от нагрузки колонны, можно обеспечить, только применяя регулируемые электромагнитные делительные устройства. Возникающие при этом погрешности регулирования могут быть обусловлены следующими причинами: а) флегмовое число превышает отношение периодов выключения и включения реле: при конденсации пара в нижней части делителя флегмы и непопадании этого конденсата в делительную воронку; при стекании части конденсата, образовавшегося в дефлегматоре, мимо делительной воронки; при попадании части паров в дефлегматор в момент отбора дистиллята, когда регулирование флегмового числа производится делением парового потока; б) флегмовое число меньше, чем отношение периодов выключения и включения реле: при наличии небольшого остаточного количества жидкости в «мертвом» объеме электромагнитного клапана; при конденсации пара в системе отбора дистиллята; при образовании в дефлегматоре капелек жидкости, не стекающих в колонну при отборе дистиллята, когда регулирование флегмового числа производится делением парового потока.
8.4.2. РЕГУЛИРОВАНИЕ СКОРОСТИ ВЫКИПАНИЯ
Необходимым условием регулирования флегмового числа по заданным периодам включения и выключения реле времени является постоянство нагрузки колонны по пару. При установке флегмового числа вручную регулирование скорости выкипания, необходимо для обеспечения постоянной разделяющей способности колонны и воспроизводимости результатов. Необходимую скорость выкипания можно поддерживать двумя методами: стабилизацией мощности электронагревателя куба (преимущественно в процессах дистилляции) и регулированием этой мощности по перепаду давления потока пара в колонне (в процессах ректификации).
Метод стабилизации мощности электронагревателя куба может быть, конечно, использован и в процессах ректификации, однако 15* 455
Рис. 387.
Устройство для регулирования нагрузки по перепаду давления в колонне: 1 — расходомер; 2 — холодильник; 3 — контактный манометр; 4 — подвижной контакт; 5 — реле.
Рис. 388.
Контактный манометр Креля-
Изготовитель: народное предприятие «Комбинат технического стекла», Ильменау.
этот метод требует частого контроля нагрузки колонны по пару, особенно при значительной разнице удельных теплот испарения компонентов кубовой жидкости. Регулирование мощности обогрева куба по перепаду давления потока пара в колонне более предпочтительно, поскольку перепад давления непосредственно связан со скоростью движения пара в колонне (см. разд. 4.11). Преимущество этого метода регулирования состоит также в том, что колебания напряжения в сети при использовании электронагревательных устройств или колебания давления и теплотворной способности газа при обогреве газовым пламенем не оказывают влияния на точность регулирования [13].
На рис. 387 показано устройство для регулирования нагрузки по перепаду давления, выполненное по нормалям «Дестинорм» [11 ]. Расходомер 1, служащий для измерения количества жидкости, стекающей из колонны в куб, имеет боковой патрубок с холодильником 2, предохраняющим от проникновения пара в контактный манометр 3. Чтобы предотвратить образование капелек конден-456
Рис. 389.
Контактный манометр с подвижным коленом:
/ — отметка Для заполнения жидкости —наполнителя;
2 — фильтр G1.
Изготовитель: фирма Шотт, Майнц,
сата в боковом патрубке или в холодильнике 2, можно продувать их азотом через счетчик пузырьков или, что еще проще, обогревать боковой патрубок с помощью электроспирали. Холодильник соединен с контактным манометром 3, снабженным подвижным контактом
4, который можно перемещать даже в процессе работы устройства. Одно колено контактного манометра при ректификации под атмосферным давлением остается открытым, а при вакуумной ректификации соединяется с вакуумированным трубопроводом, идущим к верхней части колонны.
Контактный манометр (рис. 388) в комбинации с реле, описанным в разд. 8.2.2, заполняют ртутью, а в комбинации с электронным реле — органической токопроводящей жидкостью. Эта жидкость по сравнению с ртутью повышает чувствительность прибора примерно в 10 раз. Сначала с помощью реостата устанавливают необходимую нагрузку колонны по пару, соответствующую определенному перепаду давления. Затем опускают контакт 4 (см. рис. 387) до соприкосновения его нижнего конца с мениском жидкости-наполнителя и несколько увеличивают силу тока. Реле 5 связано с электронагревателем куба и выключает его при замыкании контакта. Целесообразно использовать такую электроизмерительную схему, в которой регулируется только 1/3 общей мощности. Рекомендуется применять реле с запаздыванием включения, чтобы случайные скачки давления в кубе, возникающие при кипении, не приводили к включению электронагревателя. Можно пользоваться также контактным манометром с подвижным коленом (рис. 389), который имеет миллиметровую шкалу на колене и угловую
шкалу.
Изящный метод регулирования описал Штаге [58]. Регулирование осуществляется с помощью электронного устройства по температуре или по расходу флегмы, выраженному числом капель или миллилитров в 1 мин. Число капель флегмы определяется оптическим путем и пропорционально этому числу непрерывно регулируется мощность электрообогрева куба. Температура куба регулируется по температуре кипения кубовой жидкости в различные моменты перегонки. Нагрузка колонны по пару остается постоянной даже в период, когда имеет место смена фракций в кубовой жидкости,
457
8.5. ИЗМЕРЕНИЕ ФИЗИКО-ХИМИЧЕСКИХ
СВОЙСТВ ДИСТИЛЛЯТА
В ПРОЦЕССЕ РЕКТИФИКАЦИИ
Вследствие того что температура в верхней части колонны не определяет однозначно степень чистоты дистиллята, в процессе ректификации целесообразно определять и другие физико-химические свойства дистиллята без его отбора. Для этого необходимо проводить измерения на линии подачи флегмы в колонну, что дает возможность своевременно изменять режим процесса ректификации соответственно результатам измерений с целью получения требуемого состава дистиллята.
При перегонке на лабораторных и пилотных ректификационных установках можно непрерывно измерять следующие физико-химические свойства [59]: температуру затвердевания, показатель преломления и диэлектрическую проницаемость *. Во всех случаях, кроме определения температуры затвердевания, основная трудность состоит в обеспечении термостатирования исследуемой жидкости, температура которой при ректификации обычно непрерывно повышается.
8.5.1. ИЗМЕРЕНИЕ ТЕМПЕРАТУРЫ ЗАТВЕРДЕВАНИЯ
Применительно к ректификации высококипящих жирных кислот Янцен и Тидке предложили весьма удобное устройство для определения температуры затвердевания веществ с высокой температурой плавления (см. рис. 390). В этом устройстве, так же как и в приборе Тиле для определения температуры плавления, дистиллят (или флегма) сначала попадает через капельницу 3 в капилляр 5, омываемый термостатирующей жидкостью. Понижая температуру этой жидкости, определяют температуру затвердевания дистиллята по показаниям термометра 4. По байпасной трубке 2 в приемник 1 поступают следующие порции исследуемой жидкости. Повышая затем температуру термостатирующей жидкости, определяют таким же образом температуру плавления исследуемой жидкости.
* Наряду с указанными физико-химическими свойствами при ректификации на лабораторных и пилотных установках часто измеряют также вязкость, плотность, поверхностное натяжение и т. д. (см. разд. 8.5.4). — Прим. ред.
Рис. 390.
Устройстко Янцена—Тидке для определения температуры затвердевания веществ с высокой температурой плавления:
/ — приемник исследуемой жидкости; 2 — байпасная трубка;
3 — капельница; 4 — термометр; 5 — капилляр.
458
Мюллер и Ценшельский [60 ] разработали автоматический прибор, регистрирующий температуру плавления в течение любого периода времени перегонки с точностью ±0,25 °C.
8.5.2. ИЗМЕРЕНИЕ ПОКАЗАТЕЛЯ ПРЕЛОМЛЕНИЯ
Показатель преломления наряду с температурой кипения широко применяется для характеристики органических веществ [59]. При разделении близкокипящих веществ непрерывное определение
Рис. 391.
Рефрактометр «Ремат 10».
а — передняя панель прибора с кюветами К51 и двумя линейными показывающими шкалами; б — задняя панель прибора с кабелями для подключения к электросети.
Изготовитель: народное предприятие «Карл Цейс Иена».
459
Рис. 392.
Устройство Штаге с проточными рефрактометрами для измерения равновесных данных в системе пар—жидкость.
показателя преломления дистиллята позволяет своевременно изме-нять режим процесса ректификации, чтобы по возможности сократить объем промежуточных фракций и тем самым увеличить выход целевого продукта.
Фотоэлектрический рефрактометр позволяет фиксировать показания по шкале электрического прибора и одновременно записывать результаты измерений. Автоматические проточные рефрактометры с непрерывной регистрацией показаний разработаны Томасом с сотр. [61] и Лэтчумом [62]. Рефрактометр «Ремат 10», выпускаемый народным предприятием «Карл Цейс Иена» [63 ], также представляет собой проточный рефрактометр. Этот прибор позволяет определять показатель преломления движущейся пробы жидкости по разности между показателями преломления данной пробы и неподвижных сравнительных проб жидкостей. На рис. 391 показаны передняя и задняя панели данного прибора, работающего в интервале температур от —10 до +120 °C. Рефрактометр имеет четыре кюветы для различных пределов измерения показателя преломления.
Для определения показателя преломления удобен прибор Штаге, служащий для измерения равновесных данных в системе пар—жидкость (рис. 392). Два выпускаемых промышленностью рефрактометра устанавливают в приборе Штаге таким образом, что они позволяют определять в проточной системе концентрации жидкости и пара по показателям преломления. Для автоматической регистрации скорости изменения показателя преломления во времени можно использовать интерферометры. Прибор Кегелеса и Собера [64 ] с фотоэлектрической ячейкой диаметром 25 мм обеспе-
460
чивает точность показаний, соответствующую двум единицам в шестом знаке после запятой при использовании светового потока соответствующего зеленой линии Hg.
8.5.3. ИЗМЕРЕНИЕ ДИЭЛЕКТРИЧЕСКОЙ ПРОНИЦАЕМОСТИ
Тенденция к увеличению применения физических методов измерения привела к дальнейшей разработке техники измерения диэлектрической проницаемости. Измерение диэлектрической проницаемости (ДП) имеет особое преимущество при ректификации смесей, содержащих воду (ДП-80), а также смесей веществ с резко отличающимися значениями этого параметра. В качестве примера можно назвать смеси: уксусная кислота (ДП-6,13)--уксусный ангидрид (ДП-22,2) и метанол—толуол. Азеотропная смесь метанол—толуол, образующаяся при ректификации, имеет ДП-26,8, которая в значительной мере отличается от значений диэлектрической проницаемости исходных компонентов, равных соответственно 33,8 и 2,37 [65]. При определении концентрации толуола в бензоле данный метод измерения также оказывается наилучшим, хотя разница в значениях диэлектрической проницаемости у компонентов смеси равна всего 0,08.
На рис. 393 показано устройство Эме, служащее для измерения диэлектрической проницаемости в условиях ректификации [65]. Измерительную ячейку устройства можно помещать в трубу, подающую флегму в колонну, что обеспечивает возможность при необходимости своевременно изменять режим процесса ректификации. Монография Эме содержит ценный обзор полученных до настоящего времени значений диэлектрической проницаемости
указанием длины волны, температуры, при которой проводили измерение, и температурного коэффициента этой величины.
Преимущество мультидекаметра Сле-фогта [66 ] состоит в том, что измеряемая частота не является строго фиксированной и может изменяться в интервале от 10 кГц до 12 МГц. Грант [67] описал прибор для регистрации значений диэлектрической проницаемости, который весьма пригоден для использования в процессах ректифи-
для различных веществ с
Рис. 393.
Устройство Эме для измерения диэлектрической проницаемости в процессе ректификации:
/ — холодильник; 2 — штуцер для ввода эталонной жидкости; 3 — измерительная ячейка; 4 — экранированный кабель; 5 — регистрирующий прибор; 6 — подстарка.
4$|
кации. Области применения методов измерения диэлектрической проницаемости органических веществ исследованы Феллони и Тантилло [68].
8.5.4. ИЗМЕРЕНИЕ ДРУГИХ ФИЗИКО-ХИМИЧЕСКИХ СВОЙСТВ
При перегонке на лабораторных и пилотных ректификационных установках для непрерывного определения физико-химических свойств дистиллята и кубовой жидкости можно использовать методы измерения, применяемые в промышленности [2]. Однако во многих случаях измерительные приборы и устройства должны быть соответствующим образом модифицированы.
При аналитической разгонке можно использовать весовой плотномер Клазена [69 ], требующий 8 мл исследуемого вещества для каждого измерения. Точность измерения 10"3 г/см3. Для непрерывного измерения плотности жидкости применяют устройство, предложенное Линфордом [70].
В промышленности, особенно нефтеперерабатывающей, широкое применение нашли методы непрерывного измерения вязкости. Эти методы могут быть использованы и в условиях ректификации на лабораторных и пилотных установках. Обзор различных вискозиметров непрерывного действия, применяемых в процессах очистки нефти, представлен в работе Фрицше [71].
Проточные измерительные приборы, такие как инфракрасный спектрометр, ультрафиолетовый спектрометр и масс-спектрометр, все шире использующиеся в химической промышленности, могут найти применение и при ректификации на лабораторных и пилотных установках, особенно для аналитических разгонок [72]. Сиггиа [73 ] дал подробный обзор современных непрерывных методов анализа, применяемых при перегонке. В работе [74] можно найти сведения об аналитических анализаторах, предназначенных для исследования отдельных фракций.
В разд. 4.2 указывалось о том, какое значительное влияние оказывает на процесс разделения в ректификационной колонне поверхностное натяжение жидкости. Тензиометр «Лауда», по DIN 53914 (изготовитель: завод измерительных приборов «Лауда») позволяет автоматически измерять и регистрировать поверхностное натяжение жидкостей.
Все большее значение приобретают радиохимические методы контроля процессов перегонки. Благодаря экономии времени применение этих методов особенно целесообразно при анализе фракций в процессах ректификации многокомпонентных смесей углеводородов, спиртов или кислот с близкими физико-химическими свойствами. При радиохимическом анализе в исходную смесь вводят некоторое количество меченого вещества и затем определяют его концентрацию в выделяемых фракциях. Хьюгс и Мэлокой с помощью меченого вещества 14СН3ОН определяли
462
давление и состав в системе СН3ОН—-С2Н5ОН—Н2О и следили за режимом процесса ректификации. Очевидно, что аналогичным путем можно проводить испытания эффективности ректификационных колонн различных типов.
8.6. ИЗМЕРЕНИЕ РАСХОДА
И ДОЗИРОВАНИЕ ГАЗОВ И ЖИДКОСТЕЙ
Техника работы с газами подробно рассмотрена в монографиях Бернгауэра [76], Виттенбергера [77], а также Мюллера и Гнаука [78]. Поэтому ниже мы не будем детально обсуждать этот вопрос. Дозировать и измерять количества газа или пара в условиях перегонки на лабораторных и пилотных установках необходимо при низкотемпературной ректификации (см. разд. 5.3.1), перегонке с паром (см. разд. 6.1), а также при адсорбционной ректификации (см. разд. 6.3). Кроме того, для разделения легко воспламеняющихся или сильно гигроскопичных веществ может оказаться необходимым проводить ректификацию в атмосфере инертного газа, чаще всего азота, что также требует измерения расхода инертного газа.
Применительно к перегонке с насыщенным и перегретым водяным паром в разд. 6.1 были рассмотрены косвенные методы измерения расхода пара. Для измерения расхода газов и жидкостей при повышенных давлениях используют ротаметры с поплавками, вращающимися в потоке исследуемой среды. Расходомеры, основанные на счете пузырьков, и капиллярные реометры требуют предварительной калибровки по газовому счетчику, в то время как ротаметры поставляются заводами-изготовителями с калибровочными кривыми для определенных газов и жидкостей со шкалами соответственно в м3/ч и л/ч (О °C, 760 мм рт. ст.).
На рис. 394 показаны модели ротаметров, обычно используемые в лабораторных и промышленных установках. Применительно к пилотным ректификационным установкам подобные ротаметры выпускаются с пневматической или электроиндукционной. передачей показаний.
Для контроля за расходом охлаждающей воды при ректификации наиболее пригодны лабораторные ротаметры типа SW (рис. 395). В конические стеклянные измерительные трубки ротаметров вставляются поплавки, изготовляемые из различных материалов в зависимости от физико-химических свойств исследуемой среды. Методы автоматического регулирования расхода охлаждающей воды описаны в работе Ницпетера [79]. Промышленностью выпускаются ротаметры-регуляторы для жидкостей или газов, которые прекращают подачу исходной смеси в ректификационную колонну при выходе из строя водяного холодильника или при уменьшении расхода охлаждающей воды по сравнению с заданным значением. При восстановлении расхода охлаждающей воды регуляторы
463
Рис. 394.
Ротаметры со штуцерами для присоединения гибких шлангов (а), со штуцерами для присоединения гибких шлангов и кожухом для термостатиро-вания (б), со штуцерами в виде стандартных шлифов (в) и в специальной арматуре (г).
Изготовитель: фирма Хенига по изготовлению ротаметров, аппаратуры и машин, Аахен.
Рис. 395.
Лабораторный ротаметр типа SW16.1.
Измеряемый расход от 0,5 до 3 л/мин (интервал измерения I) и от 3 до 12 л/мин (интервал измерения II).
Изготовитель: народное предприятие «Завод исследовательских приборов», Знтц Фрайталь.
обеспечивают подачу исходной смеси в ректификационную установку [80].
При непрерывной ректификации сосуд с исходной смесью размещают на такой высоте, чтобы гидростатическое давление столба жидкости превышало давление в ректификационной колонне. Это обеспечивает непрерывную подачу разделяемой смеси в колонну. Если на линии подачи исходной смеси в колонну установлен обычный стеклянный кран, то приходится часто регулировать расход, так как по мере снижения уровня жидкости в напорном сосуде количество протекающей исходной смеси все время уменьшается. При использовании капельницы и многоходового крана можно непрерывно контролировать расход жидкости, однако практически равномерная подача жидкости обеспечивается лишь из напорного бака, работающего на принципе сосуда Мариотта. Из большого сосуда жидкость можно подавать под действием
464
сжатого воздуха с помощью резиновой ручной груши. В пленочной дистилляционной установке Креля сосуд для исходной смеси (см. рис. 196) снабжен стеклянным краном, который обеспечивает очень точное капельное дозирование жидкости. В капельнице Даммера и Крамера [81 ] (рис. 396) использованы оба указанные выше принципа: выравнивание давления по типу сосуда Мариотта и регулировка с помощью крана. Сосуд Мариотта с шестью сменными капиллярами, усовершенствованный Хенелем [82], позволяет точно дозировать по каплям малые количества жидкости.
Штаге применил в автоматизированной ректификационной установке «Лабодест» устройство для подачи исходной смеси с магнитным краном, регулируемым по времени (рис. 397). Время, в течение которого клапан закрыт, можно устанавливать в интервале от 1 до 120 с, а время, когда он открыт — от 1 до 2 с. При каждом открывании магнитного клапана определенное количество жидкости поступает в ниже расположенное устройство, измеряющее расход подаваемой исходной смеси. Клапан данного устройства устанавливается таким образом, чтобы поступившая жидкость полностью могла стечь за промежуток времени, когда магнитный
Устройство Штаге для подачи исходной смеси.
Рис. 398.
Йенский циркуляционный насос типа 100.
Изготовитель: народное предприятие «Йенский завод стекла Шотт», Иена.
Капельница [81, 88 ]:
1 — пробка клапана; 2 — отверстие; 3 — кожух для термостатироваиия;
4 — клапан с шаровым шлифом; 5 — капилляр для подачи капель; 6 — шпиндель клапана; 7 — запорная пробка с накаткой.
465
Рис. 399.
Зависимость иапора от производительности (по воде), при различных числах оборотов мотора п Йенского циркуляционного иасоса типа 100.
Рис. 400.
Электрический сильфоииый дозировочный насос для жидкостей и газов. Изготовитель: фирма Эрнст Хааге, Мюльхайм (Рур).
клапан закрыт. Бак для исходной смеси, соединенный с трубчатой вставкой, работает как сосуд Мариотта до тех пор, пока уровень жидкости в баке не превысит уровень жидкости в трубчатой вставке.
С помощью пневматического стеклянного крана Пинкафа и Вихтерле [83] можно, например, дозировать воду в интервале от 0,02 до 3500 мл/ч с точностью ±1,5 %. Вопросы дозирования веществ в лаборатории подробно обсуждаются в монографии Пинкафа [84 ].
Весьма удобен, особенно для пилотных установок, изготовляемый в Иене циркуляционный насос типа 100 (рис. 398). Он представляет собой простейшую модель центробежного насоса, выполненную из стекла. Напор и производительность этого насоса зависят от числа оборотов мотора; их можно определить по диаграмме, приведенной на рис. 399. Электрический сильфонный насос Хааге предназначен для ступенчатого дозирования газов и жидкостей при напоре до 20 м вод. ст. Стандартные модели без сальникового уплотнения имеют производительность от 15 мл/ч до 1500 л/ч. Их изготавливают из стали V4A, томпака и стеклопластика (рис. 400).
Особую группу составляют насосы перистальтического действия с гибкими шлангами из различных материалов. Их преимуще-466
ственно применяют для дозирования низкокипящих жидкостей, эмульсий, суспензий, газов и шламов различных видов [86]. Изменяя диаметр шланга (от 3 до 10 мм) и число оборотов вращения вала, можно дозировать вещества с расходом от нескольких мл/ч до 20 л/ч в интервале температур от —90 до +150 °C. На рис. 401 показан несоздающий пульсаций напора шланговый насос типа DP2-2, который можно использовать как для создания избыточного давления, так и вакуума. Этот насос имеет 16 отдельных независимых друг от друга каналов.
Для больших ректификационных установок непрерывного действия целесообразно применять поршневые насосы. В разд.
Рис. 401.
Шланговый насос типа DP2-2 с тремя различными скоростями подачи дозируемой среды.
Производительность от 3 до 2400 мл/ч.
Изготовитель: народное предприятие «Лабораторная техника», Ильменау.
Рис. 402.
Стеклянный центробежный насос с тефлоновым уплотняющим кольцом (прокладкой).
Изготовитель: фирма «Стеклянная техника», Висбаден — Ширштайн.
467
Рис. 403.
Насос для точного дозирования модели «Нормадос Р31».
Изготовитель: фирма Брана —Люббе. Гамбург.
производительность до 20 м3/ч Специализированная фирма Бран
5.1.3.1 были рассмотрены стеклянные дозирующие насосные головки с сильфонами из тефлона (см. рис. 143)188]. Для подачи различных сред можно применять также стеклянные центробежные насосы (рис. 402), имеющие и напор до 15 м. вод. ст. и Люббе (Гамбург) выпускает
для лабораторных и пилотных ректификационных установок ряд дозировочных насосов (рис. 403), которые можно использовать и для автоматического перемешивания различных жидкостей. Насосы модели «Нормадос» типов К и J с производительностью от 0,6 до 1121 л/ч применяют^ лабораторных и промышленных установках. Регулирование производительности осуществляется вручную, электрически или пневматически. Благодаря тому, что детали и
узлы насосов, имеющие одинаковое назначение, выполняют взаимозаменяемыми, из них можно собирать любые насосы данной модели. Насосы модели «Гномодос» рассчитаны на небольшую производительность (от 0,6 до 4,4 л/ч). Они имеют установочную гайку с накаткой, служащую для точной установки длины хода поршня.
Модель «Гномодос SO2» имеет несколько дозирующих насосных головок. Дозирующая насосная головка может выполняться в виде плунжерного или мембранного насоса. Передаточный механизм насоса позволяет изменять длину хода плунжера в интервале от 0 до 15 мм. К насосу можно подключать одновременно до пяти насосных головок.
9
Оборудование лаборатории. Ввод в эксплуатацию ректификационных установок
9.1. ПЛАНИРОВКА ПОМЕЩЕНИЯ
Основные нормы и правила проектирования новых лабораторий с учетом специфики техники перегонки рассмотрены в книге Бере [1], а также в работах Колемана [2] и Шрамма [3]. Планировка специальных лабораторий для аналитической дистилляции подробно описана Кинканноном и Вакером [4]. Для лаборатории, в которой проводится перегонка, необходимо помещение с протяженными и высокими стенами, удобными для крепления стендов, служащих для монтажа ректификационной аппаратуры. Примеры рациональной планировки и оборудования различных лабораторных помещений показаны на рис. 404—407.
Способы размещения пилотных ректификационных установок в промышленности демонстрируются на рис. 140, 141, 146 и 160.
Планировка помещения удлиненной формы (рис. 404). Для уменьшения шума вакуумные насосы размещают в отдельном помещении лаборатории для перегонки. Насосы 3 располагают у стены, отделяющей помещение для насосов от основного помещения лаборатории, чтобы по возможности сократить длину вакуумных коммуникаций. У противоположной стороны данной стены в основном помещении размещают стенд 6, предназначенный для монтажа вакуумных ректификационных установок. Для удобства регуляторы вакуума и вакуумметры устанавливают непосредственно за стендом. Помещение для вакуумных насосов можно одновременно использовать в качестве небольшой слесарно-механической мастерской.
У стены с окнами размещают лабораторные столы 1, к которым подводится электроэнергия и вода. На этих столах в основном проводят работы по измерению различных физико-химических свойств получаемых фракций дистиллята: показателя преломления с помощью рефрактометра Аббе или интерферометра, температур затвердевания и плавления; диэлектрической проницаемости и оптического вращения с помощью поляриметра. Рабочий стол 4, установленный в средней части основного помещения лаборатории, предназначен преимущественно для химических работ. У большей стены, выходящей в вестибюль, также размещают стенд 6. Для перегонки ядовитых веществ, вызывающих головную боль и головокружение (таких, как диэтиловый эфир, бензол, хлорированные углеводороды или органические нитросоединения) в лаборато-
469
Стена с окнами
Рис. 404.
Схема размещения оборудования лаборатории для перегонки, расположенной в комнате удлиненной формы, с помещением для вакуумных насосов (А) и боксом для работ с вредными веществами (£)<
/ — лабораторные столы; 2 — письменные столы; 3 — вакуумные насосы;
4 — рабочий стол; 5 — раковины; 6 — стенды для монтажа ректификационных установок; 7 — вытяжной шкаф.
Рис. 405.
Схема размещения оборудования лаборатории для перегонки, расположенной в укороченной комнате, с помещением для вакуумных насосов (Л) и боксом для работ с вредными веществами (Б)»
Обозначения те же, что иа рис. 404.
Рис. 406.
Лабораторный стол со штативами (в торцевой части).
Изготовитель: фирма Лейбольда, «Оборудование лаборатории», Кёльн— Байенталь.
рии выделяют специальное помещение — бокс. Это помещение должно быть снабжено хорошей вентиляционной установкой, обеспечивающей 40-кратный обмен воздуха в 1 ч. Бокс отделяют от основного помещения лаборатории прозрачной перегородкой из армированного стекла, через которую можно непрерывно наблюдать из лаборатории за работой установок в боксе. Пульты автоматизированных ректификационных установок с регистрирующую
Рис. 407.
Схема размещения оборудования лаборатории для перегонки, расположен* ной в укороченной комнате, с помещением для вакуумных насосов (Л) и высокими вытяжными шкафами.
Обозначения те же, что на рнс. 404.
щими приборами располагают в боксе у стены, обращенной к основному помещению лаборатории.
Планировка помещения укороченной формы (рис. 405, 407). Принцип планировки лабораторного помещения тот же, что и в варианте, изображенном на рис. 404. Стенды располагают вдоль свободных поперечных достаточно протяженных стен. Хорошо вентилируемый бокс, отделенный перегородкой от основного помещения лаборатории, предназначен для работы с вредными веществами. Рабочий стол 4, расположенный поперек комнаты, может
иметь на торце штативы для крепления небольших аппаратов (рис. 406).
Другой вариант планировки помещения укороченной формы показан на рис. 407. Если для определения свойств дистиллята преимущественно применяют химические методы, то в средней части основного помещения лаборатории размещают в поперечном направлении два больших рабочих стола 4. Вместо бокса устанавливают два вытяжных шкафа 7, которые снабжены стендами 6 для монтажа аппаратуры. Один из шкафов, имеющий высоту, равную высоте комнаты, размещают на полу (рис. 408). Второй шкаф
Рис. 408.
Вытяжной шкаф со штативом.
471
равен по высоте рабочему столу, его снабжают штативами для крепления небольших дистилляционных приборов. Скорость воздуха в дверном проеме вытяжного шкафа должна составлять 0,25—0,5 м/с.
9.2. СТЕНДЫ И КОММУНИКАЦИИ
Стенды для монтажа ректификационной аппаратуры могут быть как сборными, так и стационарно смонтированными. На рис. 409 показан стенд, собираемый из отдельных деталей специально для конкретной лабораторной установки. Передвижной стенд (рис. 410) позволяет размещать установку в любом месте лаборатории. Для монтажа больших ректификационных установок предпочтительно использовать стенды, крепящиеся к стене помещения. Укрепленные в стене шины Иордаля (рис. 411а) снабжены держателями (рис. 4116), которые можно устанавливать на любой высоте [5]. С помощью поперечных перекладин можно подгонять данное устройство под конкретную аппаратуру (см. рис. 408).
Исходя из многолетнего опыта автор рекомендует конструкцию стенда, показанную на рис. 412. Этот стенд состоит из отдельных секций шириной 1 м, он зацементирован в стену, покрытую метлахской плиткой снизу на высоту 1,5 м. Внизу стенда имеется ванна глубиной 80 см, облицованная кафелем. Ванна служит для сбора кубовой жидкости, которая может вылиться при поломке стеклянного куба. Кроме того, в случае загорания этой жидкости в ванне огонь будет локализован непосредственно возле установки.
Стенд, собираемый нз отдельных взаимозаменяемых деталей. Изготовитель: фирма Герард Плейгер, Анвере.
Рис. 410.
Передвижной стенд.
472
Рис. 412.
Секция стенда с ванной, облицованной кафелем, коммуникациями и электрощитом:
1 — двух- и трехполюсные розетки; 2 — общий выключатель; 3 — выключатели; 4 — контрольные лампы; 5,6 — трубопроводы для подачи воды и газа соответственно.
Рис. 413.
Стальные уголки с удлиненными прорезями для монтажа стендов с рабочими площадками.
Коммуникации для воды, газа, сжатого воздуха, водяного пара, а также линии вакуума прокладываются по стене. Отработанная вода отводится по сточному каналу. Настольные приборы и инструменты располагают на горизонтально размещенных деревянных щитах. В качестве распределителей электропитания для аппаратуры используются электрощиты, укрепляемые между двумя секциями стенда (см. рис. 412). Каждый щит снабжен 8— 10 штепсельными розетками с контрольными лампами и выключателями. По контрольным лампам легко определять, какая часть приборов установки включена. Общий выключатель дает возможность при необходимости выключать одновременно все электроприборы.
Для обслуживания ректификационных колонн, часто достигающих потолка комнаты, целесообразно применять узкие лестницы, которые можно хранить в углу за стендом.
Вентиляция стенда должна быть выполнена таким образом, чтобы воздух подавался у пола, а отсасывался у потолка. Обычно предусматривают 10-кратный обмен воздуха. Если при перегонке могут выделяться большие количества отходящих газов, то прокла
473
дывают специальные газоотводные трубопроводы, соединяющие конденсатор дистиллята с всасывающим отверстием вытяжной вентиляции.
Для обслуживания полупромышленных ректификационных установок в каждом конкретном случае создают специальные стенды с рабочими площадками (см. ниже рис. 424), которые также можно собирать из отдельных узлов и деталей. Монтаж таких стендов значительно упрощается при использовании стальных уголков с удлиненными прорезями (рис. 413).
9.3. СБОРКА УСТАНОВОК
Перед сборкой ректификационной установки, особенно состоящей из большого числа аппаратов, целесообразно составить монтажную схему, в.соответствии с которой разложить на полу узлы и детали установки. Затем необходимо проверить плотность посадки конусов и муфт в шлифовых соединениях, вставляя их без смазки. Смочив конус или нанеся на него мелом кольцевую полоску, можно легко обнаружить неравномерно отшлифованные части конуса и муфты. Шлифы со свободной посадкой конуса следует сразу же заменить. Такая предварительная проверка шлифов экономит много времени при последующей герметизации вакуумных ректификационных установок.
Монтаж установки обычно начинают от пола, причем при сборке установки необходимо предусмотреть возможность последующего снятия куба без демонтажа других аппаратов. Для этого целесообразно использовать вертикально перемещающиеся нагреватели кубов (см. рис. 327), сферические нагревательные приборы или пружинные колбодержатели (рис. 414). Отметим, что обычные
Рис. 414.
Пружнииый колбодержатель.
Рис. 415.
Стяжки с пружинами для крепления шлифовых соединений.
Рис. 416.
Клеммовый держатель для сферических шлифовых соединений.
474
Рис. 417.
Стяжка для плоских шлифовых соединений.
Рис. 418.
Держатель шлифовых соединений типа «Шлиф-кламмер» .
Изготовитель: фирма Эдвардс, Лондон.
кольцевые держатели не обеспечивают надежного закрепления куба.
Колонну устанавливают строго по отвесу. После закрепления колонны в вертикальном положении и проверки ее герметизации монтируют различные смежные аппараты и детали установки с последующей проверкой герметизации каждого отдельного узла установки.
Сборные детали установки должны иметь специальные крючки, предназначенные для крепления пружин, стягивающих шлифовые соединения. Для соединения шлифов крупных аппаратов используют пружинные стяжки (рис. 415). Клеммовый держатель для сферических шлифовых соединений показан на рис. 416- (см. разд. 3.1), а стяжка для плоских шлифовых соединений — на рис. 417. Легко разбираемым является держатель для шлифовых соединений модели «Шлиф-кламмер» (фирма Эдвардс, Лондон), крепящийся за выступ на стеклянной трубе (рис. 418). Резиновые манжеты для крепления шлифовых соединений (рис. 419) изготавливают для стандартных шлифов NS12,5—NS29. Они применимы при температурах до 130 °C [6]. Преимущество данных манжетов заключается в том, что они обеспечивают равномерное стягивающее действие по всей окружности шлифового соединения и позволяют отказаться от шлифовой смазки.
При использовании двойных муфт необходимо следить за тем, чтобы держатели не смещались в них по горизонтали или вертикали при затяжке винтов. В противном случае могут возникнуть меха-
475
Рис. 419.
Резиновые манжеты для крепления шлифовых соединений.
Рис. 420.
Двойная муфта (максимальный диаметр детали 14 мм).
Изготовитель: фирма Герард Плейгер, Анвере.
нические напряжения, которые обычно приводят к поломке стеклянных аппаратов, вследствие их термического расширения в процессе перегонки. Двойная муфта удобной конструкции показана на рис. 420. Муфта с крючками (рис. 421) предназначена специально для сборки стендов. Зажимные крючки и двойной контакт в трех точках гарантируют надежность соединения. Стандартная каркасная система, собираемая из большого числа взаимозаменяемых деталей, применяется для монтажа больших пилотных установок [7].
Нагревательные элементы, сферические сосуды с жидкостью и другие приборы можно точно устанавливать с помощью регулируемого штатива Маннхена—Риделя (рис. 422). С помощью этого штатива можно устанавливать аппараты на высоте до 15 см с точностью до 1 мм. Для крепления колонн и сосудов большого диаметра вместо обычных щековых клемм лучше использовать бандажи (рис. 423). Систематический обзор штативов различных конструкций представлен в работе Шварца [5].
Для сборки пилотных и полупромышленных ректификационных установок из стекла необходимы специальные соединитель-
Рис. 421.
Муфта с крючками.
Изготовитель: фирма Шпек, Франкфурт на Майне.
476
Рис. 422.
Лабораторный регулируемый штатив Маннхеиа-Риделя.
Рис. 423.
Бандажи для крепления колони.
Рис. 424.
Трубчатый стенд для ректификационной установки (общий объем аппаратов 200 л), изготовленный из одинаковых труб с оцинкованными соединительными муфтами.
Изготовитель: фирма «Стеклянная техника», Висбаден—Ширштайи.
ные детали, опорные и поддерживающие устройства. В каталогах предприятий технического стекла указано очень большое число таких деталей и устройств, предназначенных для сборки установок; рассмотреть их в данной книге в полном объеме не представляется возможным. Предприятия стекла выпускают также стенды для монтажа ректификационных установок. На рис. 424 показан стенд, который составлен из одинаковых труб, соединяемых с помощью трубчатых муфт, снабженных резьбой.
9.4. ГЕРМЕТИЗАЦИЯ УСТАНОВОК
В большинстве случаев возникают трудности при уплотнении шлифовых соединений и кранов. Перегоняемые жидкости обладают весьма различной растворяющей способностью по отношению к жировым смазкам шлифовых соединений, поэтому наиболее подходящую смазку следует выбирать на основе результатов предварительных испытаний (табл. 70).
477
Таблица 70.
Жировые смазки для шлифовых соединений и кранов
Смазка Максимальная рабочая температура, °C Состав или предприятие-изготовитель Область применения
Смазка Рамсэя 25 6 ч. вазелина, 1 ч. парафина, 6ч. пара-каучука Для обычных работ
Высоковакуумная 25 Предприятие Е. Лей- Для работ под высо-
смазка типа Р Высоковакуумная смазка типа R Смазка «Лителен» 30 150 больда, Кёльн ким вакуумом
Смазки типа 1 ч. растворимого Для работ с вещества-
BZ6564 и BZ6563 крахмала, 1 ч. глицерина [5]; предприятие Е. Мерка, Дармштадт ми, хорошо растворяющими жиры, например, диэтиловым эфиром и хлорированными парафиновыми углеводородами
Графитовая смазка Порошок графита, смесь графита и парафина, коллоидный раствор графита в воде Высокотемпературная перегонка
О применимости смазок на основе силикона в настоящее время нельзя сделать окончательного вывода, из-за недостатка опытных данных. Однако можно утверждать, что всегда имеется опасность попадания некоторой части смазки в колонну, что приводит к ухудшению смачиваемости насадки. Смазка «Лителен» (см. табл. 70), состоящая из литиевого мыла, практически не изменяет свою консистенцию с повышением температуры. Ее рекомендуется применять для смазки кранов в интервале рабочих температур от 0 до 150 °C. Обзор о методиках приготовления специальных смазок в лаборатории дан в работе Брооке [8].
Для кранов в основном применяют более мягкие смазки, чем для шлифовых соединений. Систематический обзор различных смазок представил Вагнер [9]. В работах Бернхауэра [10], Виттенбергера [11], Вольфа [12] и Фридрихса [13] даны подробные указания по уходу за стеклянными шлифовыми соединениями и кранами, особенно при плотной посадке муфт шлифов и сердечников кранов. Для соединения деталей, используемых в процессах молекулярной дистилляции, наиболее подходящими являются дегазированные смазки с низким давлением паров, такие как высоковакуумные смазки типов Р и R. Стандартные шлифы с ртутным затвором (рис. 425) обеспечивают практически полную герметизацию, однако их следует применять только в исключительных случаях вследствие опасности пролива ртути.
478
Шлифовые соединения без смазки даже при самой высокой степени чистоты обработки поверхностей конуса и муфты не обеспечивают достаточной герметичности при работе под вакуумом, и в частности не предохраняют от проникновения паров из ректификационной колонны. Поэтому верхнюю половину конуса шлифо-вого соединения следует всегда равномерно покрывать слоем смазки. Для этого лучше всего использовать гладкий деревянный шпатель. Пальцем можно нанести более равномерный слой, однако в этом случае имеется опасность попадания в смазку грязи, пота или влаги. Перед смазыванием, особенно при использовании смазки Рамсэя или апиезоновой смазки, конус шлифа можно слегка прогреть (но не на открытом пламени горелки!), чтобы нанесенная смазка при вращении конуса растеклась и покрыла его поверхность равномерным слоем. После этого конус вставляют в муфту и при нажатии вращают его до тех пор, пока все шлифовое соединение не станет прозрачным. Если с первой попытки это не удастся сделать, то всю процедуру повторяют снова, начиная с прогревания конуса.
Если при перегонке герметичность шлифового соединения нарушается из-за растворения или разложения жировой смазки, то уплотнение можно восстановить при помощи различных замазок. Для этого пригоден прежде всего пицеин с температурой плавления 80 или 105 °C. Хорошим заменителем пицеина является сургуч, однако он хрупок. При рабочих температурах до 250 °C применяют замазку из глицерина и свинцового глета, которая сильно затвердевает. Пицеин и сургуч не следует расплавлять на открытом газовом пламени, целесообразно нагреть уплотняемую поверхность до температуры около 100 °C большим факелом пламени горелки Бунзена, а затем наносить замазку шпателем, нагретым до той же температуры. Покрытую замазкой поверхность снова прогревают небольшим факелом пламени. При этом замазка распределяется равномерным слоем и проникает в зазоры между конусом и муфтой шлифового соединения.
Уплотнение шлифовых соединений можно обеспечивать также с помощью конусообразного тефлонового манжета, надеваемого на конус. При этом обеспечивается надежная герметизация и одновременно легкость разборки шлифового соединения. Французская фирма Гашо изготавливает манжеты типа «Родетс» для различных
, типоразмеров шлифовых соединений. Прецизионные шлифовые соединения можно применять и без смазки (см. разд. 3.1). Герметизацию сферических и плоских S шлифовых соединений в пилотных и полупромышленных ректификационных установках легко обеспечить с помощью уплотняющих тефлоновых колец и рубашек.
Рис. 425.
Стандартный шлиф с ртутным затвором.
479
9.5. ВВОД РЕКТИФИКАЦИОННЫХ УСТАНОВОК В ЭКСПЛУАТАЦИЮ
Подготовку к пуску ректификационной установки обычно начинают с заполнения куба разделяемой смесью. При обычной перегонке куб следует заполнять не более чем на 2/3 его номинальной емкости, а при вакуумной перегонке — только до половины. Одновременно рекомендуется вводить с жидкостью в куб небольшие твердые частицы, для обеспечения более равномерного кипения. Объем загружаемой жидкости следует определять с учетом повы-
шения температуры до ее ожидаемого значения в начале процесса перегонки. Для этого применяют нагреваемые мерные сосуды, которые особенно удобны при работе с веществами, находящимися при комнатной температуре в твердом состоянии *.
Токсичные и очень летучие вещества лучше подавать в куб из закрытого сосуда, создавая в последнем избыточное давление; по окончании заполнения куба его необходимо сразу же закрыть. Для обеспечения предварительного смачивания содержимого ректификационной колонны (насадки, неподвижные или движущиеся контактные устройства) рекомендуется по возможности загружать разделяемую смесь в куб сверху через дефлегматор. В тех случаях, когда необходимо предотвратить увлажнение разделяемой смеси, колонну перед загрузкой продувают теплым воздухом (феном):
при включенном электронагревателе кожуха.
Как уже указывалось, насадку перед загрузкой ее в колонну следует очень тщательно очистить. Следует иметь в виду, что
многократная промывка четыреххлористым углеродом и трихлорэтиленом не обеспечивают необходимой степени чистоты. Для
устранения прилипших к элементам насадки слоев загрязнений?
целесообразно сначала несколько раз промыть насадку горячим? бензолом, а затем сполоснуть ее трихлорэтиленом [14].
Перед проведением вакуумной перегонки необходимо сначала’ проверить герметичность установки. Для этого после достижений; в аппаратах установки заданного остаточного давления вакуумный насос отключают и в течение нескольких часов наблюдают з
изменением давления (см. разд. 5.4.1). Повторно герметичност] установки проверяют после разогрева установки, в условиях тепло вого расширения ее деталей. Электронагреватель куба включаю" по достижению заданного давления, а нагреватель кожуха ко лонны — после того, как начнется кипение жидкости. Охлажда!
щую воду подают в конденсатор во всех случаях до включени электронагревательных приборов, регулируя вручную ее расхо, по показаниям ротаметра. Следует отметить, что в пусковой перио, приходится неоднократно устанавливать расход воды. Поел
* При перегонке веществ, имеющих температуру плавления выше комиа' ной температуры, целесообразнее загружать разделяемую смесь по массе, опр< делив предварительно ее плотность при ожидаемой рабочей температуре nepi гонки и учитывая степень заполнения куба. — Прим. ред.
480
того, как пары достигнут верхней части колонны, устанавливают требуемую нагрузку (см. разд. 4.10.7) или предварительно создают режим работы, соответствующий «захлебыванию» колонны (см. разд. 4.10.8), для обеспечения лучшей смачиваемости насадки. Перед установкой заданного флегмового числа в ректификационной колонне не менее 0,5 ч проводят рабочий процесс с бесконечным флегмовым числом, что позволяет более точно определить начальную температуру отгонки (см. разд. 4.10.4).
Как уже указывалось выше, в куб необходимо вводить небольшие твердые частицы для обеспечения равномерного кипения жидкости. Обеспечивать равномерность кипения путем подачи воздуха через капилляр без необходимости не рекомендуется, так как это затрудняет точное определение рабочего давления при перегонке и в большинстве случаев вызывает частичную полимеризацию кубовой жидкости *. В качестве твердых частиц, обеспечивающих равномерность кипения, используют кусочки обожженной глины (от цветочных горшков), пемзы и тетраидальной насадки из платиновой проволоки. При перегонке веществ, обладающих сильной щелочной реакцией, в куб вводят оловянный порошок. Следует иметь в виду, что действие твердых частиц при вакуумной разгонке прекращается, если впустить в установку воздух. Облегчение парообразования в кубе при использовании твердых частиц обусловлено наличием в их порах маленьких пузырьков воздуха. Поэтому при проведении процесса вакуумной ректификации с перерывами необходимо каждый раз вводить в куб новые твердые частицы [15].
Если в начале перегонки жидкость в кубе очень сильно вспенивается, то можно уменьшить пенообразование путем введения небольшого количества противовспенивателей [16], из которых наиболее подходящими являются силиконовые масла.
При перегонке очень вязких жидкостей целесообразно непрерывно перемешивать содержимое куба с помощью магнитной мешалки или мешалки Хюбнера [17].
9.6. ТЕХНИКА БЕЗОПАСНОСТИ
Вопросам техники безопасности при лабораторных перегонках необходимо уделять особое внимание, поскольку зачастую приходится работать с токсичными, легковоспламеняющимися и взрывоопасными веществами, а при вакуумной перегонке всегда возможна опасность разрыва стеклянных сосудов [18]. Пределы взрываемости и температуры вспышки горючих газов и паров приведены в докладе Диттмара [19].
* При необходимости рекомендуется вводить в куб вместо воздуха азот, чтобы избежать вредного воздействия кислорода воздуха. — Прим. ред.
481
Электрические установки во взрывобезопасном исполнении необходимо в каждом конкретном случае предварительно испытывать.
Перегонку токсичных веществ в основном проводят в описанных выше отдельных боксах (см. разд. 9.1) или в обычных вытяжных шкафах. Взрывоопасные вещества, такие как диэтиловый эфир, лучше перегонять на открытом воздухе или при отсутствии данной возможности, в специально предназначенных помещениях. Если в разделяемой жидкой смеси растворены ядовитые газы, необходимо соединить конденсатор с всасывающим отверстием вытяжной вентиляции трубопроводом для отвода выделяющихся газов. При ректификации едких веществ следует работать в резиновых перчатках и защитных очках, поскольку всегда возможно появление капель жидкости на любых участках установки. Еще лучше пользоваться при работе специальным защитным костюмом.
Особые предосторожности необходимо соблюдать при вакуумной перегонке. Для предотвращения несчастных случаев следует пользоваться защитными очками, а еще лучше — специальными защитными приспособлениями для лица. Если ректификационная установка уже в значительной мере защищена колбонагревателем и обогревающим кожухом колонны, то надежное предохранение от осколков при разрыве стеклянных аппаратов и коммуникаций ^ожно обеспечить, закрывая неизолированные части аппаратуры полуцилиндрами из стекловолокна. В тех случаях, когда это по условиям опытов невозможно, например, при необходимости визуального наблюдения за процессом разделения, следует устанавливать защитные приспособления из проволочной сетки или листов органического стекла. Буферные сосуды целесообразно помещать в выложенные стекловолокном деревянные ящики или в корзину из проволочной сетки. Вакуумные насосы с ременным приводом обязательно закрывают деревянными ящиками.
Оборудование специальных лабораторий, в которых проводятся работы в условиях высокого давления, а также перегоняют токсичные вещества, подробно описано в работе Крейга и Дау [20].
1. ЛИТЕРАТУРА
1. RECHENBERG С. V., Einfache und fraktionierte Destination in Theorie und Praxis, Schimmer u. Co. AG, Miltitz 1923.
2. YOUNG S., Theorie und Praxis der Destination, Springer-Verlag, Berlin 1932 (Ubers, aus d. Engl.).
3. THORMANN K., Destillieren und Rektifizieren, Verlag Spamer, Leipzig 1928 (TOPMAH R. Перегонка и ректификация. Химтеоретиздат. М., 1935).
4. BADGER W. L., und McCABE W. L., Elemente der Chemie-Ingenieur-Technik, Springer-Verlag, Berlin 1932 (Ubers, aus d. Engl.). (БЕДЖЕР B., МАК-КЭБ В. Основные процессы и аппараты химических производств. Госхимтехиздат. М.—Л., 1933).
5. McCABE W. L„ und THIELE Е. L. Ind. Engng. Chem., int. Edit. 17 (1925), 605.
6. JANTZEN E., Dechema Monogr. 5 (1932), 48; Angew. Chem. 36 (1923), 592.
7. RIRSCHBAUM E., Destillier- und BektifiZiertechnik, 4. Aufl., Springer-Verlag, Berlin/Heidelberg/New York, ,1969.
8. GYULA N., Leparlas (Destination), Muszaki Konyvkiado, Budapest 1955.
9. JACOBS J., Destillier- und Rektifizieranlagen, R. Oldenbourg-Verlag, Munchen 1950.
10. ROBINSON C. S., und GILLILAND E. R-, Elements of Fraction al Distillation, McGraw-Hill Bo'k Company, New York 1950.
10a. PERRy J. H., Chemical Engineers Handbook, 3. Aufl., McGraw-Hill Book Company, New York 1953.
11. SCHULTZE Gg. R., und STAGE H. Die Kolonnendestillation im Laboratorium, VDI-Forschungsheft 424, VDI-Verlag, Berlin 1944.
12. THORMANN K\, Dechema—Erfahrungsaustausch, Laboratoriumstechnik, Arbeitsmethoden und Gerate, Destillieren und Rektifizieren, Dechema, Frankfurt/Main, September 1950.
13. THORMANN K., Dechema—Erfahrungsaustausch, Techn. Apparate und Anlagen, Trennen fliissiger Mischungen, Destillieren, Betriebstechnik, Dechema, Frankfurt/Main, August 1952.
14. STAGE H., und SCHULTZE Gg. R., Die grundlegenden Arbeiten fiber Theorie, Apparate sowie Verfahren der Destination und Rektifikation. VDI-Verlag, Berlin 1944.
15. ROSE A. und E., Distillation Literature Index und Abstracts 1941—1945, 1946—1952, Applied Science Laboratories, State College, Pennsylvania 1953.
16. ZIC Referatedienst Chemische Verfahrenstechnik, hrsg. von der Zentralstelle fur Information der chemischen Industrie, Berlin 1972 ff.
Forschritte der Verfahrenstechnik, hrsg. von der Ingenieurwissenschaftlichen Abteilung der Farbenfabriken Bayer AG, Leverkusen, Verlag Chemie GmbH, Weinheim/Bergstr. 1952 ff.; ab Band 9 (1971 ff.) hrsg. von der Verfahrenstechnischen Gesellschaft im VDI, VDI-Verlag, Dusseldorf.
Verfahrenstechnische Berichte (Vorankfindigungsblatter, Karteiblatter und halbjahrliche Sach-, Autoren- und Firmenregister), hrsg. von der Ingenieurwissenschaftlichen Abteilung der Farbenfabriken Bayer AG, Leverkusen, Verlag Chemie GmbH, Weinheim/Bergstr.
17. WALSH T. J., Ind. Engng. Chem., seit 1956.
483
16. ORLlGEK A. 6., und POLL H., Hilfsbuch fiir Mineraloitechniker, fid. 1: Die Eigenschaften von Kohlenwasserstofien, Mineralolprodukten und Hilfsstoffen, Springer-Verlag, Wien 1951.
19. SIGWART K-, Destination und Rektifikation, in: Ullmanns Encyklopadie der technischen Chemie, Bd. 1, Urban & Schwarzenberg, Miinchen/Berlin 1951, S. 429 bis 470.
20. KORTUM G„ und BUCHHOLZ-MEISEN HEIMER H., Die Theorie der Destination und Extraktion von Fliissigkeiten, Springer-Verlag, Berlin/Gottingen/Heidelberg 1952.
21. BOSNJAKOVIC Fr., Technische Thermodynamik, Bd. 1 (6. Aufl.) und Bd. 2 (5. Aufl.), Theodor Steinkopff Verlag, Dresden 1972/1971. {БОШНЯКОВИЧ Ф. Техническая термодинамика. T. 1,2. Госэнергоиздат. М.—Л., 1955, 1956).
22. GATTERMANN L., und WIELAND Н., Die Praxis des organischen Chemikers, 41. Aufl., Walter de Gruyter & Co., Berlin 1962. {ГАТТЕРМАН Л., ВИЛАНД Г. Практические работы по органической химии. Госхимиздат. М.—Л., 1948. Перевод с 31-го немецкого издания).
23. WITTENBERGER W., Chemische Laboratoriumstechnik, 6. Aufl., Springer-Verlag, Wien/New York 1963.
24. WEYGAND C., und HILGETAG G., Organisch— chemische Experimentierkunst, 4. Aufl., Johann Ambrosius Barth, Leipzig 1970. {ВЕЙГАНД K-, ХИЛЬГЕТАГ Г. Методы эксперимента в органической химии. Химия. М., 1968).
25. BERNHAUER К., Einfiihrung in die organisch—chemische Laboratoriumstechnik, Springer-Verlag, Wien 1947.
26. CARNEY Th. P., Laboratory Fractional Distillation, Mac-Millan, New York 1949.
27. ROSENGART M. J., Die Technik der Destination und Rektifikation im Laboratorium, VEB Verlag Technik, Berlin 1954 (Obers, aus d. Russ.). (Р03ЕНГАРТ M. И. Техника лабораторной перегонки и ректификации. Госхимиздат. М.—Л., 1951).
28. ROSE A., und Е., und MITARB., Technique of Organic Chemistry, Vol. IV, Distillation, Interscience Publishers, New York, 1951.
29. WEBER U. von, Gewinnung atherischer Ole durch Destination, in: GILDEMEISTER E., und HORFFMANN Fr., Die atherischen Ole (bearb. von W. Treibs), Akademie-Verlag, Berlin 1966.
30. SIGWART K-, Destillieren und Rektifizieren, in: H0UBEN-WEYL, Methoden der organischen Chemie, (hrsg. von E. Muller), Bd. 1/1, Georg Thieme Verlag, Stuttgart 1958.
31. ZUIDERWEG F. J., Laboratory Manual of Batch Distillation, Interscience Publishers, New York 1957.
32. COULSON E. A., und HERINGTON E. F. G., Laboratory Distillation Practice, George Newnes, London 1958.
33. BUKALA M„ MAJEWSKI J., und RODZINSKI W., Fraktionierte Destination im Laboratorium, Panstwowe Wydawnictwo Naukowe, Warschau 1958.
34. Industrial and Engineering Chemistry, Chemical and Process Engineering, VDI-Zeitschrift, Chemische Technik.
35. HOPPE K-, KUNNE H., und BENDIX H., Fortschritte in der Destination, Rektifikation, Sorption und Extraktion, VEB Deutscher Verlag fiir Grundstoffindustrie, Leipzig 1970.
36. TREYBAL E. R., Masstransfer Operations (International Student Edition), 2. Aufl., McGraw-Hill Book Company, New York 1968.
GEISELER G., und MITARB., Ausgewahlte physikalische Methoden der organischen Chemie, Akademie-Verlag, Berlin 1963.
KEIL B., und MITARB., Laboratoriumstechnik der organischen Chemie (hrsg. von H. Furst),- Akademie-Verlag, Berlin 1961.
WIBERG К. B., Laboratory Technique in Organic Chemistry, McGraw-Hill Book Company, New York 1960.
484
37. Organikum—Organisch—Chemisches Grundpraktikum, 13. Aufl., VEB Deutscher Verlag der Wissenschaften, Berlin 1974.
Anorganikum—Lehr- und Praktikumsbuch der anorganischen Chemie (hrsg. von L. Kolditz), 7. Aufl., VEB Deutscher Verlag der Wissenschaften, Berlin 1975.
38. ROCK H., Destination im Laboratorium—Extraktive und azeotrope Destination, Dr. Dietrich Steinkopff Verlag, Darmstadt 1960.
39. STAGE H., und BOSE K-, Die Belastungsverhaltnisse in Fflllkorpersaulen imter Destillationsbedingungen, Springer-Verlag, Berlin/Gottingen/Heidel-berg 1962.
MULLER E., und STAGE H., Experimented Vermessung von Dampf—Fliissigkeits—Phasengleichgewichten, Springer-Verlag, Berlin/Gottingen/Heidelberg 1961.
40. HALA E„ PICK J., FRIED V., und VILIM 0., Gleichgewicht— Fliissigkeit—Dampf (hrsg. von W. Mannchen), Akademie-Verlag, Berlin 1960; englische Ausgabe: Vapour—Liquid Equilibrium, 2. ed., Pergamon Press Ltd., London 1968.
41. HALA E., WICHTERLE I., POLAK J. und BOUBLIK T„ Vapour— "Liquid Equilibrium Data at Normal Pressures, Pergamon Press Ltd., London 1969.
42. КОГАН В. Б., ФРИДМАН В. М., КАФАРОВ В. В. Равновесие между жидкостью и паром. Справочное пособие. Наука. М.—Л., 1966.
ОГОРОДНИКОВ С. К., ЛЕСТЕВА Т. М., КОГАН В. Б. Азеотропные • смеси. Справочник. Химия. Л., 1971.
43. BILLET R., Grundlagen der thermischen Fliissigkeitszerlegung, Bibliographisches Institut, Mannheim 1961; BILLET R., Verdampfer-Technik, Bibliographisches Institut, Mannheim 1965.
43a. BILLET R., Industrielle Destination, Verlag Chemie GmbH, Weinhe, Bergstr. 1973.
44. BILLET R., Optimierung in der Rektifiziertechnik, unter besonderer Berflcksichtigung der Vakuumrektifikation, Bibliographisches Institut, Mannheim 1967.
45. OLIVER E. D., Diffusional Separation Processes: Theory, Design and Evaluation, John Wiley & Sons, New York 1966.
46. PRATT H. R. C., Countercurrent Separation Processes, Elsevier Publishing Company, Amsterdam 1967.
47. KRELL E., FREY H.-Р., GAW A LEK G., und WERNER G., Einfiihrung in die Trennverfahren, VEB Deutscher Verlag fiir Grundstoffindustrie, Leipzig 1975.
48. HOPPE K-, und MITTELSTRASS M., Grundlagen der Dimensionierung von Kolorinenboden, Theodor Steinkopff Verlag, Dresden 1967.
49. JUNGNICKEL H-, und OTTO W., Tiefe Temperaturen in der Verfahrenstechnik (Bd. 11 der Reihe «Beitrage zur Verfahrenstechnik»), VEB Deutscher Verlag fiir Grundstoffindustrie, Leipzig 1972.
50. FRANK W., und KUTSCHE D., Die schonende Destination, Otto Krausskopf-Verlag GmbH, Mainz 1969.
51. ОЛЕВСКИЙ В. M., РУЧИНСКИЙ В. P. Ректификация термически нестойких продуктов. Химия. М., 1972.
52* HOLLO J., KURUCZ Е„ und BORODI A., The Applications of Molecular Distillation, Akademiai Kiadd, Budapest 1971.
53. SCHUBERTH H., Thermodynamische Grundlagen der Destination und . Extraktion, Bd. I, VEB Deutscher Verlag der Wissenschaften, [Berlin 1972.
54. BRAUER H., und MEWES D., Stoffaustausch, einschl. chemischer Reaktionen, Verlag Sauerlander AG, Aarau und Frankfurt/Main 1971. BRAUER H., Grundlagen der Einphasen- und Mehrphasenstromung, Verlag Sauerlander AG, Aarau und Frankfurt/Main 1971.
485
55. HOLLAND C. D., Multicomponent Distillation, Prentice-Hall International, Englewood Cliffs 1963.
56. Chemie-Ing.-Techn. 42 (1970), 14, 945—957.
57. BILLET R., CZ-Chemie-Techn. 3 (1974) 10, 353—361.
2. ЛИТЕРАТУРА
1. SC HELEN Z_ H., Zur Geschichte der pharmazeutisch—chemischen Destilliergerate. Beilage zum Bericht April 1911, Verlag Schimmel und Co. AG, Miltitz.
2. GILDEMEISTER E., Die atherischen Ole, Verlag Schimmel und Co. AG, Miltitz 1928.
3. RECHENBERG C. von, Einfache und fraktionierte Destination in Theorie und Praxis, Verlag Schimmel und Co. AG, Miltitz 1923.
4. UNDERWOOD A. J. V., Trans. Instn. chem. Engr. 13 (1935), 34.
5. FORBES R. J., Short history of the art of destination, Verlag E. J. Bull, Leiden 1948.
6. FORBES R. J., und DIJKSTERHUIS E. J., »A History of Science and Technology, Bd. I: Ancient Times of the Seventeenth Century; Bd. 11: The Eighteenth and Nineteenth Centuries. Pelican Books Nr. A 498 und A 499, Verlag Penguin Books Ltd., Harmondsworth/Middlessex 1963.
7. SCHNEIDER IV., Lexikon alchemistisch—pharmazeutischer Symbole, Verlag Chemie GmbH, Weinheim/Bergstr. 1962.
8. LIPPMANN E. 0. von, Entstehung und Ausbreitung der Alchemie, Bd. 1 u. 2, Verlag Julius Springer, Berlin 1919, 1931; Bd. 3, Verlag Chemie GmbH, Weinheim/Bergstr. 1953.
9. LIPPMANN E. 0. von, Abhandlungen und Vortrage zur Geschichte der Naturwissenschaften 1 (1906), 2 (1913), Verlag Chemie GmbH, Weinheim/Bergstr.
10. LIPPMANN E. 0. von, Beitrage zur Geschichte der Naturwissenschaften und der Technik 1 (1923), 2 (1953), Verlag Chemie GmbH, Weinheim/Bergstr.
11. STAGE F., Angew. Chem., Ausg. B. 19 (1947), 181.
12. KREIS H., Liebigs Ann. Chem. 224 (1884), 259; YOUNG S. J. Chem. Soc. [London] 75 (1889), 679; FRIEDRICHS J., Z. angew. Chem. 32 (1919), 340.
13. Glas-Instrumenten-Techn. 14 (1970), 196.
14. KRELL E., Glas-Instrumenten-Techn. 11 (1967) 1—5, Beilage 1—20.
3. ЛИТЕРАТУРА
1. DDR-Standards und Fachbereichsstandards der DDR, Hrsg: Amt fiir Standardisierung der Deutschen Demokratischen Republik, Berlin 1968.
2. Verzeichnis der DIN-Normblatter, Stand vom 28.2.72, Beuth-Vertrieb GmbH, Berlin (West).
3. TELLE W., Chemische Laboratoriumsgerate, 2. Aufl., VEB Deutscher Verlag fiir Grunds toff Industrie, Leipzig 1969.
4. FRIEDRICHS F., Die Glashfltte 76 (1949), 3—6.
5. FLIEDNER K., Glasapparatetechnik 5 (1958), 33—37; Beilage zur Chem, Techn. 10 (1958).
6. KUEHNHANSS G., und MIT ARB., Chem. Techn. 9 (1957), 407—409.
7. TGL 10192—10200, 11641—11644, 12023—12032, 20277 DIN—проект 28800—28808.
7a. HAMPEL B., Chemiker-Ztg. 95 (1971) 7, 316—325.
8. KRELL E., Chem. Techn. 6 (1954), 452—455.
9. PADELT E., und LAPORTE H., Einheiten und Grofienarten der Naturwissenschaften, 2. Aufl., VEB Fachbuchverlag, Leipzig 1967.
10. FORSTER H., Einheiten, Grofien, Gleichungen und ihre praktische Anwendung, VEB Fachbuchverlag, Leipzig 1968.
486
11. HAEDER W.,und GARTNER E., Die gesetzlichen Einheiten in der Technik Verlag Beuth—Vertrieb, Berlin 1970.
12. KRELL E„ Chem. Techn. 7 (1955), 30—32.
13. LESSEES A. G., Chem. Engng. 63 (1956), 208—210.
14. ORLICEK A. F., P6LL H„ und WALENDA H., Hilfsbuch fflr Mineralotechniker, Bd. 1: Die Eigenschaften von Kohlenwasserstoffen, Mineralolprodukten und Hilfsstoffen, Springer-Verlag; Wien 1951.
15. BAEHR H., Chemie-Ing.-Techn. 23 (1953), 676.
16. KRAMER M. T., Glas-Instrumenten-Techn. 15 (1971), 251—254; 17 (1973), 43—48, 137—139.
4. ЛИТЕРАТУРА
1. SIGWART K-, Destination und Rektifikation, in: Ullmanns Encyklopadie der technischen Chemie, Bd. 1, Urban Schwarzenberg, Mflnchen/Berlin 1951, S. 429/470.
2. TGL 21 120.
з’. KUHN W.', Helv. chim. Acta 25 (1942), 252—295; KUHN W., und RYFFEL K., Helv. chim. Acta 26 (1943), 1693—1721.
За. НИСЕЛЬСОН Л. А., ТРЕТЬЯКОВА К- В., ЯТКО М. Е. ЖПХ, 1973, т. 46, № 7, с. 1613—1614.
3b. GRACZYK J., Nukleonika 1 (1973), 1, 25—33.
4. MEIER W., und HUBER M., Chemie-Ing.-Techn. 39 (1969), 797—800.
5. HUBER M„ und HILTENBRUNNER R., Chem. Engng. Sci. 21 (1966), gjg_____
6. KAFAROV V. V., D0R0KH0V J. N., und SHESTOPALVOV V. V., Hydrodynamische Parameter von Ftillkorperkolonnen, Tagungsheft 4, Distillation 1969, Brighton/England, Referat: Chemie-Ing.-Techn. 42 (1970) 14 953
7. JONAS, Chr., Chem. Techn. 26 (1974) 12, 87—91.
8. ZUIDERWEG F. J., und HARMENS A. Chem. Engng. Sci. 9 (1958) 2/3, 89—103.
9. KRELL E„ Chem. Techn. 16 (1964) 10, 591—600.
10. ТИТОВ А. А., ЗЕЛЬВЕНСКИЙ Я- Д- Хим. пром., 1974, Ns 9, с. 50—53.
11. KRELL Е., Plaste und Kautschuk 14 (1967) 4, 251—254.
Ila. KWASNIAK J., Verfahrenstechnik 8 (1974) 11, 315-322.
lib. YILMAZ T.,und BRAUER H., Chemie-Ing.-Techn. 45 (1973) 14, 928—934.
11c. KUNNE H.—J., Chem. Techn. 25 (1973) 9, 533—538.
lid. DIETZi H., REINEMANN G., und COMEL M„ Chem. Techn. 25 (1973)
3, 143—148.
lie. GODAU H.—J., Chem. Techn. 26 (1974) 9, 561—564.
Ilf. BILLET R., CZ-Chem. Techn. 3 (1974), 9—12.
llg. ADOLPHI H.—V., Chem. Techn. 26 (1974), 10, 643—645.
lift. АНТОНОВ Ю. E„ САФИН P. III., PENT В. И. TOXT, 1975, t. 9, № 1, c. 140—142.
12. STAUDINGER H., Anleitung zur organlschen qualitativen Analyse, 6. Aufl., Springer-Verlag, Berlin/Gottingen/Heidelberg 1955, S. 12—13.
13. STAGE H., Erdol und Kohle 3 (1950), 478—490.
14. HILDEBRAND J. H., und ROTARIU S. J., Analytic. Chem. 24 (1952), 770—773.
15. MAUSER H., und KORTUM G., Z. Naturforsch. 10 a/4 (1955), 317—322.
16. MANNCHEN W., Einftihrung in die Thermodynamik der Mischphasen, VEB Deutscher Verlag fiir Grundstoffindustrie, Leipzig 1965.
17. SCHUBERTH H., Thermodynamische Grundlagen der Destination und Extraktion, Bd. I, VEB Deutscher Verlag der Wissenschaften, Berlin 1972.
18. DREISBACH R. R., Pressure—Volume—Temperature Relationship of Organic Compounds, 3. Aufl., Handbook Publ., Inc., Sandursky, Ohio 1952.
19. DREISBACH R. R., Physical Properties of Chemical Substances, The Dow Chemical Company, Midland, Michigan 1951—1953.
487
20. STULL D. R., Ind. Engng. Chem. 39 (1947), 517/550 u. 1684.
21. STAGE H., Fette u. Seifen 53 (1951), 677—682.
22. STAGE H., Fette u. Seifen 55 (1953), 217—224.
23. STAGE H., MULLER E., und FALDIX P., Erdol u. Kohle 6 (1953), 375—380.
24. STAGE H-, MULLER E., und FALDIX P., Chem. Techn. 6 (1954), 31/36.
25. JORDAN T. E., Vapor Pressure of Organic Compounds, 2. Aufl., Interscience Publishers, New York 1954.
26. RIDDICK J. A., und TOOPS JR., E. E., Organic Solvents, Physical Properties and Methods of Purification, Interscience Publischers, New York 1955.
27. SAGE B. H., und LACEY W. N., Some Properties of the Lighter Hydrocarbons, Hydrogen Sulfide and Carbondioxide American Petroleum Institute, New York 1955.
28. GLASER F., und RU.LAND H., Chemie-Ing.-Techn. 29 (1957), 772—775.
29. JAECKEL R., Destination und Sublimation in Fein- und Hochvakuum, in: HOUBEN—WEYL, Methoden der organischen Chemie (hrsg. von Eugen Muller), Bd. 1/1, Georg Thieme Verlag, Stuttgart 1958.
30. KIENITZ H„ Bestimmung des Dampfdruckes, in: HOUBEN—WEYL, Methoden der organischen Chemie (hrsg. von Eugen Muller), Bd. III/l, Georg Thieme Verlag, Stuttgart 1958, S. 255—324.
31. MILAZZO G., Chemie-Ing.-Techn. 28 (1956), 646—654.
32. SWIETOSLAWSKI IF., Ebulliotrie Measurements, Reinhold, New York 1945. SWIETOSLAWSKI W., und ANDERSON J. R„ in: A. WEISSBERGER; Physical Methods of Organic Chemistry, Vol. I, Interscience Publishers, New York 1994, S. 107.
33. WEBER U. von, J. prakt. Chem. 1/4 (1955), 318—323.
34. EBLIN P.„ J. chem. Educat. 27 (1950), 67.
35. HERRINGTON E. F., und MARTIN J. F., Trans. Faraday Soc. 49/2 (1953), 154—162. t '
36. NATELSON S„ und ZUCKERMANN J. L., Ind. Engng. Chem., analyt. Edit. 17 (1945) 739 bis 740.
37. GOULD C., JR., HOLZMANN G., und NIEMANN C., Analytic Chem. 19 (1947), 204—206.
38. WIBERLEY F. J., SIEGERIEDT R. K-, und BENEDETTI-PICHLER A. A., Mikrochem. verein. Mikrochim. Acta 38 (1951), 417—475.
39. НЕСМЕЯНОВ A. H. Атомная энергии. 1957, т. 3, вып. 9, с. 227—237.
40. СКЛЯРЕНКО С. И., МАРКИН Б. И., БЕЛЯЕВА Л. Б. ЖФХ, 1958, т. 32, № 8, с. 1916—1921.
41. HENNING F., und STOCK A., Z. Physik 5 (1921), 226.
42. Smith und Menzies, Z. physik. Chem. 75 (1911), 501.
43. EUCKEN A., und SUHRMANN R., Physikalisch—chemische Praktikumsaufgaben, 7. Aufl., Akademische Verlagsgesellschaft Geest & Portig KG, Leipzig 1968, S. 100—106.
44. KORTUM G., MOEGLING D„ WOERNER F.; Chemie-Ing.-Techn. 22 (1950), 453—457.
45. SCHLUNDER E. U., Chemie-Ing.-Techn. 35 (1963) 7, 482—487.
46. ROCK H., Z. physik. Chem., N. F. 4 (1955), 242—245.
47. PETER M., Magyar Kemiai Folyoirat 10 (1953), 295—300, Referat in: Chem. Techn.. 6 (1954), 307.
47a. KOLLAR G., und NAGY E., Chem. Techn. 23 (1971), 1, 35—36.
48. PEARSON D. E., J. chem. Educat. 28 (1951), 60—62.
49. ENGLISH W. D., J. Amer. chem. Soc. 74 (1952), 2927—2928.
50. DUHRING U., Wiedem. Ann. 11 (1880), 163; 52 (1894), 556.
51. RECHENBERG G. von, Einfache und fraktionierte Destination in Theorie und Praxis, Verlag Schimmel & Co. AG, Miltitz 1923.
52. JANTZEN E., und ERDMANN W„ Fette u. Seifen 54 (1952), 197—20Г.
53. КИРЕЕВ В. А., ЖПХ, 1934, т. 7, № 1—2, с. 1—15.
488
54. ANTOINE С., C. R. hebd. Seances Acad. Sci. 107 (1888), 681, 863, 1143/45.
55. THOMSON G. W., Chem. Rev. 38 (1946), 1—39.
56. RIEDEL L„ Chemie-Ing.-Techn. 26 (1954), 83—89.
57. RECKHARD, H., Erdol u. Kohle 116 (1958), 234—241; Chemiker-Ztg., 84 (1960), 33—36.
58. WICHTERLE I., und LINER J., Die Antoinschen Dampfdruck-Konstanten reiner Verbindungen, Verlag Academia, Prag 1971 (in engl. Sprache).
59. Berechnung thermodynamischer Stoffwerte von Gasen und Flflssigkeiten, VEB Deutscher Verlag fur Grundstoffindustrie, Leipzig 1966, S. 77—89.
60. OTHMER D. F., und YU E.—S., Ind. Engng. Chem. 60 (1968), 1, 22—35. 60a. ZIA T., und THODOS G., Canad. J. chem. Engng. 52 (1974) 5, 630—635.
61. SCHWAB G. M„ Z. f. angew. Chem. 43 (1930), 155—156.
62. STANGE R., Chem. Techn. 5 (1953), 78—79.
63. OTHMER D. F., MAURER P. W., MOLINARY C. J., KOWALSKI R.C., Ind. Engng. Chem. 49 (1957), 125—137.
64. WITTENBERGER W., Chemiker-Ztg, 76 (1952), 371—373.
65. BERGHOLM A., Svensk kern. Tidskr. 60 (1948), 96.
66. HOFFMANN W„ und FLORIN F., Z. Ver. dtsch. Ing., Beih. Verfahfenstechn., (1943), 47—51.
67. COX E. R., Ind. Engng. Chem. 15 (1932), 592—593.
68. DAVIS D. S., Ind. Engng. Chem. 17 (1925), 435.
69. CALINGART G., und DAVIS D. S„ Ind. Engng. Chem. 17 (1925), 1287.
70. FALDIX P., und STAGE H., Chem. Techn. 5 (1953), 526—528, 718—719.
71. STAGE H„ und JUILFS J., Chemiker-Ztg. 77 (1953), 511, 538, 575; 78 (1954), 43, 112, 182, 217.
72. THORMANN K., Destillieren und Rektifizieren, Spamer-Verlag, Leipzig 1928. {TOPMAH К., Перегонка и ректификации, Химтеоретиздат, М., 1935).
73. MATZ W., Die Thermodynainik des Warme- und Stoffaustausches in der Verfahrenstechnik, Bd. 1 und 2, Dr Dietrich Steinkopff Verlag, Darmstadt. 1949/1953.
74. KORTUM G., und BUCHHOLZ-MEISENHEIMER H., Die Theorie der Destination und Extraktion von FlOssigkeiten, Springer-Verlag, Berl in/Go tt ingen/Heidelberg 1952.
75. BOSNJAKOVI^ Fr., Technische Thermodynamik, Teil II, 5. Aufl., Verlag Theodor Steinkopff, Dresden 1971.
(БОШНЯКОВИЧ Ф., Техническая термодинамика, т. 2, Госэнергоиздат, М—Л., 1956).
76. FLORIN F., Brennstoff-Chem. 33 (1952), 161—166.
77. McCABE W. L., und THIELE E. W., Ind. Engng. Chem., ind. Ed. 17 (1925), 605.
78. HALA E., PICK J-, FRIED V., und VILIM 0., Rovnovaha kapalinapara Nakladetelstoi ceskoslovenska akademie red, Prag 1955; 2. engl. Ausgabe: Vapour-Liquid-Equilibrium, Pergamon-Press, London 1968; deutsche Ausgabe: Gleichgewicht—Flfissigkeit—Dampf, Akademie-Verlag, Berlin 1960.
79. Chemie-Ing.-Techn. 41 (1969) 12, 735—736.
80. WILSON G. M., J. Amer. chem. Soc. 86 (1964), 127.
81. DONTH E. J., und WEICHMANN R., Chem. Techn. 20 (1968) 2, 93—95.
82. KRUG K., HABERLAND D., und BITTRICH H.—J., Chem. Techn. 23 (1971) 7, 410—415.
83. RENON H., und PRAUSNITZ J. M„ American Institute of Chemical Engineers Journal 14 (1968), 135.
84. Р03ЕНГАРТ M. И., Техника лабораторной перегонки и ректификации. Госхимиздат, М.—Л., 1951.
85. ORLICEK A. F., und POLL Н., Hilfsbuch ffir Mineraloltechniker, Bd. I u. Il, Springer-Verlag, Wien 1951/1955.
86. VOORWIJK J. F„ Chem. Engng. 63 (1956), 201—202.
87. HOLLO J., und LENGYEL T., Ind. Engng. Chem. 51 (1959), 957—960; Collect czechoslov. chem. Commun. 23 (1958), 1735—1743.
16 э. Крель 489
HOLLO J., Periodica polytechn. [Budapest]; chem. Ingenieurwes., 2 (1958), 113—128; Fette, Seifen, Anstrichmittel 62 (1960) 10, 913—918.
88. UMSTATTER H., und FLASCHKA H„ Erdol u. Kohle 1 (1948), 151—157.
89. WILLIAMS R. A., und HENLEY E. J., Chem. Engng. J. 1 (1970) 2, 145—151.
90. MELPOLDER F. W., und HEADINGTON С. E., Ind. Engng. Chem. 39 (1947), 763—766; ROSE A., und BILES R., Chem. Engng. Progr. 51 (1955), 138—140.
91. ROSE E„ Ind. Engng. Chem. 33 (1941), 596.
92. HAWKINS J. E., und BRENT J. A., Ind. Engng. Chem. 43 (1951), 2611—2621.
93. ЦЫПКИНА 0. ЖПХ, 1958, т. 28, № 2, с. 185—192.
94. KORTUM G„ Chemiker-Ztg. 74 (1950), 151—154.
95. CARLSON H. C., und COLBURN A. P., Ind. Engng. Chem. 34 (1942), 581.
96. ORLICEK A. F., Osterr. Chemiker-Ztg. 50 (1949), 86.
97. LAAR J. J. VAN, und LORENZ R., Z. anorg. Chem. 145 (1925), 239; Z. phys. Chem. 72 (1910), 723; 83 (1913), 599.
98. MARGULES M„ Wiener Sitz. Ber. 104 (1895), 1243.
98a. GREWER Th., und SCHMIDT A., Chemie-Ing.-Techn. 45 (1973) 17, 1063-1066.
99. LANDOLT H., und BORNSTEIN R., Physikalisch — Chemische Tabellen, 6. Aufl., Springer-Verlag, Berlin/Gottingen/Heidelberg 1950/55.
100. PERRY J. H„ Chemical Engineers' Handbook, 4. Aufl., McGraw-Hill, Book Company, New York 1963, S. 1942ff.
101. YU CHIN CHU, Distillation Equilibrium Data, 2. Aufl., Reinhold, New York 1956.
102. CHU WANG, LEVY und PAUL, Vapour Liquid Equilibrium Data, Edwards T. W., Ann. Arbor Mich., 1956.
103. KIRSCHBAUM E., Destillier- und Rektifiziertechnik, 4. Aufl., Springer-Verlag, Berlin/Heidelberg/New York 1969.
103a. HUBNER W., und SCHLUNDER E. U., Chemie-Ing.-Techn. 45 (1973) 5, 247—253.
103b. VOGELPOHL A., und CERETTO R., Chemie-Ing.-Techn. 44 (1972) 15, 936—938.
104. JACOBS J., Destillier — Rektifizier — Anlagen., R. Oldenbourg Verlag, Mflnchen 1950.
105. КОГАН В. Б., ФРИДМАН В. М., КАФАРОВ В. В., Равновесие между жидкостью и паром. Справочное пособие. Наука, М.—Л., 1966.
106. HALA Е., WICHTERLE I., POLAK J-, und BOUBLIK Т„ Vapour-Liquid Equilibrium Data at Normal Pressure, Pergamon Press, London 1969.
106a. The Institution of Chemical Engineers, London, fiihrt einen Daten-Service (PPDS-Physical Property Data Service) durch, der auch die Phasengleichgewichtsdaten Fliissig/Dampf erfa₽t.
107. KORTUM G„ und FREIER H.—J., Chemie-Ing.-Techn., 26 (1954), 670—675.
108. WEBER U. von, Z. phys. Chem., N. F. 38 (1963) 3/4, 129—139.
109. GELBIN D., Chem. Techn., 15 (1963) 1, 6—9.
110. STAGE H., MULLER E., und GEMMEKER L., Chemiker-Ztg./Chem. Apparatur 85 (1961), 11.
111. OTHMER D. F„ Ind. Engng. Chem., ind. Edit. 20 (1928), 743.
112. STAGE H., und FISCHER W. G., Glas-Instrumenten-Techn. 12 (1968), 11, 1167—1173.
113. GILLESPIE D. C„ Ind. Engng. Chem., analyt. Edit. 18 (1946), 575.
114. ROCK H., und SIEG L., Z. physik. Chem., N. F. 3 (1955), 355—364; Chemie-Ing.-Techn. 28 (1956), 133.
115. KORTUM G., und BIEDERSEE H., von, Chemie-Ing.-Techn. 42 (1970), 8, 552—560.
490
116. SCHMIDT R., WERNER G„ und SCHUBERTH H., Z. phys. Chem. 242 (1969). 381—390.
117. WICHTERLE I., und HALA E., ind. Engng. Chem. Fundamentals 2 (1963) 2, 155—157.
118. NEUMANN A., und WALCH W„ Chemie-Ing.-Techn. 40 (1968) 5, 241—244.
118a. STAGE H„ und FISCHER W-. G., verfahrenstechnik 7 (1973) 6, 1—3.
119. KUMARKRISHNA RAO, V. N„ SWAMI D. R., und
NARASINGARAO M., Amer. Ind. chem. Eng. 3 (1957), 191—197.
120. JOST W., ROCK H., SCHRODER W., SIEG W., und WAGNER H. G., Z. physik. Chem., N. F. 10 (1957), 133—136;
SCHRODER Ц7., Chemie-Ing.-Techn. 30 (1958), 523—525.
121. LYDERSEN A. L., und HAMMER E. A., Chem. Engng. Sci. 7 (1958), 241—245.
122. JUNGHANS W., und WEBER U. von, J. prakt. Chem. 4/2 (1955), 2g5______273
123. MAI K. L., und BABB A. L., Ind. Engng. Chem. 47 (1955), 1749—1757.
124. ЗЕЛЬВЕНСКИЙ И- Д-, ШАЛЫГИН В. А., ЖФХ, 1957, т. 31, № 7, с. 1501—1509.
125. REDLICH О., und KISTER А. Т., Ind. Engng. Chem. 40 (1948), 341—348.
126. HERINGTON E. F. G., Nature [London] 161 (1947), 610—611.
HAMER D. F. 0., und MITARB., Ind. Engng. Chem. 42 (1950), 120.
HAASE R., und JOST W., Z. phys. Chem., N. F. 9 (1956), 300—301.
TIERNEY J. W., Ind. Engng. Chem. 50 (1958), 707—710.
127. HERINGTON E. F. G„ J. appl. Chem. 2 (1952), 11—23.
128. BITTRICH H.—J., Chem. Techn. 14 (1962) 9, 527—533.
129. TAO L. C., Ind. Engng. Chem., Fundamentals 1 (1962) 2, 119—123.
130. ROLLING H., Chemie-Ing.-Techn. 24 (1952), 405—411.
131. BOWMAN J. R„ und CICHELLI M. T., Ind. Engng. Chem. 41 (1949), 1985.
132. JUNGE C., Chem. Techn. 8 (1956), 579—588.
133. ANSCHUTZ R„ Chem. Techn. 9 (1957), 516—519.
134. FISHER G. T., Ind. Engng. Chem., Process Design and Development 2 (1963), 284—288.
135. BILLET R., Chemie-Ing.-Techn. 30 (1958), 513—515.
136. THUM 0., Chemie-Ing.-Techn. 29 (1957), 675—678.
137. NERETNICKS I., ERICSON I., und ERIKSSON S., Brit. chem. Engng.
14 (1969) 12, 1711 bis 1712.
138. WEBER U. VON, Chem. Techn. 2 (1950), 241—246.
139. VOGELPOHL A., Chemie-Ing.-Techn. 43 (1971) 20, 1116—1121.
139a. DOMENECH S., GUICLION C., und ENJALBERT M., Chem. Engng. Sci. 29 (1974) 7, 1519—1528, 1529—1535.
140. BILLET R., Chemie-Ing.-Techn. 30 (1958), 407—416.
140a. WILCOX W. R., Ind. Engng. Chem. Fundamentals 3 (1964) 1, 81—83.
141. THORMANN K., Chem. Techn. 2 (1950), 255—256.
142. CAVERS S. D., Ind. Engng. Chem., Fundamentals 4 (1965) 2 , 229—230.
143. POHL K-, Chemie-Ing.-Techn. 28 (1956), 562—564.
144. HORVATH P. J.t und SCHUBERT R. F., Chem. Engng. 10 (1958).
145. FLATT R., Chimia [Zurich] 9 (1955), 232—237.
146. STAGE H., und SCHULTZE Gg. R., Oel u. Kohle 5/6 (1944), 90—95.
147. ZUIDERWEG F. J., Laboratory of Batch Distillation, Interscience Publishers, New York 1957.
148. SCHAFER W., Angew. Chem., Teil В 19 (1947), 251—253.
149. FISCHER W., Chemie-Ing.-Techn. 23 (1951), 116.
150. MATZ W., Angew. Chem., Teil В 19 (1947), 131—134.
151. HILBERATH F., Oel u. Kohle 39 (1943), 875—886.
152. BRAGG L. B., und LEWIS J. W„ Wld. Petroleum Rep. 14 (1943), 61.
152a. STRANGIO V. A., und TREYBAL R. E„ Ind. Engng. Chem. Process Design and Development 13 (1974) 3, 279—285.
16*
491
153. ROSE A. und E., und Mitarb., Distillation, Interscience Publishers, New York 1951.
154. ROSE A., Ind. Engng. Chem. 33 (1941), 594.
155. FENSKE M. R., TONGBERG S. 0., und QUIGGLE D., Ind. Engng. Chem. 26 (1934), 1169.
156. RICHTER H., Oel u. Kohle 40 (1944), 282—288.
157. POHL H., Erdol u. Kohle 5 (1952), 291—294.
158. ZUIDERWEG F. I., Chemie-Ing.-Techn. 25 (1953), 297—308.
159. SIZMANN R., Chemie-Ing.-Techn. 33 (1961), 659—668.
160. MANNING R. E., und CANNON M. R., Ind. Engng. Chem. 49 (1957), 346—349.
161. STUKE B., Chemie-Ing.-Techn. 25 (1953), 677—682.
162. HEISE F., HILLER G., und WAGNER H. Gg., Chemie-Ing.-Techn.
41 (1969) 20, 1100 bis 1105.
162a. KAUTER A., und WAGNER H. Gg., Chemie-Ing.-Techn. 42 (1970), 1127; 46 (1974), 997.
162b. BORCHARDT E., und WAGNER H. Gg., Chemie-Ing.-Techn. 43 (1971), 95g_______gg2
163. CHILTON T. H., und COLBURN A. P., Ind. Engng. Chem., ind. Edit. 27 (1935), 255 bis 260, 904.
164. CARNEY Th.. P., Laboratory Fractional Distillation, Mac-Millan, New York 1949.
165. DANATOS S., und OSBURN J. 0., Chem. Engng. 65 (1958), 147—150.
166. ARKENBOUT G. J., und SMIT W. M„ Separation Sci. 2 (5) (1967), 575—596.
167. MERKEL F., Arch. Warmewirtsch. Dampfkesselwes. 10 (1929), 13—17.
168. WEBER U. von, in: GILDEMEISTER E„ und [HOFMANN Fr., Die atherischen Ole (bearb. v. W. Treibs), Bd. I, Akademie-Verlag, Berlin 1966.
169. ROBINSON C. S., und GILLILAND E. R., Elements of Fractional Distillation, 6. Aufl., McGraw-Hill Book Company, New York 1950, 476—478.
170. WEBER U. von, Chem. Techn. 2 (1950), 241—246.
171. ROLLING H., Chemie-Ing.-Techn. 22 (1952), 405—411.
172. KOHRT H. U., Angew. Chem., Teil В 20 (1948), 117—124.
173. HAUSEN H„ Z. angew. Physik 4 (1952), 41—51.
174. WIJK W. R. van, und THISSEN H. A. C., Chem. Engng. Sci. 1 (1952), 121/123; 3 (1954), 145—152.
175. ЛЬВОВ С. В. — ДАН СССР, 1947, т. 57, № 4, с. 375—378.
175а. MOSTAFA Н. A., Chemie-Ing.-Techn. 47 (1975), 2 , 63.
175b. CHIEN Н. Н. J., Chem. Engng. Sci. 8 (1973) 11, 1967—1974.
175c. SEROV V. V., ABRAMENKO V. P„ und ZYKOV D. D„ Int. chem. Engng. 13 (1973) 3, 514—516.
176. KRELL E„ Chem. Techn. 4 (1952), 200—207.
176a. FALDIX P.,und STAGE H., Chemie-Ing.-Techn. 41 (1969) 23, 1265—1269.
176b. MULLER W„ Chem. Techn. 26 (1974) 1, 16—20.
176c. BILLET R., Verfahrenstechnik 8 (1974) 3, 65—72.
177. THORMANN K-, Dechema-Erfahrungsaustausch, Techn. Apparate und Anlagen, Trennen flussiger Mischungen, Destillieren, Betriebstechnik, Dechema, Frankfurt/Main, August 1952.
178. ELLIS S. R. M., und FRESHWATER D. C., Perfum. essent. Oil. Rec.
45 (1954), 380 bis 386.
179. COLBURN A. P., Trans. Amer. Inst. chem. Engr. 37 (1941), 805.
180. UNDERWOOD A. J. V., Chem. Engng. Progr. 44 (1948), 603.
181. FENSKE M. R„ Ind. Engng. Chem. 24 (1932), 482.
182. HARBERT W. D., Ind. Engng. Chem. 37 (1945), 1162.
183. BRUUN P. J., Meded. Landbouwhogeschool, Wagningen 61 (9), (1961), 1—94.
184. UNDERWOOD A. J. V., Trans. Instn. chem. Engr. [London] 10 (1932), 112—152.
185. MURCH D. P., Ind. Engng. Chem. 45 (1953), 2616—2621.
492
186. MULLIN J. W., Ind. Chemist, 33 (1957), 390, 408—417.
187. KIRSCHBAUM E., Angew. Chem., Teil В 19 (1947), 13—14; Chemie-Ing.-Techn. 28 (1956), 639—644.
188. КАЗАНСКИЙ Б. А. и др., ЖОХ, 1942, t. 12, № 1—2, c. 112—123.
189. БУШМАКИН И. H., ЛЫЗЛОВА P. В. — ЖПХ, 1952, t. 25, l№ 3, c. 303—312.
190. PERKTOLD F„ Angew. Chem., Teil В 19 (1947), 184—185.
191. DAVID A., Dechema-Monogr., 23 (1954), 126—175.
192. MYLES M., und MITARB., Ind. Engng. Chem. 43 (1951), 1452—1456.
193. ALLENBY О. C. W., und L'HEUREUX C„ Analytic. Chem. 22 (1950), 1340.
194. H ALDEN W ANGER K-, Chemie-Ing.-Techn. 23 (1951), 437—440.
195. ZUIDERWEG F. J. (edit.), Recommended Test Mixtures for Distillation Columns, The Institution of Chemical Engineers, London 1969.
196. BRANDT H., und ROCK H., Chemie-Ing.-Techn. 29 (1957), 397—402.
197. FELDMANN J., und MITARB., Ind. Engng. Chem. 45 (1953), 214.
198. ЗЕЛЬВЕНСКИЙ Я- Д., ШАЛЫГИН В. A. — Хим. пром., 1962, № 6, с. 38—41.
199. SCHULTZE Gf. R., und STAGE H., Oel u. Kohle 40 (1944), 68.
200. COLLINS F. C., und LANTZ V., Ind. Engng. Chem., analyt. Edit. 18, (1946), 673—677.
201. NAUMANN K., und LEIBNITZ E., Chem. Techn. 8 (1956), 458—471.
202. ОБОЛЕНЦЕВ P. Д„ ФРОСТ A. B. — Ytefyr. хозяйство, 1947, т. 25, № 8, с. 35—45.
203. STRUCK R. T., und KINNEY C. R., Ind. Engng. Chem. 42 (1950), 77—82.
204. WEBER U. von, Personliche Mitteilung.
205. ZUIDERWEG F. H„ Chem. Engng. Sci. 1 (1952), 174—193.
206. KIRSCHBAUM E., BUSCH W., und BILLET B„ Chemie-Ing.-Techn. 28 (1956), 475—480.
207. SZAPIRO S., Zeszyty naukowe Politechn. Lodzkiej, Chem. 2 (1955), 33—37.
208. BRAUER H„ Chemie-Ing.-Techn. 29 (1957), 520-530, 785—790.
209. ЗЕЛЬВЕНСКИЙ fl. Д„ ТИТОВ А. А., ШАЛЫГИН В. A. — Хим. пром., 1963, № 2, с. 36—43.
210. WEYGAND С., und HILGETAG G., Organisch-chemische Experimentierkunst, 4 Aufl., Johann Ambrosius Barth, Leipzig 1970. (ВЕЙГАНД К., ХИЛЬГЕТАГ Г. Методы эксперимента в органической химии, Химия. М., 1968).
211. BERNHAUER К., Einfiihrung in die organische-chemische Laboratoriumstechnik, Springer-Verlag, Wien 1947.
212. PESTEMER M., Angew. Chem. 63 (1951), 118—122.
213. BRANDT P. L., PERKINS R. B., und HALVERSON L. K„ Oil Gas J. 7 (1946).
214. LENTZ H., Chemie-Ing.-Techn. 41 (1969) 15 , 855—861.
214a. LEUNG L. S., HUTTON В. E. T., und NICKLIN D. J., Ind. Engng. Chem. Fundamentals 14 (1975) 1, 63—67.
215. STAGE F„ Angew. Chem., Teil В 19 (1947), 182.
216. SCHULTZE Gg. R., und STAGE fH., Uber die Wirksamkeit der neuen Normalkolonne des «Ausschusses fiir Laboratoriumsdestillation», Fiat-Report Nr. 1 133 vom 23.12.1947.
217. SIGWART K., Destination und Rektifikation, in: Ullmanns Encyklopadie der technischen Chemie, Bd. 1, Urban und Schwarzenberg, Miinchen/Berlin 1951, S. 429—470.
218. THORMANN K., Dechema-Erfahrungsaustausch, Laboratoriumstechnik, Arbeitsmethoden und Gerate, Destillieren und Rektifizieren, Dechema, Frankfurt/Main, September 1950.
219. MACH E., VDI-Forschungsheft 375 Nov./Dez. 1935.
220. BARTH W., Chemie-Ing.-Techn. 23 (1951), 289—293; 26 (1954), 29—34.'
221. LEVA M., Flow through Packings and Beds, Part I, New York 1957.
493
222. KAST W., Chemie-Ing.-Techn. 36 (1964) 5, 464—468.
223. TEUTSCH T„ Chemie-Ing.-Techn. 36 (1964), 5, 496—523.
224. GELBE H., Chemie-Ing.-Techn. 40 (1968), 528—530.
225. GLASER H., Chemie-Ing.-Techn. 41 (1969) 7, 444—450
226. HECKMANN C. J., und KRELL E., Chem. Techn. 5 (1953), 699—704.
227. NEUMANN F., Chemie-Ing.-Techn. 33 (1961), 439—442.
228. REED T. M., und FENSKE M. R., Ind. Engng. Chem. 42 (1950), 654—660.
229. STAGE H., Aus einem Prospekt liber Labodest-Baukasten-Destillationsgerate, Zeichnung Nr. 741, 98, 156. STAGE H., und BOSE K., Chemie-Ing.-Techn. 27 (1955), 439.
230. U MHOLTZ C. L., und WINKLE M. van, Petroleum Refiner 34 (1955), 114—117.
231. SCHUMACHER R., Erdol u. Kohle 2 (1949), 501—508.
232. BORCHERT R., und JUBITZ W., Infrarotstrahler zur Erwarmung, Trocknung und Aushartung, SVT Band 5, VEB Verlag Technik, Berlin 1951.
233. JUNGE C., Chem. Techn. 6 (1954), 37—38.
234. GROSSE-OETRINGHAUS H., Erdol u. Kohle 1 (1948), 273—280.
235. REID R. C., und SHERWOOD T. K-, The Properties of Gases and Liquids, 2. Aufl., McGraw-Hill Book Company, New York 1966.
236. HECHT G., und MIT ARB., Berechnung thermodynamischer Stoffwerte von Gasen und Fliissigkeiten, VEB Deutscher Verlag fiir Grundstoffindustrie, Leipzig 1966.
237. STAGE H., Fette u. Seifen 55 (1953), 284—290.
238. KHARBANDA 0. M. P., Nomograms for Chemical Engineers, Heywood & Company, London 1958.
239. ORLICEK A. F., und POLL. H., Hilfsbuch fiir Mineraloltechniker, Bd. I u. II, Springer-Verlag, Wien 1951/55.
240. HAUSSLER W„ Technik 9 (1954), 51—52.
241. COLE P. J., Analytic, chem. 22 (1950), 946—947.
242. KRELL E., Chem. Techn. 4 (1952), 200—207.
243. MEIER-GROLMAN F. W., und WESOLOFSKY F., Oel u. Kohle 39 (1941), 297—300.
244. BECK R., Chemie-Ing.-Techn. 44 (1972), 8, 596—598.
245. GEMMEKER L., und STAGE H-, Glas-Intrumenten-Techn. 7 (1963) 1, 18—23.
246. KAPLICK K., und SCHUBERT M., Datenverarbeitung in der Verfahrenstechnik—Digitale Rechenautomaten, Bd. 6 der Reihe: Beitrage zur Verfahrenstechnik, VEB Deutscher Verlag fiir Grundstoffindustrie, Leipzig 1969.
247. WAGNER M., Analogrechner in der Verfahrenstechnik, Bd. 10 der Beihe: Beitrage zur Verfahrenstechnik, VEB Deutscher Verlag fiir Grundstoffindustrie, Leipzig, 1972.
248. ADLER H., Elektronische Analogrechner, 3. Aufl., VEB Deutscher Verlag der Wissenschaften, Berlin 1970.
249. SAVAS E. S., Computer Control of Industrial Processes, McGraw-Hill Book Company, New York/Toronto/London 1965.
250. ROTH H., Chem. Techn. 20 (1968), 2, 77—82.
251. SCHUBRING A., Chemie-Ing.-Techn. 32 (1960) 3 , 223—227.
252. SEALEY C. J., Chem. Engng. Sci. 25 (1970), 561—568.
253. LEIBNITZ E„ und SCHUHLER C., Z. physik. Chem. 217 (1961), 217—229.
254. NAGEL 0., SMITS H., WOLF D., und GUNZLER H., Chemie-Ing.-Techn. 41 (1969) 17, 983—987.
255. PILHOFFER Th., Chemie-Ing.-Techn. 42 (1970), 7J509—515. JOHNSON M. L„ und LUPFER D. E., Chem. Engng. Progr. 62 (1966) 6, 75—79.
256. WANG I. C., und HENKE G. E., Hydrocarb. Process, 45 (1966), 8, 155.
257. SARGENT R. W. H., und MURTAGH B. A., Trans. Instn. chem. Engr. 47 (1969), 85.
258. STAINTHORP F. P., und M1TARB. in: P1RIC J. M.: Efficient
494
Computer Methods for the Practising Chemical Engineer, London 1967, S. 181—189.
259. BLATT E., und FLEISSNER H., Glas-Instrumenten-Techn. 15 (1971), Sonderheft 1333 bis 1337.
260. BRAUER H., und MEWES D„ Chemie-Ing.-Techn. 44 (1972), 1/2, 93—96.
261. BITTER R. C., Chemie-Ing.-Techn. 40 (1968) 13, 651—656.
262. WAGNER H., und BLASS E„ Chemie-Ing.-Techn. 45 (1973), 63—67.
263. REICHELT W., und BLASS E., Chemie-Ing.-Techn. 43 (1971) 17, 949________956.
REICHELT W., Chemie-Ing.-Techn. 43 (1971) 22, 1196—1203; 44 (1972) 18, 1068—1071.
264. KOHLER R., und SCHOBER G., Chem. Techn. 25 (1973) 12, 718—721.
265. KIRBACH V., FRENZEL L„ und ST RANKMULLER H., Chem. Techn.
25 (1973), 12, 721 bis 722.
5. ЛИТЕРАТУРА
1. STAGE H.,und GEMMEKER L., Chem. Labor Betrieb 15 (1964) 5, 177—188;
6, 232 bis 242; 7, 290—298; 8, 325—338.
la. STAGE H., CZ-Chemie-Technik 1 (1972), 263—272.
2. BODENHEIMER W., Nature [London] 173 (1954), 124.
3. JANTZEN E„ und WITGERT H., Fette u. Seifen 46 (1939), 563.
3a. FISCHER W. G., GIT Fachz. Lab. 18 (1974) 7, 689—691.
3b. BERTELE E„ und HUMBEL F. A., GIT Fachz. Lab. 17 (1973) 6, 667—674.
4. BABCOCK M. J., Analytic. Chem. 21 (1949), 632—633.
5. SHRADER S. A., und RITZER J. E., Chem. Fabrik 12 (1935), 173.
6. KLENK E., Hoppe-Seyler’s Z. physiol. Chem. 242 (1936), 250.
7. FISCHER W. S., Chemiker-Ztg. 94 (1970) 5, 157—161; Glas-Instrumenten-Techn. 13 (1969), 535—540.
8. WINTERS J. C., und DINERSTEIN R. A., Analytic. Chem. 27 (1953), 546—550.
NERHEIM A. G., und DINERSTEIN R. A., Analytic. Chem. 28 (1956), Ю29__1033.
9. SPlEGELBERG C., Dissertation, F. U., Berlin 1956.
10. GOULD JR., C. W., HOLZMANN G., und NIEMANN C., Analytic. Chem. 20 (1948), 361.
11. BREGER J. A., Analytic. Chem. 20 (1948), 980.
12. PASCHKE R. F., und MITARB., J. Amer. Oil Chemists’Soc. 31 (1954), 5—7.
13. PFEIL E„ Angew. Chem. 54 (1941), 161—184.
14. EIGENBERGER E., Mikrochemie 26 (1939) 204.
15. CLASEN H., Chemie-Ing.-Techn. 28 (1956), 45—48.
16. STAGE H., Fette u. Seifen 55 (1953), 217—224.
17. GUNTHER G., KUHNHANSS K., und HUTTIG E., Chem. Techn. 7 (1955), 656—662.
18. PROFFT E„ und BUCHMANN G„ Chem. Techn. 8 (1956), 417—418. BUCHMANN G.; Chem. Techn. 14 (1962) 10, 617—620; 12, 741—744.
19. FURST H., und SCHWETLICK K-, Chem. Techn. 8 (1956), >458—471.
20. OLECHNOWITZ A. F., Dissertation, Halle-Wittenberg 1956.
21. FI EBACH K., Dissertation, Leipzig 1957.
22. GUTWASSER H.,und MULLER A., Chem. Techn. 21 (1969) 11,756—759. GUTWASSER H., Chem. Techn. 22 (1970) 8, 467—471.
23. MAIR B. J., und MITARB., Analytic. Chem. 30 (1958), 1814—1817.
24. NAUMANN K., und LEIBNITZ E., Chem. Techn. 8 (1956), 458—471.
25. OLDROYD D. M., und GOLDBLATT L. A., Ind. Engng. Chem., analyt. Edit. 18 (1946), 761—763.
495
26. PETROCELLI J. A„ PUZNIAK T. J., und CLARK R. 0., Analytic. Chem. 36 (1964), 1008—1011.
27. Prospekt «Aerograph Research Notes 1965». Wilkens Instrument u. Research fAG, Basel/Schweiz.
28. STAGE H., CZ-Chemie-Technik 2 (1973) 1, 35—40.
29. HAMPEL B„ Chemiker-Ztg. 95 (1971), 7 , 316—325.
30. RIEGER H., Chem. Techn. 22 (1970) 1, 36—40.
31. CARPENTER J. K-, und HELWIG R. W., Ind. Engng. Chem. 42 (1950), 571—578.
32. JORDAN D. G., Chemical Pilot Plant Practice, Interscience Publishers, New York 1955.
33. DIETZ H., und KARDOS J., Chem. Techn. 19 (1967) 12, 713—719.
34. BILLET R., Chemie-Ing.-Techn. 42 (1970) 14, 910—917.
35. KULLMANN W. W., Pharmaz. Ind. 15 (1953), 159—162.
36, JOB C., und ZELLNER H., Pharmaz. Ind. 17 (1955), 161—163. Hersteller: Schott u. Gen., Mainz.
36a. Prospektblatt «Quickfit, Einstufige Wasserdestillationsapparatur».
Leistung 3,5 bis 4 1/h dest. Wasser.
37. ШПОЛЬСКИЙ Э. ВЛ Атомная физика т. 1, 2. Наука. М., 1974.
38. БРОДСКИЙ А. И. Химия изотопов, Изд. АН СССР, М., 1957.
39. BRUNNEE К., und V0SHAGE И., Massenspektrometrie—Physikalische und apparative Grundlagen sowie Anwendungen, Verlag Karl Thi'emig, Mflnchen 1964.
BIRKENFELD H., HAASE G., und ZAHN H., Massenspektrometrische Isotopenanalyse, 2. Aufl., VEB Deutscher Verlag der Wissenschaften, Berlin 1969.
WEITZEL K., HUBNER H., HUBNER G., und MAASS /., Anwendung stabiler Isotope in Forschung und Technik, VEB Deutscher Verlag fiir Grundstoff Industrie, Leipzig 1963.
40. CONN A. L., und WOLF J. E., Ind. Engng. Chem. 50 (1958), 1231—1234, DBP Nr. 944848, ausgegeben am 28.6.1956.
41. MERCEA V., Isotopenpraxis 2 (1966), 8, 313—322.
42. MULLER G„ MAUERSBERGER K., und SPRINZ H„ Analyse stabiler Isotope durch spezielle Methoden, Akademie-Verlag, Berlin 1969.
43. BAERTSCHI P., KUHN W., und KUHN H., Nature London 171 (1953), 1018.
44. SELLERS E. S., und AUGOOD D. R., Trans. Instn. chem. Engr. 34 (1956), 53—78. •
45. TIMMERHAUS K. D., WEITZEL D. H., und FLYNN T. M., Chem. Engng. Progr. 54 (1958), 35—46.
46. WEISSER R. H„ Techn. Rundschau Sulzer 40 (1958), 71—73.
47. SCHAVORONKOV N. M., und SAKODYNSKIJ K- L, Kernenergie 4 (1961) 12, 893—904.
48. CLUSIUS K., und MEYER H„ Helv. chim. Acta 36/7 (1953), 2045—2055.
49. GROTH W., IHLE H., und MURRENHOFF A., Angew. Chem, 68 (1956), 644—648.
50. GLUSIUS K., und SCHLEICH K-, Helv. chim. Acta 42 (1959), 232—239. McINTEER В. B., und POTTER R. M., Ind. Engng. Chem. Proc. 4 (1965) 1, 35—42.
51. ДЕВЯТЫХ Г. Г., ЗОРИН А. Д., НИКОЛАЕВ Н. И. — ЖПХ, 1958, т. 31, № 3, с. 368—375.
52. СЕВР ЮГОВА Н. Н., УВАРОВ О. В., ЖАВОРОНКОВ Н. М. Атомная энергия, 1956, т. 4, с. 113—117.
53. KISTEMAKER J., BIGELEISEN J. В., und NIER А. О., Proceedings of the International Symposium on Isotope Separation (Amsterdam, April 23—27, 1967), North-Holland Publishing Comp., Amsterdam 1958.,
54. ЯГОДИН Г. А., УВАРОВ О. В., ЖАВОРОНКОВ Н. М. — ДАН СССР, 1956, т. 111, № 2, с. 384—387.
55. CLUSIUS К., und SCHLEICH К., Helv. chim. Acta 41 (1958), 1342—1358.
496
56. KIRSHENBAUM J., Physical Properties and Analysis of Heavy Water, McGraw-Hill Book Company, New York 1951.
57. UREY H. C., J. chem. Soc. [London] 1947, 562.
58. BENEDICT M., Survey of Heavy Water Production Processes, A/Conf., 8/P/819, Schweiz, 30.6.1955.
{БЕНЕДИКТ M., Обзор методов получения тяжелой воды. Мирное использование атомной энергии. Материалы Международной конференции в Женеве, август 1955, том. 8, стр. 819. Гос. Научно-технич. изд. химии. литературы. М., 1955).
59. KVHN W., Chemie-Ing.-Techn. 25 (1953), 12—18, 29 (1957), 6—16.
KVHN W., BAERTSCHI Р., und THURKAUF М., Chimia [Zurich] 8 (1954), 109—122, 145—156.
KUHN W„ Helv. chim. Acta 35 (1952), 1684—1736.
KUHN W., Helv. chim. Acta 37 (1954), 1407—1422.
60. BAERTSCHI P., und KUHN tW., Final^Concentration of Heavy Water by Rectification, A/Conf. 8/P/927, Schweiz, 8.7.1955.
61. BECKER E. №., Angew. Chem. 68 (1956), 6—13.
HUBER M., Die Rektifikation als Speziaifall der Theorie der Isotopentrennung, Sulzer-Verfahrenstechnik, Achema 1961.
62. HANNY J., HARTMANN F., und HILTBRUNNER R., Techn. Rundschau Sulzer 4 (1961), 21—26.
63. DIXON 0. G., J. Soc. chem. Ind. 68 (1949), 88—91.
64. KRELL E., Chemie-Ing.-Techn. 32 (1960), 233—240.
65. УВАРОВ) О. В., СОКОЛЬСКИЙ BaA., ЖАВОРОНКОВ H. М. — Хим. пром., 1956, № 7, с. 20—21.
66. DOSTROVSKY J., LLEWELLYN D. R„ und VR0MEN В. Н., J. chem. Soc. 4 (1952), 3509—3525.
67. JACKSON R. F., und PIGFORD R. L., Ind. Engng. Chem. 48 (1956), 1020—1026.
68. РОЗЕН A. M. — ДАН СССР, 1956, т. 107, № 2, с. 295—298.
69. LONDON Н., Separation of Isotopes, Verlag George Newnes Ltd., London 1961.
COHEN K., Theory of Isotopic Separations, McGraw-Hill Book Company, New York 1951.
70. STASCHEWSKI D., Chemie-Ing.-Techn. 40 (1968) 748; 43 (1971) 14, 810—816.
71. SUROWIEC A. J., Ind. Engng. Chem. 52 (1960) 9, 798.
GUPTA D., und RAY S. N., Ind. Engng. Chem. ProC. 1 (1962), 255—262. ПЕТЛЮК Ф. Б., ПЛАТОНОВ В. M., КИРСАНОВ И. В. — Хим. пром., 1964, № 6, с. 45—53.
72. BAERTSCHI Р„ und KUHN W., Helv. chim. Acta 40 (1957), 1084—1103.
73. STAGE H., Chemische Arbeitsgemeinschaft Verfahrenstechnik, Jan./Febr. 1970, 44—54.
73a. SCHWAB H„ Chemie-Ing.-Techn. 44 (1972) 11, 704—707.
74. GELBIN D„ Chem. Techn. 21 (1969) 6, 329—334.
75. CANNON N. R., Ind. Engng. Chem. 53 (1961), 629.
76. MCWHIRTER I. R., und LLOYD W., Chem. Engng. Progr. 59 (1963), 7, 58.
77. BURSTIN H., und WINKLER J., Chemiker-Ztg. 22 (1931), 212.
78. ROLLING H., und TRAMM H., Chemie-Ing.-Techn. 21 (1949), 9.
79. CAPITANI C., und MILANI E„ Ind. chimica 36 (1954), 671—684.
80. STAGE H., Dechema-Monogr. 15 (1950), 156—170. -4*
81. SIGWART K., Destillieren und Rektifizieren, iny HOUBEN-WEYL., Methoden der organischen Chemie, hrsg. von E. Muller, Bd. 1/1, Georg Thieme Verlag, Stuttgart 1957. ч Л- «Ф
82. HAUSEN H., und SCHLATTERER R., Chemie-Ing.-Techn.’21 (1949), 453—460.
KIRSCHBAUM E., und LIPPHARDT G., Chemie-Ing.-Techn. 29 (1957), 393—397.
497
83. OTTO IF., Chem. Techn. 14 (1962) 4, 228—231.
84. KIRSCHBAUM E., Destillier- und Rektifiziertechnik, 4. Aufl., Springer-Verlag, Berlin/Heidelberg/New York 1969.
85. FABUSS B., Z. Ver. ung. Chemiker 8 (1953), 323—324.
86. FISCHER V., Ann. Physik 6 (1947), 139—152.
87. HERRMANN E., Chemiker-Ztg. 80 (1956), 102—104, 131—134, 169—173.
88. TROSTER E., Chemie-Ing.-Techn. 32 (1960) 8, 525—531.
89. JUNGE C., Chem. Techn. 6 (1954), 37—38.
90. TRENNE H., Erdol u. Kohle 2{(1951), 63—66.
91. KUHN W., Helv. chim. Acta 25 (1952), 252—295.
92. WEBER U. von, Chem. Techn. 2 (1950), 241—246.
93. VOIGT G., Kernenergie 2 (1959), 14—39.
93a. BLASS E., Chemie-Ing.-Techn. 45 (1973) 13 , 865—872.
93b. WONDRAK A. M., und KOHLER F., Chemie-Ing.-Techn. 46 (1974), 1, 37.
94. PODBIELNIAK W. J., Ind. Engng. Chem., analyt. Edit. 3 (1931), 177;
5 (1933), 119.
95. GROSSE-OETRINGHAUS H., Erdol u. Kohle 2 (1949), 286—299.
96. WUSTROW W., Z. analyt. Chem. 108 (1937), 305.
97. STOKES Ch. S., und HAUPTSCHEIN M„ Analytic. Chem. 24 (1952), 1526.
DRAW IN H. W., Vakuum-Techn. (1961), 2.
98. LERER E., und KUSS E., Z. physik. Chem. Abt. A, Bd. 163 (1933), 73.
99. MILLER G. H., Operations with Gases in Technique of Organic Chemistry, Vol. Ill, Part. II, Chapter V, Interscience Publishers, New York 1957.
100. MULLER G., und GNAuCK G., Reinste Gase, VEB Deutscher Verlag der Wissenschaften, Berlin 1965.
101. KOCH H., und HILBERATH F„ Brennstoff-Chem. 21 (1940), 197—203.
102. GEMMEKER W., und STAGE H., Glas-Instrumenten-Techn. 6 (1962), 10, 372—376.
103. KLEIN E.,und WAIZ H., Glas-Instrumenten-Techn. 11 (1967), 5, 405—411.
104. PODBIELNIAK W. J., und PRESTON S. T., Petroleum Engr. 5 (1955), 205—211.
105. Hersteller: Ernst Haage, Miihlheim—Ruhr.
106. SIMONS G., Ind. Engng. Chem., analyt. Edit. 10 (1938), 648.
107. PODBIELNIAK W. J., Apparatus und Methods for Precise Fractional Distillation Analyses, American Chemical Society, Atlantic City Meeting 1941.
108. ROSE A. und E., und MITARB., Distillation, Interscience Publishers, New York 1951.
109. PRESTING W., und MITARB., Freiberger Forschungshefte 5, Akademie-Verlag, Berlin 1953, S. 231—238.
110. STAGE H., Destillieren und Rektifizieren, Abschnitt 19 in: Fortschritte der Verfahrenstechnik 1952/53, Verlag Chemie GmbH, [Weinheim/Bergstr. 1954.
111. BERNHAUER K., Einfiihrung in die organisch—chemische Laboratoriumstechnik, Springer-Verlag, Wien 1947.
112. GEMMEKER L., und STAGE H., Glas-Instrumenten-Techn. 6 (1962) 11, 455—458.'
113. HOLLAND-MERTEN E. L., Tabellenbuch der Vakuumverfahrenstechnik in der Grundstoffindustrie, VEB Deutscher Verlag fiir Grundstoffindustrie, Leipzig 1968.
114. JAECKEL R., Kleinste Driicke, ihre Messung und Erzeugung, in: Technische Physik in Einzeldarstellungen, Springer-Verlag, Berlin/Gottingen/ Heidelberg 1950.
LEYBOLD, Vakuum-Taschenbuch, hrsg. von K- DIELS und R. JAECKEL, Springer-Verlag, Berlin/Gottingen/Heidelberg 1962.
KEUNECKE E., Chemiker-Ztg. 81 (1957), 239—244.
YARWOOD J., Hochvakuumtechnik, 3. Aufl., Rudolf A. Lang Verlag, Berlin 1955 (Obers, aus d. Engl.).
498
PUPP W., Vakuumtechnik, Grundlagen und Anwendungen, Thiemig-Taschenbuch Bd. 43, Verlag Karl Thiemig, Munchen 1973.
115. MULLER L., Freiberger Forschungshefte, Ausg. В 6, Akademie-Verlag, Berlin 1954, S. 81—115; Freiberger Forschungshefte 11, Akademie-Verlag, Berlin 1957, S. 575.
116. SPENDLOVE M. J., Vakuum-Techn. 6 (1957), 15—18 u. 36—44.
117. HORSLEY W., J., appl. Chem. 8 (1958), 13; Chemie-Ing.-Techn. 30 (1958), 736.
118. PARKER S. G., und WILSON 0. W„ Ind. Engng. Chem. Proc. 4 (1965) 4, 365—368.
118a. БЕЗОБРАЗОВ E. Г. и др.. Хим. пром., 1974, № 4, с. 61—62.
119. LAPORTE H., Hochvakuum, VEB Wilhelm Knapp Verlag, Halle 1951.
120. ECHOLS Jr., L. S„ und GELUS E., Analytic. Chem. 19 (1947), 668.
121. ULUSOY E., und SAKALOZ T., Theoretische Grundlagen der Isotherm— Destination (Mitt. I—III), Revue de la faculte des sciences de I’universite d’Istanbul, Serie C, Tome XXHI/XXIV, Fasc. 3—4 (1958/59).
122. FRANK W., und KUTSCHE D., Die schonende Destination, Otto Krausskopf-Verlag GmbH, Mainz 1969.
123. BILLET R„ und RAICHLE L., Chemie-Ing.-Techn. 39 (1967) 3, 133—137.
124. GELBE H„ Chemie-Ing.-Techn. 40 (1968) 11, 528—530.
125. HARRIES W., Chemie-Ing.-Techn. 21 (1949), 139—142.
126. MON CH G. C., Hochvakuumtechnik, Rudolf A. Lang Verlag, Popneck 1950; Neues und Bewahrtes aus der Hochvakuumtechnik, 2. ber. Nachdruck, VEB Verlag Technik, Berlin 1961.
127. HOLLAND-MERTEN E. L., Die Vakuum-Pumpen in der Verfahrenstechnik, VEB Wilhelm Knapp Verlag, Halle 1950.
128. ARDENNE M„ von, Technik 13 (1958), 275—285.
129. KIENEL G., Glas-Instrumenten-Techn. 10 (1966) 9, 761—770.
130. KRELL E„ Chem. Techn. 2 (1950), 252—255; 4 (1952), 443—450.
131. WITTKA F., Angew. Chem. 53 (1940), 557.
132. GEMMEKER L„ und STAGE H., Glas-Instrumenten-Techn. 8 (1964) 6, 413—428; 7, 503—508.
STAGE H., Chemieanlagen + Verfahren (1972), Heft 2.
133. UTZINGER G. E., Chem. Techn. 7/8, (1943), 61.
134. JANOSFIA P., Dechema—Monogr. 55 (1965).
135. FURST H„ und SCHIMKE D., Chem. Techn. 22 (1970) 9, 553—555.
136. DIETER K., Chemiker-Ztg. 94 (1970) 12, 445—451.
137. BILLET R„ Chemie-Ing.-Techn. 29 (1957), 733—742.
138. EGLI R., Glas-Instrumenten-Techn. 11 (1967) 6, 509—515.
139a. KRAMER M. T., GIT Fachz. Lab. 18 (1974) 7, 862—868.
139. BURROWS G., Molecular—Distillation, Oxford—Clarendon—Press, Oxford 1960.
HOLO J., und MITARB., The Applications of Molecular Distillation, Akademiai Kiado, Budapest 1971.
140. RIDGWAY-WATT P„ Chem. and Ind., 27, 5. 1961, 680—692.
140a. HABENDORFF R„ CZ-Chem.-Techn. 2 (1973), 21—24.
141. HICKMAN К. C. D., und TREVOY T. J., Chemie-Ing.-Techn. 25 (1953), 672—676.
142. LANGMUIR S„ Physic. Rev. 8 (1916), 176.
143. BURROWS G., J. appl. Chem. 7 (1957), 375—384.
144. JAECKEL R., Erdol u. Kohle 4 (1951), 1761.
145. PERRY E. S„ Methods in Enzymol 3 (1957), 383—391.
JAECKEL R„ und OETJEN G.—W., Chemie-Ing.-Techn. 21 (1949), 169—176.
JAECKEL R., und MITARB., Vakuumtechnik und Molekulardestillation, in: Fortschritte der Verfahrenstechnik, Bd. 2, 1954/55, S. 29—43; Bd. 3, 1956/57, S. 62—73, Verlag Chemie GmbH, Weinheim/Bergstr. 1954/57.
JAECKEL R., Destination und Sublimation im Fein- und Hochvakuum, in: HOUBEN-WEYL, Methoden der organischen Chemie, hrsg. von
499
E. MULLER, Bd. 1/1, Georg Thieme Verlag, Stuttgart 1958.
146. FRANK W., Glas-Instrumenten-Techn. 9 (1965) 11, 777—782.
147. UTZINGER G. E„ Chemie-Ing.-Techn. 26 (1954), 129—131.
148. MELPOLDER F. W„ und MITARB., Analytic. Chem. 27 (1955), 974—977.
149. МАЛЮСОВ В. А., ЖАВОРОНКОВ H. M„ Collect. Czechoslow. Chem. Commun, 23, № 9, 1720—1726 (1958).
150. МАЛЮСОВ В. А., УМНИК H. H., ЖАВОРОНКОВ Н. М., Хим. пром., 1958, № 5, с. 36—42.
151. HICKMAN К. С. D., Ind. Engng. Chem. 39 (1947) 686—694.
152. . HICKMAN К. C. D.; Ind. Engng. Chem., analyt. Edit. 29 (1937), 968.
153. HICKMAN К. C. D., Патенты США № 2117802, 1938; № 2210928, 1940 г.
154. EMBREE N. D., Ind. Engng. Chem. 29 (1937), 975.
155. GORRIZ A. M., und MITARB., An. Real Soc. espan. Fosica Quim., Ser. В 48 (1952), 825 bis 850; Ser. В 49 (1953), 19—22, 107—114, 579—586.
156. FAWCETT E. W., Kolloid-Z. 86 (1939), 34—91.
157. MASCH L.—W., Chemie-Ing.-Techn. 22 (1950), 141—146.
158. HICKMAN K., Ind. Engng. Chem. 48 (1956), 496—499.
159. CARNEY T. P., Laboratory Fractional Distillation, MacMillan, New York 1949.
160. МАЛЮСОВ В. А., МАЛАФЕЕВ H. А., ЖАВОРОНКОВ H. М., ЖФХ, 1958, т. 32, № 10, с. 2403—2409.
.161. SCHNEIDER К. W., Erdol u. Kohle 4 (1951), 4—9.
162. SIMONS J. Н., Ind. Engng. Chem. analyt. Edit. 10 (1938), 30.
163. UY EHA H.,und HAGIHARA Y„ J. chem. Engng. Japan 3 (1970) 1, 13—17.
164. BEWILOGUA L., VERGES P., und VINZELBERG H., Isotopenpraxis 9 (1973) 3, 97—100.
. 165. LI EDEL К. H., Gias in der Chemie—Laborgerate, Gro₽apparate, Rohrleitungen, Otto Krausskopf-Verlag GmbH, Mainz 1973.
6. ЛИТЕРАТУРА
1. FINDLAY A., Die Phasenregel und ihre Anwendungen, Verlag Chemie GmbH, Weinheim/Bergstr. 1958 (Ubers, aus d. Engl.).
2. BADGER W. L., und McCABE W. L., Elemente der Chemie-Ingenieur-Technik, Springer-Verlag, Berlin 1932 (Obers, aus d. Engl.).
3. STAGE H., Fette u. Seifen 55 (1953), 513—516.
4. STAGE H., Erdol u. Kohle 3 (1950), 478—490.
5. HOFFMANN E. J., Azeotropic and Extraktive Distillation, John Wiley & Sons, London 1965.
6. Патент Германии N 767 162 3.12.1951 от 27.11.1936.
7. SIZMANN R., und STUKE B., Chemie-Ing.-Techn. 27 (1955), 669—675.
8. ALGERMISSEN J., Chemie-Ing.-Techn. 30 (1958), 502—510.
9. MERKEL H„ Chemie-Ing.-Techn. 24 (1952), 511—512.
10. STAGE H.,BUNGER R.,und JOHAS A., Fette u. Seifen 55 (1953), 580—583.
11. POZZI und ESCOT, Bull. Soc. chim. France 31 (1904), 932.
12. PFEIL E., Angew. Chem. 54 (1941), 161—184.
13. BERNHAUER K., Einfuhrung in die organisch—chemische Laboratoriumstechnik, Springer-Verlag, Wien 1947.
14. THORMANN K., Chem. Fabrik 13 (1940), 3—9.
15. WEBER U. von, Gewinnung atherischer Ole durch Destination, in; GILDEMEISTER, E., und HOFFMANN, Fr., Die atherischen Ole (bearb. von W. Treibs), Akademie-Verlag, Berlin 1966.
16. RIGAMONTI R., Chemiker-Ztg. 80 (1956), 628—631.
17. ROCK H-, Destination im Laboratorium, kExtraktive und azeotrope Destination, Dr. Dietrich Steinkopff Verlag, Darmstadt 1960.
18. KUMMERLE K., Chemie-Ing.-Techn. 32 (1960), 513—517.
19. GERSTER J. A., Chem. Engng. Progr. 65 (1969) 9, 43.
19a. MULLER E., Verfahrenstechnik 8 (1974) 3, 88—93.
500
20. LECAT M., Tables azeotrophiques, Azeotropes binaires orthobares, 2. Aufl., Brussel 1949 (Eigenverlag).
21. ОГОРОДНИКОВ С. K-, ЛЕСТЕВА T. M., КОГАН В. Б., Азеотропные смеси. Справочник, Л., Химия, 1971.
22. STUKE В., Chemie-Ing.-Techn. 25 (1953), 133—136.
23. COULSON Е. A., und HERRINGTON Е. F., J. Chem. Soc. [London] 1947, 597—607.
24. KUHN W., und MASSIN I P., Helv. chim. Acta 33 (1950), 737.
KUHN W., und KUHN H. J., Helv. chim. Acta 40 (1957), 2433—2454.
25. ENiiSTUN В. V., Commun. Fac. Sci. Univ. Ankara, Ser. В 5 (1954) 1—21.
26. ЛИТВИНОВ H. Д. — ЖФХ, 1953, т. 27, № 4, с. 476—490.
27. SWIETOSLAWSKI W., Przemysl. chem. 7 (1951), 363—370; Bull. Soc. chim. beiges 62 (1953), 10—20; Bull. Acad, polon. Sci., Cl. Ill, Vol. I, 1—2, 1953.
SWIETOSLAWSKI W., Physikalische Chemie des Steinkohlenteers, N. J. Hoffmann Verlag, Koln 1954.
SWIETOSLAWSKI W., Azeotropie und Polyazeotropie, Bd. 1, Panstwowe Wydawnictwo Naukowe, Warshau 1957.
28. LECAT M., Azeotropisme et Distillation, in: Grignard, Traite^de Chimique Organique I, Paris 1935.
28a. OTHMER D. F., Verfahrenstechnik 8 (1974) 3, 94—99; 4, 118—123.
29. SCHUBERTH H., Thermodynamische Grundlagen * der Destination und Extraktion, Bd. I, VEB Deutscher Verlag der Wissenschaften, Berlin 1972.
30. ZIEBORAK K-, Prace Glownego Inst. Chem. przemosloweje, (1951), 1—44.
31. ORSZAGH A., Przemysl. chem. 9 (32) (1953), 333—336.
32. MALESINSKI I- W., Bull. Acad, polon. Sci., Ser. Sci. chim., geol., geogr. 6 (1958), 105 bis 109.
33. STCKI J-, Bull. Acad, polon Sci., Ser. Sci. chim., geol., geogr. 6 (1958), 47—53.
34. BERG L., und MITARB., Hydrocarbon Processing 45 (1966), 103; Ind.. Engng. Chem. 37 (1945), 585; 38 (1946), 1249.
BERG L., Chem. Engng. Progr. 65 (1969) 9, 52—57.
35. ROSE A. und E., und MITARB., Distillation, Interscience Publischers, New York 1951.
36. FRANCK H. G., Angew. Chem. 63 (1951), 260—263.
37. COULSON E. A., und JONES J. J., J. Soc. chem. Ind. 65 (1946), 163—175.
38. DUMMETT G. A., Petroleum [London] 18 (1955), 41—45.
39. HUNSMANN W., und SIMMROCK К. H„ Chemie-Ing.-Techn. 38 (1966) 10, 1053—1059.
39a. ПЕТЛЮК Ф. Б., АВЕТЬДН В. С. — TOXT, 1973, т. 7, № 3, с. 307—312.
40. LINO М„ und MITARB., J.rchem. Engng. Japan 4 (1071) 1, 33—36.
41. MAIR B. J., GLASGOW A. R., und ROSSINI F. D., J. Res. nat. Bur. Standards 27 (1941), 39.
42. MAIR B. J., und MITARB., Analytic. Chem. 30 (1958), 393—400.
43. FELDMAN J., und ORCHIN M. Патент США № 2583554, 1952.
44. SCHUBERTH H., Chem. Techn. 21 (1969) 1, 28—34, 51.
45. ШЕЙНКЕР Ю. H., ПЕРЕСЛЕНИ E. M. — ЖФХ, 1952, t. 26, № 8, c. 1103—1109.
46. RUETHER J. A., und LU В. C. Y., Canad. J. chem. Engng. 50 (1972) 2, 266—270.
47. NUTTING H. S., und HORSLEY L. H., Ind. Engng. Chem., analyt. Edit. 19 (1947), 602—603.
48. JOFFE J., Ind. Engng. Chem. 47 (1955), 2533—2535.
49. MALES I NSKI W„ Bull. Acad, polon. Sci., Cl. HI 4 (1956), 371—379.
50. KORTUM G„ und BUCHHOLZ-MEISENHEIMER H„ Die Theorie der Destination und Extraktion von Fliissigkeiten, Springer-Verlag, Berlin/Gottingen/Heidelberg 1952.
51. HORSLEY L. H., Azeotropic Data, Bd. I und II, American Chemical
501
Society, Washington 1952/1962; Advances in Chem., Series No. 6 and 35 (Хорсли Л. Таблицы азеотропных смесей, ИИЛ, М., 1951).
52. EWELL R. И., HARRISON J. М., und BERG L., Ind. Engng., Chem. 36 (1944), 871.
53. MARSCHNER R. F., und GROPPER W. P., Ind. Engng. Chem. 38 (1946), 262—268.
54. HORSLEY L. H„ Analytic. Chem. 19 (1947), 603.
55. MEISSNER H. P„ und GREENFELD S. H., Ind. Chemist 40 (1948), 438_____<12
56. НОВИКОВ К- E., НАТРАДЗЕ А. Г. Хим. пром., 1958, № 2, с. 38—41.
57. SKOLNIK H„ Ind. Engng. Chem. 40 (1948), 442.
58. NAKANISI K-, ICHINOSE S„ und SKIRAI H., Ind. Engng. Chem., Fundamentals 7 (1968), 3 , 381—387.
58a. BRANDANI V., Ind. Engng. Chem. Fundamentals 13 (1974) 2, 154—156.
59. KUHN W., Helv. chim. Acta 37 (1954), 1585—1605.
60. SZAPIROj S., Przemysl chem. 12 (35) (1956), 106—110.
61. KORTUM G., und BITTEL A., Chemie-Ing.-Techn. 28 (1956), 40—44, 282—284.
61a. HUNSMANN W„ Chemie-Ing.-Techn. 33 (1961) 8, 537—540.
61b. HEGENER B., HESSE D., und WOLF D., Chemie-Ing.-Techn. 45 (1973) 14, 942—945.
62. HOUDRY PROCESS CORP. Патент США Ns 2776936, 1957.
63. GREWER, Th., Chemie-Ing.-Techn. 43 (1971), 655—658.
63a. ПАВЛОВ С. Ю., СКОРИКОВА В. В., БОЙКОВА С. 5. — ТОХТ, 1972, т. 6, Ks 3, с. 474—477.
64. STAGE Н., Erdol u. Kohle 3 (1950), 478—490.
65. SCHEIBEL Е. G., Chem. Engng. Progr. 44 (1948) 927—931.
65а. ГАРБЕР Ю. Н., МИРОНЕНКО В. Ф. — ТОХТ, 1974, т. 8, № 6, с. 846—852.
66. КОГАН В. Б. — ЖФХ, 1955, т. 29, № 8, с. 1470—1476.
67. КАФАРОВ В. В., ГОРДИЕВСКИЙ Л. А. — ЖПХ, 1956, т. 29, № 2, с. 176—183.
68. КОГАН В. Б. —Хим. пром., 1957, № 6, с. 36—41.
69. ROCK Н., Chemie-Ing.-Techn. 28 (1956), 489—495.
HORSLEY L. Н., Analytic. Chem. 19 (1947), 508—600; 21 (1949), 831.
70. PORTER R. S„ und JOHNSON J. F., Ind. Engng. Chem. 52 (1960) 8, 691—694.
71. HILDEBRAND, Solubilities, 2. Ausg., New York 1936 (Гильдебранд Д. Г. Растворимость неэлектролитов, ГОНТИ НКТП, М., 1938).
72. Патент Великобритании № 613867, 1948.
73. Патент США № 2549586, 1951; GRISWOLD J., und MITARB., Ind. Engng. Chem. 38 (1946), 65—70.
74. Патенты США №№ 2610704, 1952; 2588063, 1952; 2371342, 1945.
75. GARNER F. H„ ELLIS S. R. M., und GRANVILLE W. H., J. Inst. Petroleum 42 (1956), 148—154.
76. NULL H. W., und PALMER D. A., Chem. Engng. Progr. 65 (1969) 9, 47.
77. GELBIN D., Chem. Techn. 11 (1959), 70—73.
78. NAGEL und SINN R„ Chemie-Ing.-Techn. 39 (1967) 6/e, 275—282.
79. KORTUM G., und FALTUSZ E., Chemie-Ing.-Techn. 33 (1961), 599—606.
80. GARWIN L., und HUCHINSON К- E., Ind. Engng. Chem. 42 (1950), 727—730.
81. DAUBACH E., Diplomarbeit, Marburg 1948.
82. JOST W., Chemie-Ing.-Techn. 23 (1951), 64.
83. TURSI R. R., und THOMPSON R., Chem. Engng. Progr. 47 (1951), 304—308.
84. NOVELLA E. C., und TARASSO J. M., An. Real. Soc. espan. Fisika Quim., Ser. В 48 (1952), 441—448.
85. SCHIER К. H., Dissertation, Martin-Luther-Universitat, Halle—Wittenberg 1959.
502
86. BELCK L., Chemie-Ing.-Techn. 23 (1951), 90—91.
87. BOGART M. J. P., und BRUNJES A. S., Chem. Engr. Progr. 44 (1948), 95.
88. RIEDER R. M., und THOMPSON A. R., Ind. Engng. Chem. 42 (1950), 379
89. GUYER A., GUYER JR. A., und KARTH JOHNSON B., Helv. chim. Acta 38 (1955), 946 bis 954.
90. MIRO A. R., de la GANDARA J. L. 0., und GONZALES J. R. A., An. Real Soc. espan. Fisika Quim., Ser. В 53 (1957), 171—184, 185—194.
91. FURTER W. F., Chem. Engng. (1958) Juni, CE 173—CE 177.
92. SCHREIBER E., SCHUTTAU E„ RANT D., und SCHUBERTH H., Z. physik. Chem. [Leipzig] 247 (1971) V2, 23—40.
SCHUBERTH H„ Z. physik. Chem. [Leipzig] im Druck.
93. FUCHS 0., und ROTH F. H., Chem. Fabrik 11 (1938), 401—406.
94. INO M., und MITARB., J. chem. Engng. Japan 4 (1971) 1, 22—26.
95. JAQUES D., und FURTER W. F„ Canad. J. chem. Engng. 50 (1972) 4, 502—505.
96. FURTER W. F., und MERANDA D„ GVC/AIChe-J., Joint Meet. 2 (1974), D 1—3, 1—10.
97. SADA E., KITO Sh„ YAMAJI H., und KIMURA M., J. appl. Chem. Biotechnol. 24 (1974) %, 229—238.
7. ЛИТЕРАТУРА
1. LIEDEL К- H., Gias in der Chemie-Laborgerate, Groflapparate, Rohrleitungen, Otto Krausskopf-Verlag GmbH, Mainz 1973.
2. MONCH G. CH., Hochvakuumtechnik, Rudolf A. Lang Verlag, Popneck 1950.
3. EITEL W., und MITARB., Glastechnische Tabellen, Springer-Verlag. Berlin 1932.
4. VVB Westglas, Ilmenau, Technik 8 (1953), 675—680.
5. REIGER H., Silikattechnik 4 (1953), 339—342; Chem. Techn. 22 (1970), 1, 36—40.
6. POMEROY H. H., Analytic. Chem. 24 (1952), 1076.
7. KAMMEL H., Silikattechnik 2 (1951), 227—231.
8. KRELL E., Chem. Techn. 4 (1952), 200—207.
9. Kataloge der Glasgerateindustrie
FRIEDRICHS F., Das Gias im chemischen Laboratorium, 3. Aufl., Springer-Verlag, Berlin/Gattingen/Heidelberg 1960.
TELLE W., ABC der Laboratoriumsgerate, VEB Fachbuchverlag Leipzig 1953.
TELLE W., Chemische Laboratoriumsgerate, 2. Aufl., VEB Deutscher Verlag fiir Grundstoffindustrie, Leipzig 1969.
10. STAGE H., Erdol u. Kohle 3 (1950), 377—383.
11. KRELL E„ Chem. Techn. 4 (1952), 443—450.
12. KINZA V., DDR-Gebrauchsmuster, 10175.
13. WOLF F. W. D., Glas-Instrumenten-Techn. 4 (1960), 43—52, 126—131, 343_____349.
14. HECKMANN C. J., Chem. Techn. 24 (1972) 9, 533—536; 25 (1973) 1, 20—23.
14a. REICHELT W., Chemie-Ing.-Techn. 45 (1973) 1, 25—29.
15. MALEWSKI W., Chemie-Ing.-Techn. 42 (1970) 6, 385—390.
16. SIGWART K-, Destination und Rektifikation in: Ullmanns Encyklopadie der technischen Chemie, Bd. 1, Urban & Schwarzenberg, Munchen/Berlin 1951.
17. RAY J. D„ Rev. sci. 28 (1957), 200—201.
18. GARNEY Th. P., Laboratory Fractional Distillation, MacMillan, New York 1949.
19. THORMANN K-, Dechema-Erfahrungsaustausch, Arbeitsmethoden und Gerate, Destillieren und Rektifizieren, Dechema, Frankfurt/Main, September 1950.
503
20. FENSKE M. R., A. P. 2 037 316 vom 4.4.1936.
21. ROSE A. und E., und MITARB., Technique of Organic Chemistry, Vol. IV Interscience Publishers, New York 1951.
22. KUHN W., Chemie-Ing.-Techn. 25 (1953), 12—18.
KUHN W„ und MITARB., Chimija [Zurich] 8 (1954), 109—122.
23. CRAIG L. C., Ind. Engng. Chem., analyt. Edit. 8 (1936), 219; 9 (1937), 441.
24. KUHN W., Helv. chim. Acta 25 (1942), 252—295.
25. KUHN W„ und RYFFEL K-, Helv. chim. Acta 26 (1943), 1693—1721.
JOST W., Angew. Chem., Teil В 20 (1948), 231.
COENEN A., Dissertation, T. H. Aachen 1941.
MAIR B. J., und WILLINGHAM Ch. B., J. Res. nat. Bur. Standards
22 (1937), 519—527.
26. WEST HAVER C. J., Ind. Engng. Chem. 34 (1942), 126.
27. DONNELL С. K-, und KENNEDY R. M., Ind. Engng. Chem. 42 (1950), 2327______2332
28. JANTZEN E., und WIECKHORST 0., Chemie-Ing.-Techn. 26 (1954), 392______396
29. KOCH R., und FEIND K-, Chemie-Ing.-Techn. 30 (1958), 577—584.
30. BECK F., Chemie-Ing.-Techn. 35 (1963), 837—844.
31. FISCHER W. G., Glas-Instrumenten-Techn. 13 (1969) 5, 535—540; 14 (1970)
8, 933 bis 936; 15 (1971) 7, 753—759; 15 (1971) 8, 832—838.
Chemiker-Ztg. 94 (1970) 5, 157—161.
Fette, Seifen, Anstrichmittel 72 (1970), 444—454.
Fette, Seifen, Anstrichmittel 75 (1973), 131—133.
32. MACURA H., und GROSSE-OETRINGHAUS H., Brennstoff-Chem. 19 (1938), 437—439.
33. MANNING R. E., und CANNON M. R., Ind. Engng. Chem. 49 (1957), 346—349.
34. "HOPPE K., und MITTELSTRASS M., Grundlagen der Dimensionierung
i . von Kolonnenboden (Technische Fortschrittsberichte, Bd. 61), Verlag Theodor iSteinkopff, Dresden 1967.
35. STAGE H., Glas-Instrumenten-Techn. 13 (1969) 12, 1285—1299.
STAGE H., Chemieanlagen und-verfahren (1970), Heft 1/2.
36. KEESOM W. H., und MITARB., Physica 1 (1934), 1109/14.
37. BRUUN J. G„ und SCHICKTANZ sS. T., J. Res. nat. Bur. Standards 7 (1931), 851—882.
38. STAGE F., Angew. Chem., Teil’B 19 (1947), 182.
39. Патент ФРГ № 862598, 1952; ~ KIRSCHBA UM E., Chemie-Ing.-Techn.
25 (1953), 73—80.
40. FRITZ P. W., Glas-Instrumenten-Techn. 11 (1967) 3, 161—162; Патент ФРГ на полезную модель 1957085.
41. OLDERSHAW С. F., Ind. Engng. Chem., analyt. Edit. 13 (1941), 265—268.
42. HILBERATH F., Oel u. Kohle 39/40 (1943), 875—880.
43. WIJK W. R. van, und THISSEN H. A. C„ Chem. Engng. Sci. 3 (1954), 153—160.
44. UMHOLTZ C. L., und WINKLE M. van. Petroleum Refiner 34 [(1955), 114—117.
45. JONES P. D., und WINKLE M. van, Ind. Engng. Chem. 49 (1957), 232—238.
46. Entwickelt in dem Forschungslabor von C. F. BRAM u. Co. Hersteller: the Emil Griner u. Co.
47. McGURE G. V., und'MADDOX R. N., Ind. Engng. Chem. Proc. 6 (1971), 1, 9.
48. WAGNER C. D., LUKENS JR., H. R., und HUGHES R..R., Nucleonics 14 (1956), 78—83.
49. NEWITT D. M., DOMBROWSKI N., und KHELMAN F. H., Trans. Instn. Chem. Engr. 32 (1954), 244—261.
50. МЕЛИКЯН P. А., ЖПХ, 1958, t. 31, № 4, c. 550—558.
51. WIDMER G., Helv. chim. Acta 7 (1924), 194—196.
52. GOLODETZ A., Chem. Industrie (niss.) 35 (1912), 102—108, 141—145.
504
53. LECKY H. S., und EWELL J?. H., Ind. Engng. Chem., analyt. Edit., 12 (1940), 544—547.
54. STALLCUP W. D., und MITARB., Ind. Engng. Chem., analyt. Edit. 14 (1942), 503—547.
55. BOWER JR., J. R., und COOKE L. M., Ind. Engng. Chem. 15 (1943), 290—293.
56. PODBIELNIAK W. J., Ind. Engng. Chem. 13 (1941), 639—645.
57. BREZINA V., Glas-Instrumenten-Techn. 14 (1970) 5, 493—494.
58. STEDMAN D. F., Trans. Amer. Inst. chem. Engr. 33 (1937), 153—161.
59. KOCH H„ und RAAY H., van, Chemie-Ing.-Techn. 8 (1950), 172.
60. BRAGG L. B., Ind. Engng. Chem. 45 (1953), 1676.
61. DRP nach Prospekt Ernst Haage, Miihlheim/Ruhr.
62. ELLIS S. R. M., und VARJAVANDI J., Chem. Process Engng. 39 (1958), 239.
63. JUCHHEIM G„ Glas-Instrumenten-Techn. 10 (1966) 10, 914—917.
64. SPERANDIO A., RICHARD M., und HUBER M., Chemie-Ing.-Techn.
37 (1965), 3, 322 bis 328.
ZOGG M., Chemie-Ing.-Techn. 45 (1973) 2 , 67.
65. ТИМОФЕЕВ А. В., АЭР0В M. E„ Хим. пром., 1971, Ns'5, c. 61—62.
65a. KWASNIAK J., Verfahrenstechnik 8 (1974) 11,315—318. '
66. SHAWORONKOW N. M., und MALJUSOW W.’A., Chem. Techn. 24 (1972) 7, 397—401.
67. ZUlDERWEGiF. T., ChemAEngng. Sci. 1 (1952), 164—174, 184—194.
68. PODBIELNIAK} W. J., Патент Германии № 595250, 1934.
69. BENNER F. C., DINARDO A., und TOBIN D. J., Ind. Engng. Chem. 43 (1951), 722 bis 727.
70. BYRON E. S„ BOWMAN J. R., und COULL J., Ind. Engng. Chem. 43 (1951), 1002—1010.
71. WILLINGHAM С. B., SEDLAK V. A., ROSSINI F. D., und ESTHAVER J. W., Ind. Engng. Chem., Ind. Edit. 39 (1947), 706—712.
72. JOST W., Chemie-Ing.-Techn. 25 (1953), 356.
JOST W., Rektifikation unter extremen Bedingungen, in: Dechema-Monogr., 22 (1954), 30—72.
73. JOST W., SIEG L„ und BRANDT H., Chemie-Ing.-Techn. 25 (1953), 291_____292.
74. ГЕЛЬПЕРИН Н.И.,ХАЦЕНКО M. С., ЖПХ, 1952, т. 25, № 6, с. 610—615.
75. MURRAY К. Е., J. Amer. chem. Soc. 28 (1951), 235.
76. ИРЛИН А. Л., БРУНС Б. П., ЖАХ, 1950, т. 5, № 1, с. 44—47.
77. HOHER G., Untersuchungen uber die Trennfahigkeit einer Labor-Rektifikationskolonne mit rotierender Wendel als Austauschflache, Ingenieur-Arbeit, Ingenieurschule fiir Chemie, Leipzig 1958.
78. NEUMANN F., Chemie-Ing.-Techn. 33 (1961) 7, 485—491.
78a. МАМИН В. H. u др. Химико-фармацевтический журнал, 1974, т. 8, № 3, с. 51—55.
79. TOELDTE W., Seifen—Ole—Fette—Wachse 74 (1948), 126.
80. N. N., Glas-Instrumenten-Techn. 10 (1966) 4, 259/260.
81. KRAMER M. T., Glas-Instrumenten-Techn. 15 (1971) 8, 856—860.
82. BREZINA V., Glas-Instrumenten-Techn. 10 (1966), 3, 162—172.
83. FRIEDRICHS F., Z. angew. Chem. 33 (1920), 29—32.
84. FRIEDRICHS J., und v. KRUSKA H., Chem. Fabrik 7 (1934), 284—287.
85. MACH F„ und HERRMANN R., Chem. Fabrik 6 (1933), 211—213.
86. EICHHORN E., Untersuchung der Wirkung von Laboratoriumskiihlern, Ihgenieur—Hauptarbeit, Ilmenau 1955.
87. GEMMEKER L., und STAGE H., Glas-Instrumenten-Techn. 6 (1962) 3, 69—78; 4, 114 bis 119; 5, 169—174; 6, 206—208; 9, 327—328; 10, 372—376; 12, 494—497.
88. KRELL E„ Chem. Techn. 5 (1953), 581—587.
89. SCHNEIDER K. W., und SCHMID Ch., Erdol u. Kohle 2 (1949), 508—510.
89a. KLOSS H.—J., CZ-Chem.-Techn. 3 (1974), 89—93.
505
90. KRAMER M. T., Glas-Instrumenten-Techn. 16 (1972) 4, 445—448; 5, 571—574; 8, 943—946.
91. Prospekt Normag, Hofheim/Ts., Liste 10; Chemie-Ing.-Techn. 26 (1954), 231.
92. SCHULZ E. 0., Glasinstrumentenkunde, Wagner u. Sohn. Weimar 1937.
93. HUBNER G., Chemiker-Ztg. 83 (1959), 581—583.
94. LLOYD L. E„ und HORNBACHER H. G., Analytic. Chem. 19 (1947), 120______122. ' '
95. COLLINS F. C., und LANTZ V., Ind. Engng. Chem., analyt. Edit. 18 (1946), 673—677.
96. K1ESELBACH K-, Analytic. Chem. 19 (1947), 815.
97. STAGE H., und GEMMEKER L., Glas-Instrumenten-Techn. 6 (1962) 12, 494—497.
98. N. N., Glas-Instrumenten-Techn. 10 (1966) 3, 178—179.
99. STAGE H., und GEMMEKER L., Glas-Instrumenten-Techn. 7 (1963)
11, 608—614; 12, 687—704; 8 (1964) 6, 413—428; 7 , 503—508.
STAGE H., Glas-Instrumenten-Techn. 14 (1970) 3, 213—220; 4, 337—349.
100. Hersteller: Glaswerk Schott und Gen., Mainz.
101. ROCK E. J., und JANZ G. F., Analytic. Chem. 22 (1950), 626.
102. GRASSMANN W., und DEFFNER C., Chemiker-Ztg. 76 (1952), 623.
103. SCHRAM E., Analytic. Chem. 25 (1953), 1424.
104. STAGE H., und GEMMEKER L., Glas-Instrumenten-Techn. 7 (1963)
11, 608—614; 12, 687—704 8 (1964) 6 , 413—428; 7, 503—508.
STAGE H., Glas-Instrumenten-Techn. 14 (1970) 3, 213—220; 4, 337—349.
105. HUBNER G„ Chemiker-Ztg. 76 (1952), 67.
106. Hersteller: Edmund Buhler, Tubingen.
107. Hersteller: Dr. Hofmann und Roth, Stuttgart-Hdedelfingen; s. auch KRELL E., Chem. Techn. 7 (1955), 599—608.
108. Hersteller: W. C. Heraeus GmbH, Hanau.
109. BORCHERT R., und JUB1TZ W., Infrarotstrahler zur Erwarmung, Trocknung und Ausbartung, VEB Verlag Technik, Berlin 1951.
110. KLEES K. F., Chem. Rdsch. [Solothurn] 7 (1954), 358—359.
111. SCHULZE H., Chemie-Ing.-Techn. 23 (1951), 565—569.
112. WEBER U. von, Chem. Techn. 1 (1949), 159—160.
113. ZAMPACHOVA L., Chem. Prumysl (tschech.) 7/32 (1957), 408—413.
114. K0LL1NG H., und TRAMM H., Chemie-Ing.-Techn. 21 (1949), 9—14.
115. Hersteller: Bronwill Scientific Ind., New York.
116. KORTUM G., und B1TTEL A., Chemie-Ing.-Techn. 28 (1952), 40—44.
117. ROSE A. und E., und MITARB., Technique of Organic Chemistry, Vol, IV, Interscience Publisners, New York 1951.
118. Hersteller: W. C. Heraeus GmbH, Hanau, Elektrothermal Engineering Ltd. London und VEB Asbestdraht, Berlin.
118a. SELLIN J., Chemie—anlagen + verfahren 4 (1975), 87—91.
119. Alleinvertrieb fiir «Calorex»: Heraeus—Wittmann GmbH, Heidelberg.
120. LEVA M., Tower Packings and Packed Tower Design, 2. Aufl., The United States Stoneware Company, Akron/Ohio 1953.
121. KERENYI E„ und SCHULTHE1SZ I., Proc, and Conf, on Appl. Phys.
Chem., Veszprem, Aug. 2—5 1971, Akademiai Kiado, Bp 1971, Vol. 2, S. 67—74.
121a. REICHELT W., Chemie-Ing.-Techn. 45 (1973) 2, 53—57.
122. Hersteller: Podbielniak Inc., Chicago 11.
123. BRAUN R., Chemie-Ing.-Techn. 33 (1961), 350—352.
124. DIXON O. G., J. Soc. chem. Ind. 68 (1949), 88, 119, 299.
125. HUBER M.,und SPERANDIO A., Chem.-Ing.-Techn. 36 (1964) 3,221—227.
126. ELLIS S. R. M., und VARJAVAND1 J., Chem. Process Engng. 7 (1958), 239—243.
127. Unveroffentlichte Mefiergebnisse des Institutes fiir Apparatebau und Verfahrenstechnik^der TH Karlsruhe (pers. Mitteihing).
128. MacMAHONl H. 0., Ind. Engng. Chem., ind. Edit. 39 (1947), 712—718.
129. ’Hersteller:;The United States Stoneware Company, Akron/Ohio.
130. Herstellen^Hydronil Ltd., Chemical Plant Division, Fenton/England.
506
131. KIRSCHBAUM E„ Z. Ver. Dtsch. Ing. 98 (1956), 1801—1802.
ALT C., MEIER A., und RICHTER G., Chemie-Ing.-Techn., 27 (1955), 502—506.
132. Hersteller: Vereinigte Fiillkorperfabrik GmbH u. Co., Baumbach/Westerwald.
133. FUCHS 0., Chemie-Ing.-Techn. 23 (1951), 537—540.
134. FORSYTHE W. L., und MITARB., Ind. Engng. Chem. 39 (1947), 714.
135. WOLF F., und GUNTHER W., Dechema-Monogr. 29 (1957), 364—377.
136. KRELL E., Chem. Techn. 16 (1964) 10, 591—600.
137. KRELL E., Plaste und Kautschuk 14 (1967) 4, 251—254.
138. TELLER A. J., Chem. Engng. Progr. 50 (1954) 2, 65—71; Ind. Engng. Chem.
50 (1958), 8, 1201—1206.
139. N. N., Chem. Engng. 80 (1973) 19, 88.
140. EGBERONGBE S. A., Process Engng. (1973) 2, 82—83, 85.
141. STURMANN H., Leistungssteigerung von Fiillkorperkolonnen durch neue Wandformen, Dissertation, Hamburg 1937.
142. KRELL E. Патент ГДР № 63761, 1968. «Kolonne mit verminderter Randgangigkeit».
8. ЛИТЕРАТУРА
1. ROLFSON F. B„ und MITARB., Analytic. Chem. 20 (1948), 1014—1019.
2. BAYERL V., und QUARG M., Taschenbuch des Chemietechnologen, 3. Aufl., VEB Deutscher Verlag fiir Grundstoffindustrie, Leipzig 1968.
3. KAHTEPMAH Л.Б., ДУХАН Б.С., ИВАНОВ 17. Г. Химия и технология топлив и масел. 1958, т. 3, № 5, с. 57—60.
4. SALZER F„ Chemie-Ing.-Techn. 31 (1959), 405—409.
5. J1RMANN R„ Glas-Instrumenten-Techn. 14 (1970) 1, 11—12.
6. STAGE H., CZ-Chemie-Techn. 1 (1972), 263—272.
7. ZAPPE D., HECKMANN M., GUTWASSER H., und DORNBUSCH E., Heckmann-Mitt. Nr. 5 (1968).
8. ULUSOY E., Chemiker-Ztg. 79 (1955), 46—48.
9. STAGE H., GEMMEKER L., FISCHER G., und RICHTER R., Glas-Instrumenten-Techn. 5 (1961), 138—145 und 204—211.
10. FISCHER W. G., und WEYAND H., Glas-Instrumenten-Techn. 14 (1970) 1, 23—26.
11. KRELL E., Chem. Techn. 9 (1957), 266—274.
12. KORTUM G., und BITTEL A., Chemie-Ing.-Techn. 28 (1956), 40—44.
12a. KOUKOL H., KUPKA H. J., und HENNING G., GIT Fachz. Lab. 19 (1975), Sonderheft Labormedizin, 493—497.
13. BRANDT H., und ROCK H., Chemie-Ing.-Techn. 25 (1953), 511—514. BRANDT H., ROCK H., und LANGERS F., Chemie-Ing.-Techn. 29 (1957), 86—91.
14. ABEGG H., Chimia [Zurich] 6 (1952), 258—267.
15. LAPORTE H., Messung, Erzeugung und Konstanthaltung hoher bis tiefer Temperaturen, VEB Fachbuchverlag Leipzig 1961.
16. K'OSSLER I., und VODEHNAL J., Chem. Prumysl (tschech.) 7 (32) (1957), 292______297.
17. KADLEC M., Chem. Listy (tschech.) 51 (1957), 987—990.
17a. STAGE H., CZ-Chemie-Technik 5 (1973), 203—208.
18. VANVOR H., Glas-Instrumenten-Techn. 8 (1964), 415.
19. TELLE W., Chemische Laboratoriumsgerate, 2. Aufl., VEB Fachbuchverlag, Leipzig 1969.
20. PIATTI L., und MARTI W., Chemie-Ing.-Techn. 28 (1956), 473—475.
21. Hersteller: Siemens AG, Erlangen — Dr. Neumann GmbH, Mtinchen 80.
22. LOFFLER H. J., und HENRICI H., Chemie-Ing.-Techn. 30 (1958), 718. SOBLE A. B., Chem. Engng. Progr. 56 (1957), 444—451.
23. Hersteller: VEB Thermometerwerk, Geraberg.
507
24. Hersteller: Juchheim KG, Solingen.
25. KLEINTEICH R., Glas-Instrumenten-Techn. 3 (1959), 89—93.
26. WALTER M., Relaisbuch, Franckh’sche Verlagshandlung, Stuttgart 1951.
27. FISCHER W., Glas-Instrumenten-Techn. 14 (1970), 5, 513—516.
28. THORMANN K-, Dechema-Erfahrungsaustausch, Arbeitsmethoden und Gerate, Destillieren und Rektifizieren, Dechema, Frankfurt/Main, September 1950.
29. HUTLA V., Chem. Listy (tschech.) 51 (1957), 1964—1965.
30. JAECKEL R., Dechema—Erfahrungsaustausch, Vakuumtechnik, Dechema, Frankfurt/Main 1956.
31. LECK J- H., Pressure Measurement in Vakuum Systems, The Institute of Physics, London 1957.
32. NICKEL E., Gas- und Wasserfach, Beilage Bau und Betrieb 6 (1954), Nr. 5 (Ausg. Gas).
33. FARQUARSON J., und KERMICLE H. A., Rev. sci. Instr. 28 (1957), 324 325
34. DOSCH R., Glas-Instrumenten-Techn. 4 (1960), 9—17.
35. HOLLAND-MERTEN E. L., Tabellenbuch der Vakuumverfahrenstechnik in der Grundstoffindustrie, VEB Deutscher Verlag fiir Grundstoffindustrie, Leipzig 1964.
36. HOHLSCHUTTER H. W., und KAMPF G„ Angew. Chem. 66 (1956), 355.
36a. Hersteller: Airflor—Lufttechnik GmbH, Rheinbach.
37. CROMPTON R. W., und ELFORD M. T., J. sci. Instruments 34 (1957), 405—407.
38. STORZBACH W., Glas-Instrumenten-Techn. 14 (1970) 1, 36—37.
39. KRELL E., Chem. Techn. 3 (1951), 333—336.
40. GILMONT R., Ind. Engng. Chem., analyt. Edit. 18 (1946), 633—636.
41. Hersteller: Dr. Friedrichs — Dr. Matschke KG Normschliff-Glasgerate, Wertheim/Main.
42. YOUNG C. G., und MITARB., Canad. J. Technol. 29 (1951), 447—450.
43. KRELL E., Chem. Techn. 10 (1958), 327.
44. COULSON E.A.,und WARNE R. J., J. sci. Instruments 21 (1944), 122—123.
45. MONCH G. C-, Hochvakuumtechnik, Rudolf A. Lang Verlag, PofJneck 1950; Neues und Bewahrtes aus der Hochvakuumtechnik, VEB Wilhelm Knapp Verlag, Halle 1959.
46. HOCH H., Vakuum-Techn. 16 (1966) 1/2, 8—13.
47. STECKELMACHER W., J. sci. Instruments 1 (1951), 10—19.
48. GAEDE W., Z. techn. Physik 15 (1934), 664.
49. JAECKEL R., Kleinste Driicke, ihre Messung und Erzeugung, in: Technische Physik in Einzeldarstellungen, Springer-Verlag, Berlin/Gottingen/Heidelberg 1950.
LAPORTE H., Hochvakuum, seine Erzeugung, Messung und Anwendung im Laboratorium, VEB Wilhelm Knapp Verlag, Halle 1951;
Vakuummessungen, VEB Verlag Technik, Berlin 1955.
Y ARWOOD J., Hochvakuumtechnik, Rudolf A. Lang Verlag, Berlin 1955. • LEYBOLD, Vakuum-Taschenbuch, hrsg. von K. Diels und R. Jaeckel, 2. Aufl., Springer-Verlag, Berlin/Gottingen/Heidelberg 1962.
FRANK W., und KUTSCHE D., Die schonende Destination, Otto Krausskopf-Verlag GmbH, Mainz 1969. .,
50. MULLER B., Glas-Instrumenten-Techn. 14 (1970) 7 , 798—807.
51. EBERT H., Kompressionsvakunmmeter, Friedr. Vieweg-J- Sohn GmbH, Braunschweig 1951.
52. PECHE G., Glas-Instrumenten-Techn. 7 (1963) 1, 4—7; 2, 48—49.
53. Hersteller: Rudolf Brand, Wertheim/M., Glashiitte.
54. NISBET J. S., J. sci. Instruments 26 (1949), 271—273.
55. MELPOLDER F. W., Ind. Eng. Chem. 39 (1957), 617.
56. FISCHER H. E„ Analytic. Chem. 20 (1948), 982.
57. GEMMEKER L., und STAGE H., Glas-Instrumenten-Techn. 6 (1962) 12, ; 494—497 ; 7 (1963) 1, 18—23.
508
58. STAGE H., Chemie-Ing.-Techn. 22 (1950), 374—375.
STAGE H., GIT Fachz. Lab. 19 (1975) 3, 182.
59. HOUBEN-WEYL, Methoden der organischen Chemie, hrsg. von E. Miiller, Physikalische Methoden, Teil 1 und 2, Georg Thieme Verlag, Stutgart 1955.
60. MULLER R. H., und ZENCHELSKY S. T., Analytic Chem. 24 (1952), 844—847.
61. THOMAS G. R., und MITARB., Analytic. Chem. 22 (1950), 1221—1223.
62. LATCHUM J. W., Патент США № 2529030, 1950.
63. NEBE W., RIEGLER H., und MENZEL W., Jenaer Rundschau 18 (1973), Messe—Sonderheft 87—89.
64. KEGELES G., und SOBER H. A., Analytic. Chem. 24 (1952), 654—660.
65. OEHME F., Chemische Analyse durch Messung von Dielektrizitats-Konstanten, Verlag VEB Laborchemie, Apolda 1953.
OEHME F., Dielektrische MefJmethoden, 2. Aufl., Verlag Chemie GmbH, Weinheim/Bergstr, 1962.
66. SLEVOGT К. E., Dechema-Monogr. 14 (1950), 97—120; 17 (1951), 95—107.
67. GRANT R. A., J. appl. Chem. 8 (1958), 136; Chem.-Ing.-Techn. 30 (1958), 738.
68. FELLONI L„ und TANTILLO E., Ann. Chimica 47 (1957), 751—758. 69. CLASEN H., Chemie-Ing.-Techn. 28 (1956), 45-48; 29 (1957), 110—111. 70. LINFORD A., Ind. Chemist. 34 (1958) 403, 481—488.
71. FRITSCHE R. W., Petroleum Processing 7 (1952), 1138—1143.
72. KOCH 0., Chemie-Ing.-Techn. 30 (1958), 181—187.
KIENITZ H., Chemie-Ing.-Techn. 32 (1960) 10, 641—650.
73. SIGGIA S., Continons Analysis of Chemical Process Systems, John Wiley &Sons, London 1959.
74. Chemie-Ing.-Techn. 36 (1964) 9 A, 1455—1457.
75. HERFORTH L.,und KOCH H., Praktikum der angewandten Radioaktivitat, 2. Aufl., VEB Deutscher Verlag der Wissenschaften, Berlin 1972.
76. BERN HAU ER K-, Einfiihrung in die organisch—chemische Laboratoriumstechnik, Springer-Verlag, Wien 1947.
77. WITTENBERGER W., Chemische Laboratoriumstechnik, 6. Aufl., Springer-Verlag, Berlin/Gottingen/Heidelberg 1963.
78. MULLER G., und GNAUCK G., Reinste Gase, VEB Deutscher Verlag der Wissenschaften, Berlin 1965.
79. HINZPETER G„ Vakuum-Techn. 11 (1962), 2.
80. Hersteller: Fischer, Labor- u. Verfahrenstechnik, Bonn-Bad Godesberg.
81. DAMMER H. J., Glas-Instrumenten-Techn. 15 (1971) 2, 115—116.
82. HANEL R., Chem. Techn. 15 (1963) 3, 176—177.
83. PINKAVA J., und WICHTERLE 0., Collect, czechoslov. chem. Commun. 20 (1955), 697 bis 707.
PINKAVA J., und KALAB V., Collect, czechoslov. chem. Commun. 20 (1955), 707—712.
84. PINKAVA J., Laboratoriumstechnik kontinuierlicher chemischer Prozesse, Nakladatelstvi technicke Literatury (SNTL), Prag 1958.
85. SCHMIDT W., Chem. Techn. 10 (1958), 131—134.
86. HEUSCH R., Fette, Seifen, Anstrichmittel 72 (1970), 11, 969—977.
87. POHLENZ W., Pumpen fiir Flfissigkeiten, VEB Verlag Technik, Berlin 1970.
88. LI EDEL К. H., Gias in der Chemie — Laborgerate, GrofJapparate, Rohrleitungen, Otto Krausskopf-Verlag GmbH, Mainz 1973.
9. ЛИТЕРАТУРА
1. BEHRE A., Chemisch-physiktalische Laboratorien und ihre neuzeitlichen Einrichtungen, Akademische Verlagsgesellschaft Geest & Portig,
’ Leipzig 1950.
2. COLEMANN H. S., und MITARB., Laboratory Design., Reinhold, New York 1952.
3. SCHRAMM W., Chemische und biologische Laboratorien, 3. Aufl., Verlag Chemie GmbH, Weinheim/Bergstr. 1969.
509
' 4. KINCANNON С. В., und BAKER M. 0., Analytic. Chem. 29 (1957), 1189—1193.
5. SCHWARZ H., Stative, Aufhangung von Apparaturen, Bn: Houben-Weyl, Methoden der organischen Chemie (hrsg. von E. Miiller), Bd. 1/1, Georg Thieme Verlag, Stuttgart 1958.
6. Hersteller: Ernst Haage, Miilheim-Ruhr.
7. Hersteller: Geru, Geroldwill/Schweiz.
8. BROOKE M„ J. Chem. Educat. 28 (1951), 602—603.
9. WAGNER G., Osterr. Chemiker-Ztg. 43 (1940), 229.
10. BERN HAUER K-, Einftihrung in die organised—chemische Laboratoriumstechnik, Springer-Verlag, Wien 1947.
11. WITTENBERGER W., Chemische Laboratoriumstechnik, 7. Aufl., Springer-Verlag, Berlin/Heidelberg/New York 1973.
12. WOLF F. W. D., Glas-Instrumenten-Techn. 4 (1960), 34—52, 126—131.
13. FRIEDRICHS F., Das Gias im chemischen Laboratorium, 3. Aufl., Springer-Verlag, Berlin/Gottingen/Heidelberg 1960.
14. BRAUER H., Chemie-Ing.-Techn. 29 (1957), 520—530.
15. GREINACHER H., Ausgewahlte Aufgaben und Probleme aus der Experimentalphysik (§ 28: Der Siedeverzug), Springer-Verlag, Wien 1953.
16. Патенты США № 2580880, 1952 ; 2580922, 1952.
17. HUBNER G., Chemiker-Ztg. 75 (1951), 229—230.
18. The General Safety Committee of the Manufacturing Chemist’s Association. Guide for Safety in the Chemical Laboratory, Van Nostrand, New York 1954.
19. DITTMAR P., Erdol u. Kohle 11 (1958), 25—30.
20. CRAIG L. E., und DEW J. E., Ind. Engng. Chem. 51 (1959), 1249—1252.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Азеотроп гетерогенный (гетероазеотроп) 85, 303, 306
гомогенный (гомоазеотроп) 85, 303, 306
определение состава 311 сл.
отрицательный 52, 85, 299 сл., 307, 309
положительный 52, 85, 299 сл., 307, 309
Азеотропии 300 сл.
Азеотропная смесь 299 точка 76
Анероид 439
Аргон, обогащение методом ректификации 222 сл.
Бани 398
для высокотемпературной ректификации 258
Бар 33
Баровакуумметры 440 сл.
Барометры 439
корректировка показаний 181 сл. Бензол, давление паров при различных температурах 78 Бидистиллятор воды 217 сл.
Бора фторид, концентрирование методом ректификации 222 сл.
Бюретка 328
Вакуум
измерение 267, 440 сл., 446 сл.
получение 267
регулирование 443 сл., 448, 451 сл. Вакуумметры 441 сл., 447, 451 сл. Вакуумные коммуникации, определение размеров 265 сл.
Вакуускопы 447, 449
Вентиль газовый проходной 214 Вода — фенол, зависимость состава азеотропа от давления 306
Время разгонки 127
пусковое 127
Высота единицы переноса (ВЕП) 99, 124
Высота, эквивалентная теоретической ступени (ВЭТС) 96 сл., 124, 130, 141
зависимость от диаметра колонны 137
----рабочей высоты колонны 138 сл.
колонн безнасадочиых 336
— с вращающимися контактными устройствами 363
колонн насадочных 136, 162 сл.
— с неподвижными контактными устройствами 354 сл.
— для полумикроректификации 200
насадок 120, 130 сл., 412, 414
Вытяжной шкаф 471
Вязкость, определение 462
Газовая смесь
давление, общее и парциальное 72 сл., 77 сл., 81 сл., 295 сл.
разделение ректификацией 250, 262, 292
Герметичность
кранов и шлифовых соединений 31 сл., 268, 477 сл.
установок 268 сл., 480
Гидравлическое сопротивление колонны 152, 164 сл. — насадочной 368 — тарельчатой 345, 349 — трубчатой щелевой 341 насадки 166 сл.
Головки ректификационных колони
206, 248, 378—386
азеотропные 320 дистилляционные 24 для парциальной конденсации 248
Градус Кельвина 33
Давление 33
газовой (паровой) смеси
----общее и парциальное
72 сл., 77сл., 81 сл., 295сл.
512
измерение и регулирование 433 сл., 438 сл., 443 сл.
насыщенных паров, определение 54 сл., 60 сл.
остаточное, измерение и регулирование 267, 440 сл., 443 сл.,
446 сл„ 448, 451 сл.
рабочее при перегонке 53 сл.,
151 сл., 262, 438
Дефлегматоры 26, 245 сл., 369
Дефлегмации 23, 245
Джоуль 33
Диаграмма
равновесия пар—жидкость 51 сл., см. также Кривая равновесия пар — жидкость разгонки 185 сл. — бензиновой фракции 180 — смеси жирных кислот 184, 244, 270, 275
--------- растворителей 206 Дистиллят 38
определение физико-химических свойств 179 сл., 458 сл.
отбор 206, 329 сл., 390 сл., 453 — при низкотемпературной ректификации 250, 253
Дистилляторы воды 216 сл. для молекулярной дистилляции 283 сл.
пленочный Креля 273 сл.
Дистилляционная способность 290 Дистилляционные
приборы, с колбой Кляйзена 326 сл.
-----Энглера 326 установки 25, 328 сл. — автоматизированные 419, 422 — для молекулирной дистилляции 286 сл.
— для перегонки веществ с высокой температурой плавления 259 — с пленочным испарителем 277сл.
— с трубчатой колонной 241 Дистилляция 19 сл., 39 сл., 175 сл., 327 сл.
вакуумная 263—269
с восходящим или нисходящим потоком паров 21
молекулярная 262 сл., 280—292, 438
«на коротком пути» 284 ртути 260 сл.
тепловой баланс 175 сл.
по Энглеру 40, 207, 326
Дозирование газов 463 сл. жидкостей 463 сл.
Жидкость
взаимная растворимость 50 измерение расхода 463 сл. Жирные кислоты
разделение непрерывной ректификацией 133 сл., 243 сл.
— парциальной конденсацией 248 сл.
— перегонкой с водяным паром 296 сл.
— пленочной дистилляцией 275 — расширительной перегонкой 270 сл.
Закон
Дальтона 72
Рауля 77, 81, 308
«Захват» жидкости 149
Захлебывание колонны 158 сл.
Идеальная смесь
летучесть 81 — относительная 84 сл. построение кривой равновесия 77 сл., 86 сл.
Изотенископ 58 сл.
Изотопы, разделение методом ректификации 219 сл.
Испарение 70
скорость 280 сл.
пленочное 272 сл.
Испарители для молекулирной дистилляции 286
для пленочной перегонки 273 сл. циркуляционные 211 сл., 400
513
Каналообразование 43, 119 сл.
Капельницы 380, 465
Кипение
бинарных смесей 52
способы стабилизации 330, 481
температура, определение 55 сл., 61 сл., 182 сл., 295 сл.
Кипятильники погружные 396
Клапаны 334 сл.
Колбы для перегонки 26, 387 сл.
Кляйзена 26, 326 сл.
Кольца Рашига 27, 120, 144, 151, 409
Конденсаторы 369
Конденсация 22 сл., 369
при высокотемпературной ректификации 258 сл.
краевая 245
парциальная 245 сл.
Концентрация 34 сл.
Коэффициент
активности 84 сл.
использования насадочной колонны 44
неравномерности орошения 44
обогащения 112 сл.
полезного действия колонны 148, 349
-----тарелки 136, 161
разделения 112
теплопередачи для холодильников 373 сл.
Краны 332 сл.
вакуумные 267
герметизация 478
для работы под давлением 292 регулирующие 381 сл.
Кривая
«выделении» 290 сл.
давления паров 53, 74
----- построение 63 сл.
равновесия пар—жидкость 74 сл.
------- дифференциальная 109
-------построение
---------графический способ 79 ---------- расчет 76 сл.
--------- экспериментальное определение 86 сл.
------- смеси бензол—гептан
109
Кривая
--------- бензол—толуол 98
---------жирных кислот 105
---------ж-крезол—и-крезол
109
---------толуол—н-октан 109
разгонки 93
Куб 387 сл., 481
загрузка 480
обогрев 22, 394 сл., 455 сл.
— при высокотемпературной ректификации 258
для периодической разгонки 209
Лаборатория для перегонки, планировка помещения 469 сл.
Летучесть 81
относительная 81 сл., 115
Манометры 439 сл., 447 сл., 456 сл.
Мак-Леода 442, 447 сл.
Массовые
доля 34
проценты 34 сл.
Массообмен 38
в насадочной колонне 99, 119
в тарельчатой колонне 99, 119 фактор интенсивности 127 сл.
«Мертвый» объем 205 сл.
Металлы, очистка высокотемпературной ректификацией 259
Мнкродистилляция 196 сл.
молекулярная 199 сл.
Мнкроперегонка 195 сл.
вакуумная 198
Микроректификация 196 сл.
Многокомпонентная смесь 131 сл.
расширительная перегонка 271 ректификация 131 сл., 239
— аналитическая 203
— расчет процесса на ЭВМ 193
средняя масса моля 34
Молекулярная дистилляция 262 сл.,
280 сл., 438
микродистилляции 199 сл.
514
Мольные
доля 34 сл.
проценты 34 сл.
Муфты 331
Нагревание исходной смеси н кубовой жидкости 398 сл.
при мнкроперегонке 201
Нагревательные банн 258, 398 приборы электрические 393 сл. элементы 403 сл., 406
Нагрузка ректификационных колонн 153 сл., 164
расчет для периодической ректификации 188
регулирование 456 сл.
Насадка 120, 165, 352 сл., 407—415 влияние материала на эффективность разделения 415 гидравлическое сопротивление 166 сл.
доля активной и смоченной поверхности 46
очистка 155, 480 пакетная 42, 358 пленочная 359 пленочное течение жидкости 48 сл.
для полумикроперегонкн 200 распределение жидкости 41 сл. регулярная 42, 48 смачивание 48, 121, 415 Стедмана 355 сл.
укладка 139, 155, 343 сл. «Хэли-грид» 170, 250, 354 сл. Насосы
вакуумные 267 сл. дозировочные 213 сл., 466 сл. установка в помещении 469 Номограмма
для корректировки показаний барометра 181 сл.
----термометра 182 сл.
для определения размеров вакуумных коммуникаций 265 сл.
— скорости паров для насадок 172
Номограмма
— числа теоретических ступеней разделения 116, 256 сл.
для пересчета концентраций 35 сл.
Ньютон 33
Обогрев
кожуха колонны 403 сл.
куба 22, 394 сл., 455 сл.
при высокотемпературной ректификации 258
Объем паров, расчет 71
Объемные
доля 34
проценты 34 сл.
Орошающая жидкость 148 сл.
Отбор дистиллята 329 сл., 390, 453 при аналитической ректификации 206
при низкотемпературной ректификации 250, 253
Относительная летучесть 81 сл.
для бинарной смеси 115
Параметр разделения 226 сл.
Парциальная конденсация 245 сл. Паскаль 33
Перегонка 19 сл., 32, 38
азеотропная 299 сл., 304—314, 320
вакуумная 263—269, 482
взрывоопасных веществ 482
с водяным паром 51, 294 ' сл. высокотемпературная 257—262 давление рабочее 53 сл., 151 сл., 262, 438
изотермическая 249, 262
наработочная 207 сл., 216 сл. непрерывная 40, см. также Ректификация непрерывная
в парах носителя 51, 294—299 периодическая 40, 234 сл.
пленочная 262, 272—280, 438
полунепрерывная 235 препаративная 207 сл.
простая 38 сл., 195, 234
515
расчет процесса на ЭВМ 190 сл. расширительная 262, 269—271, 438 солевая 321—323 стандартизация методов 28 сл. тепловой баланс 175 сл.
токсичных веществ 469 сл., 482 фракционная 195
экстрактивная 299, 303, 314—321 Перепад давления
в вакуумном трубопроводе 265 в колонне 152, 164, 341, 364, см.
также Гидравлическое сопроти-леиие колонны
Переходы 331
Пилотная установка 11, 208, 211—216 Плотность
жидкости, определение 462 паров, расчет 71 сл.
Показатель преломления, определение 180, 459 сл.
Полумикроперегонка 195—202 Полумикроректификация, см. Полуми-кроперегонка
установки 342 сл., 422
Полюсное расстояние 95
Правило Дюринга 61
Приемники дистиллята 387, 390 сл.
Приставки для дистилляционных аппаратов 328 сл., 376—378
Пристеночный эффект 43, 137 Пробоотборники 156
Рабочая задержка 149
Разгонка 40
.. время 127
фракционная 234
Разделяющая способность ректификационной колонны 93, 137, 338 сл.
зависимость от нагрузки и рабочего давления 153
связь с гидравлическим сопротивлением 166 сл.
Разделяющий агент 301, 304 для азеотропной перегонки 304, 309 сл.
для экстрактивной перегонки 314 сл.
Разъемные соединения 30, 32
516
Растворы, классификация 53
Расходомеры 463
Регуляторы давления 443 сл., 451 сл.
Ректификатор-автомат 420 сл.
Ректификационная колонна 25 сл., 335 сл.
безнасадочная 335—343
с вращающей лентой 200, 203, 360, 362, 364 сл.
— спиралью 361
с вращающимися и неподвижными пластинами 360 сл.
— контактными устройствами 359—369
— щетками 277
с вращающимся конденсатором
360 сл., 363 сл.
— цилиндром 360, 363 сл.
высота рабочая 138 сл., 171, 189 гидравлическое сопротивление
152, 164 сл., 341, 345, 349, 368
диаметр 164, 137 сл., 189 динамические характеристики
49 сл.
с елочным дефлегматором 336 сл.
захлебывание 158 сл.
испытание 136, 145, 155 сл.,
189 сл.
исчерпывающая часть 103
с колпачковыми тарелками 26, 42, 210 сл., 241, 345 сл.
с кольцевой щелью, см. трубчатая щелевая
многотрубчатая 338 сл.
для молекулярной дистилляции
283 сл.
нагрузка 153 сл., 164, 188, 456 сл.
с насадкой Стендмана 353 , 356 насадочная 42, 99, 118 сл., 162 сл., 344—345
— для низкотемпературной ректификации 250 сл.
с неподвижными контактными устройствами 352—359
для низкотемпературной ректификации 252 сл.
с орошаемой насадкой 42
Ректификационная колонна
с орошаемыми пластинами 353, 356 сл.
с перемешивающейся пленкой 275 сл.
перепад давления 152, 164, 168, 341, 364, см. также гидравлическое сопротивление
пленочные 42, см. также Испарители для пленочной перегонки с проволочной спиралью 353 сл. разделяющая способность 93, 137,
153, 166 сл., 338 сл.
с регулярной насадкой 42, 352 роторная 278, см. также с вращающимися контактными устройствами
роторно-спиральная 361
с ситчатыми тарелками 42, 348 сл.
«со смоченными стенками» 353
со спиралью из металлической сетки 353 сл.
со стеклянной спиралью 353 тарельчатая 26, 42, 99, 150, 161, 164, 209 сл., 240 сл., 345—352 тарельчато-насадочная 240, 335
> тепловая изоляция 177 сл., 401 сл. трубчатая змеевиковая 337 сл. — щелевая 200, 204, 338, 340 сл., 365
удерживающая способность 127, 149 сл., 161, 200, 335, 354 сл., 363
с фонтанирующими тарелками 346 Ректификационные установки
автоматизированные 417 сл., 422 сл., 342
для азеотропной ректификации 302 для аналитической ректификации 205 сл.
вакуумные
— непрерывного действия 25, 104, 214, 236
— полунепрерывного действия 235 герметизация 477—479 конструкционные материалы
324 сл.
Ректификационные установки непрерывного действия 21, 25, 104, 214, 236, 240—243
для низкотемпературной ректификации 223, 251 сл., 423
пилотные 11, 208, 211—216 для полумикроректификации 342 сл.
полупромышленные 208—211, 215 для получения тяжелой воды
229 сл.
для ректификации под давлением 292 сл.
— с перегретым водяным паром 298
сборка 330 сл., 474 сл. с трубчатой колонной 241 эксплуатация 244 сл., 480 сл.' для экстрактивной ректификации 303
Ректификация 32, 38 сл.
азеотропная 51, 299 сл., см. также
Перегонка азеотропная азеотропноэкстрактивная 299 аналитическая 202—207 вакуумная 263—269 высокотемпературная 249, 258 сл. газов 250, 262, 292 под давлением 250,292 сл., 438сл. изотопов 219 сл.
непрерывная 103 сл., 133, 235— 245, 398 сл.
низкотемпературная 249, 250— 256, 422 сл. .
— разделение йзотопов 222 Сл. «обменная» 221
периодическая — расчет процесса разделения 186 сл., 239 сл.
--- числа теоретических ступеней . разделения 100 сл., 111 сл., 186
пленочная 438 по принципу «падающей пленки» 286 сл.
пробная 207 расчет процесса на ЭВМ 190 сл* тепловой баланс 175 сл.. ,
517
«термическая» 365 циклический метод 240 экстрактивная 299, 303, см. также Перегонка экстрактивная
Реле времени 453 сл.
Реометры 463
Рефрактометр 459 сл.
Ротаметры 463 сл.
Ртуть, дистилляция 260 сл.
Сборники дистиллята 328 сл., 387, 390 сл.
Скорость паров в колонне 154 сл., 164 сл., 172, 343 константа 170 сл.
Смазки жировые 478 сл.
Среднее относительное обогащение 136
Стандартизация единиц измерения 33 методов перегонки 31 термометров 429 физических величин 33 шлифов 28 сл.
Стекло как конструкционный материал 211, 324 сл.
Стенды для ректификационной аппаратуры 472 сл.
Степень перемешивания 44 сл.
Тарелка ректификационной колонны 39 сл.
барботажная 345
колпачковая 345, 348
ситчатая 345, 348
теоретическая 95 сл.
фонтанирующая 345
Температура
измерение 428—434
затвердевания, определение 458 сл.
кипения, определение 55 сл.,
61 сл., 182 сл., 295 сл.
регулирование 434—438
Теоретическая ступень разделения 96, 123 сл.
Теплоизоляция 393 сл., 401 сл.
Теплоноситель 397 сл.
518
Теплообмен в колонне 119
Термометры
контактные 434 сл.
корректировка показаний 181 сл. проверка градуировки 431 сл. стеклянные 429 сл.
сопротивления 428 сл., 432 сл.
Термопары 432 сл.
Течеискатели 268 сл.
Толуол, давление паров при различных температурах 78
Трубопроводы для ректификационных установок соединительные элементы 406 теплоизоляция 406
Тяжелая вода, получение ректификацией 224 сл.
Углерод, разделение изотопов ректификацией 233 сл.
Удерживающая способность ректификационной колонны 127, 161 динамическая 149 сл.
колонн безнасадочных 335 сл.
— с вращающимися контактными устройствами 363
— с неподвижными контактными -устройствами 354 сл.
— тарельчатых 150, 161 колонок для полумикроректификации 200
общая 149, 151 статическая 149 сл.
Уравнение
Антуана, зависимость между давлением насыщенных паров и температурой 62 сл.
Ваи-дер-Ваальса 70
Вебера для расчета пускового времени 127
Клаузиуса—Клапейрона 64 сл.
Мерча для расчета ВЭТС 141, 144 состояния 70 сл.
рабочей линии укрепляющей части колонны 98 сл.
Фенске для определения числа теоретических ступеней разделения 115 сл., 135
Фазовая диаграмма
р—х 70 сл.
/—х—у 74 сл.
Фазовое равновесие, методы исследования 86 сл.
Фактор интенсивности массообмена 127 сл.
Фенолы, разделение ректификацией 243 сл.
Флегма 39
«дикая» 178, 403
Флегмовое число 97, 145 сл.
бесконечное 97, 145
измерение 379
для низкотемпературной ректификации 255, 257
расчет для непрерывной ректификации 103 сл.
----периодической ректификации 100 сл., 187
регулирование 379, 453 сл.
Форштос 328, 376
Хладоагент 369
Хлор, получение изотопов ректификацией 233 сл.
Холодильники 22 сл., 328, 369—375
Число единиц переноса 122
определение 123 сл.
Число теоретических ступеней разделения
определение, аналитические методы 111 сл., 160
— при конечном флегмовом числе 145
— метод Мак-Кэба и Тиле 100 сл., 103 сл., 107 сл.
-----Оболоицева и Фроста 160 ---- Роуза 113 сл.
Число теоретических ступеней разделения
— для насадки Стедмана 356
— для непрерывной ректификации 103 сл.
— для низкотемпературной ректификации 255 сл.
— для периодической ректификации 100 сл., 186 сл.
— для ректификации азеотропных смесей 313
---многокомпонентных смесей 131 сл.
— по уравнению Фенске 115 сл., 135
— по фактору обогащенйя 112 сл.
— на ЭВМ 192
— для экстрактивной перегонки 318
Число флегмовое см. Флегмовое число эквивалентных теоретических ступеней разделения 147
Шлифы 28 сл.
для вакуумных работ 267 герметизация 478 сл.
смазки 478 сл.
соединения 31, 330, 474 сл.
Штативы 476 сл.
Эбуллиометр 55 сл.
Эбуллиоскоп 55 сл.
ЭВМ для расчета ректификационных колонн 190
Электронагреватели 394
Эталонная смесь 140 сл., 156
Этанол, абсолютирование азеотропной ректификацией 313 сл.
Этанол — вода, зависимость состава азеотропа от давления 306
Эффект пристеночный 43, 137
Эрих Крель
РУКОВОДСТВО ПО ЛАБОРАТОРНОЙ ПЕРЕГОНКЕ
Редактор
Л. Н. ЛАРИЧЕВА
Художественный редактор
Н. В. НОСОВ
Технический редактор
В. М. СКИТИНА
Художник
А. Я. МИХАЙЛОВ
Корректоры:
О. И. ГОЛУБЕВА, И. М. АВЕЙДЕ
ИБ № 854
Сдано в наб. 17.08.79. Подп. в печ. 24.03.80.
Формат бумаги 60X 90*/,,.
Бумага тип. № 2. Гари, литературная.
Печать высокая. Усл. печ. л. 32,5.
Уч.-изд. л. 34,87. Тираж 7000 экз.
Заказ № 1589. Цена 3 р. 30 к.
Изд. № 1780.
Ордена «Знак Почета» издательство «Химия».
107076, Москва, Стромынка, 13
Ленинградская типография № 6 Ленинградского производственного объединения «Техническая книга» Союзполиграфпрома при Государственном комитете СССР по делам издательств, полиграфии и книжной торговли.
193144, Ленинград, С-144, ул. Моисеенко, 10.
Э. Кроль