ГОСТ ISO/IEC 17025-2019
МЕЖГОСУДАРСТВЕННЫЙ СТАНДАРТ
ОБЩИЕ ТРЕБОВАНИЯ К КОМПЕТЕНТНОСТИ ИСПЫТАТЕЛЬНЫХ И КАЛИБРОВОЧНЫХ ЛАБОРАТОРИЙ
General requirements for the competence of testing and calibration laboratories
______________________________________________________________________
Текст Сравнительного анализа ГОСТ ISO/IEC 17025-2019 c ГОСТ ИСО/МЭК 17025-2009 см. по ссылке:
— .
______________________________________________________________________
МКС 03.120.20
Дата введения 2019-09-01
Предисловие
Цели, основные принципы и общие правила проведения работ по межгосударственной стандартизации установлены ГОСТ 1.0 «Межгосударственная система стандартизации. Основные положения» и ГОСТ 1.2 «Межгосударственная система стандартизации. Стандарты межгосударственные, правила и рекомендации по межгосударственной стандартизации. Правила разработки, принятия, обновления и отмены»
Сведения о стандарте
1 ПОДГОТОВЛЕН Республиканским унитарным предприятием «Белорусский государственный центр аккредитации» (Государственное предприятие «БГЦА») на основе собственного перевода на русский язык англоязычной версии стандарта, указанного в пункте 4
2 ВНЕСЕН Государственным комитетом по стандартизации Республики Беларусь
3 ПРИНЯТ Межгосударственным советом по стандартизации, метрологии и сертификации (протокол от 28 июня 2019 г. N 55)
За принятие проголосовали:
|
Краткое наименование страны по МК (ИСО 3166) 004-97 |
Код страны по МК (ИСО 3166) 004-97 |
Сокращенное наименование национального органа по стандартизации |
|
Армения |
AM |
Минэкономики Республики Армения |
|
Беларусь |
BY |
Госстандарт Республики Беларусь |
|
Казахстан |
KZ |
Госстандарт Республики Казахстан |
|
Киргизия |
KG |
Кыргызстандарт |
|
Россия |
RU |
Росстандарт |
|
Таджикистан |
TJ |
Таджикстандарт |
|
Узбекистан |
UZ |
Узстандарт |
4 Приказом Федерального агентства по техническому регулированию и метрологии от 15 июля 2019 г. N 385-ст межгосударственный стандарт ГОСТ ISO/IEC 17025-2019 введен в действие в качестве национального стандарта Российской Федерации с 1 сентября 2019 г.
5 Настоящий стандарт идентичен международному стандарту ISO/IEC 17025:2017* «Общие требования к компетентности испытательных и калибровочных лабораторий («General requirements for the competence of testing and calibration laboratories», IDT).
________________
* Доступ к международным и зарубежным документам, упомянутым в тексте, можно получить, обратившись в Службу поддержки пользователей. — .
В настоящем стандарте слово customer переведено как «заказчик» и применяется в качестве одного из значений термина «потребитель» (см. ГОСТ Р ИСО 9000-2015, пункт 3.2.4; СТБ ISO 9000-2015, пункт 3.2.4), поскольку, как правило, лаборатории взаимодействуют непосредственно с заказчиком услуг по испытаниям и калибровке, а не с конечным пользователем. Кроме того, в практике осуществления лабораторной деятельности общеупотребительным является слово «заказчик».
Международный стандарт разработан Комитетом по оценке соответствия (CASCO) Международной организации по стандартизации (ISO).
При применении настоящего стандарта рекомендуется использовать вместо ссылочных международных стандартов соответствующие им межгосударственные стандарты, сведения о которых приведены в дополнительном приложении ДА
6 ВЗАМЕН ГОСТ ИСО/МЭК 17025-2009
7 ПЕРЕИЗДАНИЕ. Март 2021 г.
Информация о введении в действие (прекращении действия) настоящего стандарта и изменений к нему на территории указанных выше государств публикуется в указателях национальных стандартов, издаваемых в этих государствах, а также в сети Интернет на сайтах соответствующих национальных органов по стандартизации.
В случае пересмотра, изменения или отмены настоящего стандарта соответствующая информация будет опубликована на официальном интернет-сайте Межгосударственного совета по стандартизации, метрологии и сертификации в каталоге «Межгосударственные стандарты»
Введение
Настоящий стандарт разработан с целью укрепления доверия к деятельности лабораторий. В настоящем стандарте содержатся требования к лабораториям, выполнение которых позволит им продемонстрировать компетентность и способность получать достоверные результаты. Лаборатории, которые соответствуют требованиям настоящего стандарта, также будут в целом функционировать в соответствии с принципами ISO 9001.
Согласно требованиям настоящего стандарта лаборатория должна планировать и осуществлять действия по управлению рисками и возможностями. Управление рисками и возможностями создает основу для повышения результативности системы менеджмента, достижения лучших результатов и предотвращения негативных последствий. Лаборатория несет ответственность за принятие решения о том, какие риски и возможности необходимо рассматривать.
Использование настоящего стандарта упростит сотрудничество между лабораториями и другими органами и поможет в обмене информацией и опытом, а также в гармонизации стандартов и процедур. Признание результатов лабораторной деятельности между странами упрощается, если лаборатории соответствуют требованиям настоящего стандарта.
В настоящем стандарте используются следующие глагольные формы:
— «должен» — обозначает требование;
— «следует» — обозначает рекомендацию;
— «может» — обозначает разрешение;
— «способен» — обозначает возможность.
Более подробную информацию можно найти в Директивах ISO/IEC, часть 2.
Для целей исследования пользователям рекомендуется делиться своим видением настоящего стандарта и предложениями по внесению изменений в последующие издания. Чтобы принять участие в онлайн-опросе, необходимо пройти по ссылке: https://www.surveymonkey.com/r/6MQ95ZC.
1 Область применения
Настоящий стандарт устанавливает общие требования к компетентности, беспристрастности и стабильному функционированию лабораторий.
Настоящий стандарт применим ко всем организациям, занимающимся лабораторной деятельностью, независимо от численности персонала.
Заказчики лабораторий, регулирующие органы, организации и схемы, использующие паритетную оценку, органы по аккредитации, а также другие стороны применяют настоящий стандарт при подтверждении или признании компетентности лабораторий.
2 Нормативные ссылки
При применении настоящего стандарта рекомендуется использовать вместо ссылочных международных стандартов соответствующие им межгосударственные стандарты, сведения о которых приведены в дополнительном приложении ДА
ISO/IEC Guide 99, International vocabulary of metrology — Basic and general concepts and associated terms (VIM) (Международный словарь по метрологии. Основные и общие понятия и соответствующие термины (VIM))
ISO/IEC 17000, Conformity assessment — Vocabulary and general principles (Оценка соответствия. Словарь и общие принципы)
3 Термины и определения
В настоящем стандарте применены термины по ISO/IEC Guide 99 и ISO/IEC 17000, а также следующие термины с соответствующими определениями.
ISO и IEC поддерживают терминологические базы данных для использования в области стандартизации, которые доступны по следующим ссылкам:
— онлайн-платформа ISO для поиска доступна по ссылке: http://www.iso.org/obp;
— электропедия IEC: доступна по ссылке: http://www.electropedia.org/
3.1 беспристрастность (impartiality): Наличие объективности.
Примечание 1 — Объективность означает, что отсутствуют конфликты интересов или они разрешаются таким образом, что не оказывают негативного влияния на последующую деятельность лаборатории (3.6).
Примечание 2 — Другими терминами, используемыми при передаче сути составляющих беспристрастности, являются «отсутствие конфликтов интересов», «отсутствие предвзятости», «отсутствие предубеждений», «нейтралитет», «справедливость», «открытость», «объективность», «отстраненность» и «паритет».
[ИСТОЧНИК: ISO/IEC 17021-1:2015, 3.2, изменено — в примечании 1 слова «органа по сертификации» заменены на «лаборатории», в примечании 2 слово «независимость» исключено из списка.]
3.2 жалоба (претензия) (complaint): Выражение неудовлетворенности любым лицом или организацией в отношении лаборатории (3.6), касающееся деятельности или результатов этой лаборатории, по которому ожидается ответ.
[ИСТОЧНИК: ISO/IEC 17000:2004, 6.5, изменено — слова «в отличие от апелляции» были исключены, а слова «орган по оценке соответствия или орган по аккредитации, относящийся к деятельности этого органа» были заменены словами «лаборатории, касающейся деятельности или результатов этой лаборатории».]
3.3 межлабораторное сличение (interlaboratory comparison): Организация, выполнение и оценивание измерений или испытаний одного и того же или нескольких подобных образцов двумя или более лабораториями в соответствии с заранее установленными условиями.
[ИСТОЧНИК: ISO/IEC 17043:2010, 3.4]
3.4 внутрилабораторное сличение (intralaboratory comparison): Организация, выполнение и оценивание измерений или испытаний одного и того же или нескольких подобных образцов в пределах одной лаборатории (3.6) в соответствии с заранее установленными условиями.
3.5 проверка квалификации (proficiency testing): Оценивание характеристики функционирования участника по заранее установленным критериям посредством межлабораторных сличений (3.3).
[ИСТОЧНИК: ISO/IEC 17043:2010, 3.7, изменено — удалены примечания к пункту.]
3.6 лаборатория (laboratory): Орган, который осуществляет один или несколько из следующих видов деятельности:
— испытания;
— калибровка;
— отбор образцов, связанный с последующими испытаниями или калибровкой.
Примечание 1 — В контексте настоящего стандарта понятие «лабораторная деятельность» относится к трем вышеуказанным видам деятельности.
3.7 правило принятия решения (decision rule): Правило, которое описывает, как учитывается неопределенность измерений при принятии решения о соответствии установленному требованию.
3.8 верификация (verification): Предоставление объективных свидетельств того, что данный объект соответствует установленным требованиям.
Примеры
1 Подтверждение того, что данный стандартный образец является, как заявлено, однородным для значения величины и соответствующей методики измерений, вплоть до образца массой 10 мг.
2 Подтверждение того, что эксплуатационные характеристики измерительной системы или законодательные требования к ней соблюдены.
3 Подтверждение того, что может быть достигнута целевая неопределенность измерений.
Примечание 1 — Когда применимо, следует учитывать неопределенность измерений.
Примечание 2 — Объектом может быть, например, процесс, методика измерения, материал, вещество или измерительная система.
Примечание 3 — Установленные требования могут заключаться, например, в соблюдении спецификации производителя.
Примечание 4 — В законодательной метрологии, как это определено в VIML, и при оценке соответствия в целом верификация относится к исследованию и клеймению и/или выдаче свидетельства о поверке измерительной системы.
Примечание 5 — Поверку не следует путать с калибровкой. Не каждая верификация является валидацией (3.9).
Примечание 6 — В химии верификация идентичности рассматриваемого объекта или реакции требует описания структуры или свойств этого объекта или реакции.
[ИСТОЧНИК: ISO/IEC Guide 99:2007, 2.44]
3.9 валидация (validation): Верификация (3.8), при которой установленные требования связаны с предполагаемым использованием.
Пример — Методика измерения, обычно используемая для измерения массовой концентрации азота в воде, может быть валидирована также для измерения массовой концентрации азота в сыворотке крови человека.
[ИСТОЧНИК: ISO/IEC Guide 99:2007, 2.45]
4 Общие требования
4.1 Беспристрастность
4.1.1 Лабораторная деятельность должна осуществляться беспристрастно, а также структурироваться и управляться таким образом, чтобы обеспечивать беспристрастность.
4.1.2 Руководство лаборатории должно принять обязательства по беспристрастности.
4.1.3 Лаборатория должна нести ответственность за беспристрастность своей лабораторной деятельности и не должна допускать коммерческое, финансовое или иное давление, ставящее беспристрастность под угрозу.
4.1.4 Лаборатория должна идентифицировать риски для своей беспристрастности на постоянной основе. Это должно включать риски, которые возникают в процессе ее деятельности, в результате ее отношений или отношений ее персонала. Вместе с тем такие отношения не обязательно представляют собой риск для беспристрастности лаборатории.
Примечание — Отношения, которые угрожают беспристрастности лаборатории, могут основываться на праве собственности, управлении, руководстве, персонале, общих ресурсах, финансах, договорах, маркетинге (включая брэндинг) и комиссионных выплатах или на других видах стимулирования в отношении новых заказчиков и т.п.
4.1.5 При обнаружении риска для беспристрастности лаборатория должна быть в состоянии продемонстрировать то, как она устраняет или минимизирует такой риск.
4.2 Конфиденциальность
4.2.1 Лаборатория должна на основе юридически значимых обязательств нести ответственность за управление всей информацией, поступившей извне или полученной в процессе выполнения лабораторной деятельности. Лаборатория должна заранее информировать заказчика об информации, которую она намерена разместить в свободном доступе. Исключение составляет информация, которая становится общедоступной по решению заказчика либо по согласованию между лабораторией и заказчиком (например, с целью реагирования на жалобы). Вся иная информация считается представляющей коммерческую тайну и должна рассматриваться в качестве конфиденциальной.
4.2.2 Если в соответствии с законодательством или договорными отношениями лаборатория должна раскрыть конфиденциальную информацию, она должна уведомить заказчика или иное заинтересованное лицо о раскрытой информации, в случае если это не запрещено законодательством.
4.2.3 Информация о заказчике, полученная не от самого заказчика (например, лица, направившего жалобу, регулирующих органов), должна быть конфиденциальной между заказчиком и лабораторией. Сведения о поставщике (источнике) этой информации должны быть конфиденциальными для лаборатории и не должны передаваться ее заказчику, если это не согласовано с источником данной информации.
4.2.4 Персонал, включая любых членов комитетов, подрядчиков, персонал внешних органов или отдельных лиц, действующих от имени лаборатории, должен соблюдать конфиденциальность всей информации, полученной или созданной в ходе выполнения лабораторной деятельности, за исключением случаев, предусмотренных законодательством.
5 Требования к структуре
5.1 Лаборатория должна быть юридическим лицом или подразделением юридического лица, которое несет юридическую ответственность за ее деятельность.
Примечание — Для целей настоящего стандарта правительственная лаборатория считается юридическим лицом на основе ее правительственного статуса.
5.2 Лаборатория должна определить руководство, которое несет полную ответственность за лабораторию.
5.3 Лаборатория должна определить и документировать область лабораторной деятельности, при осуществлении которой она соответствует настоящему стандарту. Область, в отношении которой лаборатория заявляет о соответствии настоящему стандарту, не должна включать лабораторную деятельность, осуществляемую на постоянной основе внешними поставщиками.
5.4 Лаборатория должна осуществлять свою деятельность таким образом, чтобы соответствовать требованиям настоящего стандарта, своих заказчиков, регулирующих органов и организаций, обеспечивающих признание. Лаборатория должна нести ответственность за деятельность, осуществляемую во всех местах ее постоянного размещения, вне мест ее постоянного размещения, на временных или передвижных площадях и на объектах заказчика.
5.5 Лаборатория должна:
a) определить организационную и управленческую структуру лаборатории, ее место в головной организации и взаимосвязи между управленческими, техническими и вспомогательными службами;
b) установить ответственность, полномочия и взаимоотношения всех сотрудников, занятых в управлении, выполнении или проверке работ, влияющих на результаты лабораторной деятельности;
c) документировать свои процедуры в объеме, необходимом для обеспечения стабильного осуществления своей деятельности и достоверности результатов.
5.6 Лаборатория должна иметь персонал, который, независимо от других обязанностей, имеет полномочия и ресурсы, необходимые для выполнения своих обязанностей, в том числе:
a) внедрение, поддержание и совершенствование системы менеджмента;
b) выявление отклонений от системы менеджмента или от процедур для осуществления лабораторной деятельности;
c) инициирование мер по предотвращению или минимизации таких отклонений;
d) представление руководству лаборатории отчетов о функционировании системы менеджмента и необходимости ее улучшения;
e) обеспечение результативности лабораторной деятельности.
5.7 Руководство лаборатории должно обеспечить:
a) обмен информацией о результативности системы менеджмента и важности удовлетворения требований заказчиков и других требований;
b) сохранение целостности системы менеджмента при планировании и внесении изменений в нее.
6 Требования к ресурсам
6.1 Общие требования
Лаборатория должна располагать персоналом, помещениями, оборудованием, системами и вспомогательными службами, необходимыми для управления лабораторной деятельностью и для ее осуществления.
6.2 Персонал
6.2.1 Весь персонал лаборатории, как постоянный, так и привлекаемый, который может повлиять на деятельность лаборатории, должен действовать беспристрастно, быть компетентным и должен работать в соответствии с системой менеджмента лаборатории.
6.2.2 Лаборатория должна документировать требования к компетентности персонала для каждой функции, влияющей на результаты лабораторной деятельности, в том числе требования к образованию, квалификации, профессиональной подготовке, техническим знаниям, навыкам, опыту.
6.2.3 Лаборатория должна гарантировать, что персонал обладает компетентностью для выполнения лабораторной деятельности, за которую он несет ответственность, и для оценки значимости отклонений.
6.2.4 Руководство лаборатории должно довести до каждого сотрудника его обязанности, ответственность и полномочия.
6.2.5 Лаборатория должна иметь процедуру(ы) и вести записи по:
a) определению требований к компетентности;
b) подбору персонала;
c) подготовке персонала;
d) наблюдению за персоналом;
e) наделению персонала полномочиями;
f) мониторингу компетентности персонала.
6.2.6 Лаборатория должна уполномочить персонал на выполнение конкретной лабораторной деятельности, включая (но не ограничиваясь) следующее:
a) разработку, изменение, верификацию и валидацию методов;
b) анализ результатов, в том числе заявлений о соответствии или мнений и интерпретаций;
c) подготовку отчетов о результатах, их проверку и утверждение.
6.3 Помещения и условия окружающей среды
6.3.1 Помещения и условия окружающей среды должны быть пригодными для осуществления лабораторной деятельности и не должны оказывать негативное влияние на достоверность получаемых результатов.
Примечание — Воздействия, которые могут негативно влиять на достоверность результатов, включают (но не ограничиваются) следующие: микробиологическое загрязнение, пыль, электромагнитные помехи, излучение, влажность, электроснабжение, температура, шум и вибрация.
6.3.2 Требования, предъявляемые к помещениям и условиям окружающей среды, необходимым для осуществления лабораторной деятельности, должны быть документированы.
6.3.3 Лаборатория должна осуществлять мониторинг условий окружающей среды, управление ими и их регистрацию в соответствии с техническими требованиями, методами и методиками или в случаях, когда они влияют на достоверность результатов.
6.3.4 Меры по управлению помещениями должны быть внедрены, подвергаться мониторингу и периодическому пересмотру и включать (но не ограничиваться) следующее:
a) доступ и использование участков, оказывающих влияние на лабораторную деятельность;
b) предотвращение загрязнений, взаимного влияния или неблагоприятных воздействий на лабораторную деятельность;
c) эффективное разграничение зон, в которых проводится несовместимая лабораторная деятельность.
6.3.5 При осуществлении лабораторией деятельности на объектах, находящихся вне ее постоянного управления, она должна обеспечить соответствие помещений и условий окружающей среды требованиям настоящего стандарта.
6.4 Оборудование
6.4.1 Лаборатория должна иметь доступ к оборудованию (включая, но не ограничиваясь, средства измерения, программное обеспечение, эталоны, стандартные образцы, справочные данные, реактивы, расходные материалы или вспомогательные устройства), которое необходимо для надлежащего осуществления лабораторной деятельности и которое может повлиять на ее результаты.
Примечание 1 — Существует множество названий для стандартных образцов и сертифицированных стандартных образцов, например эталоны, калибровочные эталоны, стандартные образцы, контрольные образцы. В ISO 17034 приведена дополнительная информация о производителях стандартных образцов (RMP). Производители стандартных образцов, соответствующих требованиям ISO 17034, считаются компетентными. Стандартные образцы от производителей, соответствующие требованиям ISO 17034, поставляются с паспортом/сертификатом, который определяет среди прочих характеристик однородность и стабильность для указанных свойств, а для сертифицированных стандартных образцов — указанные свойства с сертифицированными значениями, их неопределенность измерений и метрологическую прослеживаемость.
Примечание 2 — В ISO Guide 33 даны рекомендации по выбору и использованию стандартных образцов. В ISO Guide 80 приведены указания по изготовлению образцов, применяемых для внутреннего контроля качества.
6.4.2 В тех случаях, когда лаборатория использует оборудование, находящееся вне зоны ее постоянного управления, она должна обеспечить его соответствие требованиям настоящего стандарта.
6.4.3 Лаборатория должна иметь процедуры обращения с оборудованием, его транспортировки, хранения, эксплуатации и планового обслуживания в целях обеспечения надлежащего функционирования и предотвращения загрязнения или повреждения.
6.4.4 Лаборатория должна подтвердить соответствие оборудования установленным требованиям перед вводом его в эксплуатацию или после возврата в эксплуатацию.
6.4.5 Оборудование, используемое для измерений, должно обеспечивать точность и/или неопределенность измерений, требуемые для обеспечения достоверного результата.
6.4.6 Измерительное оборудование должно быть калибровано, если:
— точность и неопределенность измерений влияют на достоверность представляемых результатов; и/или
— калибровка оборудования требуется для установления метрологической прослеживаемости представляемых результатов.
Примечание — К видам оборудования, оказывающим влияние на достоверность представленных результатов, можно отнести оборудование, служащее для:
— прямого измерения определяемой величины, например применение весов для измерения массы;
— внесения поправок в измеренные значения, например измерения температуры;
— получения результата измерения путем вычислений на основе значений нескольких величин.
6.4.7 Лаборатория должна разработать программу калибровки, которая должна пересматриваться и корректироваться по мере необходимости с целью поддержания доверия к статусу калибровки.
6.4.8 Все оборудование, которое требует калибровки или имеет определенный срок годности, должно быть маркировано, закодировано или иным образом идентифицировано, чтобы позволить пользователю оборудования быстро идентифицировать статус калибровки или срок годности.
6.4.9 Оборудование, которое было подвергнуто перегрузке или неправильному обращению, выдает сомнительные результаты, а также в случае, если было замечено, что оно является дефектным или не соответствует установленным требованиям, должно быть выведено из эксплуатации. Оно должно быть изолировано, чтобы предотвратить его использование, или четко обозначено или промаркировано как неисправное, пока не будет проверено, что оно работает правильно. Лаборатория должна исследовать влияние дефекта или отклонения от установленных требований и приступить к рабочей процедуре по управлению несоответствующей работой (см. 7.10).
6.4.10 Если промежуточные проверки необходимы для поддержания уверенности в исправности оборудования, то эти проверки должны проводиться в соответствии с установленной процедурой.
6.4.11 Если результаты калибровки и сведения о стандартных образцах включают в себя опорные значения или поправочные коэффициенты, то лаборатория должна обеспечить, что опорные значения и поправочные коэффициенты обновляются и применяются должным образом в соответствии с установленными требованиями.
6.4.12 Лаборатория должна принимать практические меры по предотвращению непреднамеренных регулировок оборудования, которые могут привести к признанию результатов недействительными.
6.4.13 Должны вестись записи о состоянии оборудования, которое может повлиять на лабораторную деятельность. Записи должны включать следующее, когда это применимо:
a) идентификацию оборудования, включая версию программного обеспечения, в том числе встроенного;
b) наименование изготовителя, идентификацию типа, серийный номер или другую уникальную идентификацию;
c) данные верификации о том, что оборудование соответствует установленным требованиям;
d) текущее местонахождение;
e) даты и результаты калибровок, регулировок, критерии приемки и планируемую дату следующей калибровки или межкалибровочный интервал;
f) документацию на стандартные образцы, результаты, критерии приемки, соответствующие даты и сроки годности;
g) план технического обслуживания и техническое обслуживание, выполненное к настоящему моменту времени, если это требуется для работы оборудования;
h) подробную информацию о любых повреждениях, неисправностях, модификациях или ремонте оборудования.
6.5 Метрологическая прослеживаемость
6.5.1 Лаборатория должна установить и поддерживать метрологическую прослеживаемость результатов своих измерений, связывая их с соответствующей основой для сравнения посредством документированной непрерывной цепи калибровок, каждая из которых вносит свой вклад в неопределенность измерений.
Примечание 1 — В ISO/IEC Guide 99 метрологическая прослеживаемость определяется как «свойство результата измерения, в соответствии с которым результат может быть соотнесен с основой для сравнения посредством документированной непрерывной цепи калибровок, каждая из которых вносит вклад в неопределенность измерений».
Примечание 2 — См. приложение А для получения дополнительной информации о метрологической прослеживаемости.
6.5.2 Лаборатория должна обеспечить прослеживаемость результатов измерений к Международной системе единиц (СИ) посредством:
a) калибровки, предоставляемой компетентной лабораторией; или
Примечание 1 — Лаборатории, удовлетворяющие требованиям настоящего стандарта, считаются компетентными.
b) сертифицированных значений сертифицированных стандартных образцов компетентного производителя с указанной метрологической прослеживаемостью к СИ; или
Примечание 2 — Производители стандартных образцов, выполняющие требования стандарта ISO 17034, считаются компетентными.
c) непосредственной реализации единиц СИ, подтвержденной сличениями, прямыми или косвенными, с национальными или международными эталонами.
Примечание 3 — Подробная информация о практической реализации определений некоторых важнейших единиц приведена в Брошюре СИ.
6.5.3 Если установление метрологической прослеживаемости к единицам СИ с технической точки зрения не представляется возможным, лаборатория должна продемонстрировать метрологическую прослеживаемость к соответствующей основе для сравнения, например к:
a) сертифицированным значениям сертифицированных стандартных образцов, предоставленных компетентным изготовителем;
b) результатам, полученным с применением референтных методик измерений, установленных методов или согласованных стандартов (эталонов), если они четко описаны и признаны в качестве обеспечивающих результаты измерений, которые отвечают своему предполагаемому назначению и подтверждаются соответствующими сличениями.
6.6 Продукция и услуги, предоставляемые внешними поставщиками
6.6.1 Лаборатория должна обеспечить пригодность используемых продукции и услуг, предоставляемых внешними поставщиками, которые влияют на деятельность лаборатории, когда они:
a) предназначены для использования в собственной лабораторной деятельности;
b) предоставляются лабораторией, частично или полностью, напрямую заказчику в том состоянии, в котором они были получены от внешнего поставщика;
c) используются для поддержания работы лаборатории.
Примечание — Продукция может включать, например, эталоны и оборудование, вспомогательные устройства, расходные материалы и стандартные образцы. Услуги могут включать, например, услуги по калибровке, отбору образцов, испытаниям, обслуживанию помещений и оборудования, проверке квалификации, оценке и аудиту.
6.6.2 Лаборатория должна иметь процедуры и вести записи для:
a) определения, рассмотрения и утверждения требований лаборатории к продукции и услугам, предоставляемым внешними поставщиками;
b) определения критериев для оценивания, выбора, мониторинга деятельности и периодического оценивания внешних поставщиков;
c) обеспечения того, чтобы продукция и услуги, поставляемые внешними поставщиками, соответствовали установленным требованиям лаборатории или, когда это применимо, требованиям настоящего стандарта, прежде чем они будут использованы в работе или непосредственно переданы заказчику;
d) осуществления каких-либо действий по результатам оценивания, мониторинга деятельности и периодического оценивания внешних поставщиков.
6.6.3 Лаборатория должна информировать внешних поставщиков о своих требованиях в отношении:
a) предоставляемых продукции и услуг;
b) критериев приемки;
c) компетентности, включая требования к квалификации персонала;
d) деятельности, которую лаборатория или ее заказчик намерены осуществить на территории внешнего поставщика.
7 Требования к процессу
7.1 Рассмотрение запросов, тендеров и договоров
7.1.1 Лаборатория должна иметь процедуру для рассмотрения запросов, тендеров и договоров. Процедура должна обеспечивать, что:
a) требования надлежащим образом определены, документированы и правильно понимаются;
b) лаборатория располагает возможностями и ресурсами для выполнения требований;
c) в случае привлечения внешних поставщиков выполняются требования 6.6 и лаборатория предлагает заказчику, чтобы конкретная лабораторная деятельность была выполнена внешним поставщиком, и получает одобрение заказчика.
Примечание 1 — Принято, что лабораторная деятельность может осуществляться внешним поставщиком в тех случаях, когда:
— лаборатория располагает ресурсами и компетентностью для осуществления деятельности, однако в силу непредвиденных обстоятельств она не в состоянии выполнить ее частично или полностью;
— лаборатория не располагает ресурсами или компетентностью для осуществления деятельности.
d) выбраны соответствующие методы или методики и они способны удовлетворить требования заказчиков.
Примечание 2 — Для внутренних или постоянных заказчиков рассмотрение запросов, тендеров и договоров может быть выполнено в упрощенном виде.
7.1.2 Лаборатория должна информировать заказчика, когда метод, запрашиваемый заказчиком, является неприменимым или устаревшим.
7.1.3 Когда заказчик запрашивает заключение о соответствии спецификации или стандарту на испытания или калибровку (например, годен/не годен, в пределах допуска/за пределами допуска), то спецификация или стандарт и правила принятия решений должны быть четко определены. Если правило принятия решения не определено в спецификации или стандарте, то оно должно быть сообщено заказчику и согласовано с ним.
Примечание — Дополнительную информацию о заявлениях о соответствии см. в ISO/IEC Guide 98-4.
7.1.4 Любые разногласия между запросом или тендером и договором должны быть устранены до начала лабораторной деятельности. Каждый договор должен быть приемлемым как для лаборатории, так и для заказчика. Отклонения от положений договора по запросу заказчика не должны влиять на объективность лаборатории или достоверность ее результатов.
7.1.5 Заказчик должен быть проинформирован о любом отклонении от условий договора.
7.1.6 Если в договор вносятся изменения после того, как работа началась, анализ договора должен быть проведен повторно и любые изменения должны быть доведены до сведения всех сотрудников, на деятельность которых влияют данные изменения.
7.1.7 Лаборатория должна сотрудничать с заказчиками или их представителями для уточнения запросов заказчика и наблюдения за деятельностью лаборатории, выполняющей работу.
Примечание — Такое сотрудничество может включать:
a) обеспечение приемлемого доступа к соответствующим зонам лаборатории для наблюдения за лабораторной деятельностью, выполняемой для конкретного заказчика;
b) подготовку, упаковку и отправку объектов, необходимые заказчику с целью проверки.
7.1.8 Записи по анализу, включая любые значительные изменения, должны сохраняться. Также должны сохраняться записи соответствующих переговоров с заказчиком, касающиеся требований заказчика или результатов лабораторной деятельности.
7.2 Выбор, верификация и валидация методов
7.2.1 Выбор и верификация методов
7.2.1.1 Лаборатория должна применять соответствующие методы и методики для всех видов лабораторной деятельности и при необходимости для оценивания неопределенности измерений, а также статистические методы для анализа данных.
Примечание — Термин «метод», используемый в настоящем стандарте, и термин «методика измерений», приведенный в ISO/IEC Guide 99, могут рассматриваться как синонимы.
7.2.1.2 Все методы, методики и сопутствующие документы, такие как инструкции, стандарты, руководства по эксплуатации и справочные данные, имеющие отношение к лабораторной деятельности, должны поддерживаться в актуальном состоянии и быть легкодоступными для персонала (см. 8.3).
7.2.1.3 Лаборатория должна обеспечить применение последней действующей редакции метода, за исключением случаев, когда ее применение является нецелесообразным или невозможным. При необходимости для применения метода должны быть разработаны дополнительные уточнения, чтобы обеспечить его непротиворечивое применение.
Примечание — Международные, региональные или национальные стандарты или другие признанные технические требования, содержащие достаточную и точную информацию о том, как осуществлять лабораторную деятельность, не требуется дополнять или переписывать в качестве внутренних процедур лаборатории, если эти стандарты написаны таким образом, что могут применяться производственным персоналом лаборатории. Для вариативных этапов метода или для дополнительного подробного описания может потребоваться предоставление дополнительной документации.
7.2.1.4 Когда заказчик не определяет метод, который необходимо применять, лаборатория должна выбрать подходящий метод самостоятельно и проинформировать об этом заказчика. Рекомендуется использовать методы, опубликованные в международных, региональных или национальных стандартах, либо рекомендованные авторитетными техническими организациями, либо описанные в соответствующих научных статьях или журналах, либо установленные изготовителем оборудования. Также могут применяться методы, разработанные лабораторией или модифицированные.
7.2.1.5 До внедрения методов в работу лаборатория должна подтвердить, что она может надлежащим образом применять выбранные методы, обеспечивая требуемое исполнение. Записи о верификации должны сохраняться. Если изменения в метод были внесены организацией-разработчиком, то верификация должна быть проведена повторно в необходимом объеме.
7.2.1.6 При необходимости разработки нового метода должен быть составлен план работ и назначен квалифицированный персонал, обеспеченный необходимыми ресурсами. В процессе разработки метода должна проводиться периодическая оценка работ с целью подтверждения того, что требования заказчика все еще выполняются. Любые изменения, вносимые в план работ, должны быть одобрены и утверждены.
7.2.1.7 Отклонение от методов для всех видов лабораторной деятельности должно допускаться только тогда, когда это отклонение оформлено документально, технически обосновано, утверждено и принято заказчиком.
Примечание — Согласие заказчика на отклонения может быть заранее оговорено в договоре.
7.2.2 Валидация методов
7.2.2.1 Лаборатория должна проводить валидацию нестандартных методов, методов, разработанных лабораторией, и стандартных методов, используемых за пределами их области применения или каким-либо иным образом модифицированных. Валидация должна быть настолько полной, насколько это необходимо, чтобы отвечать потребностям данного применения или области применения.
Примечание 1 — Валидация может включать процедуры отбора образцов, обращения с объектами испытания или калибровки и их транспортировки.
Примечание 2 — Для валидации метода может применяться один из следующих способов либо их комбинация:
a) калибровка или оценивание смещения и прецизионности с использованием эталонов или стандартных образцов;
b) систематическая оценка факторов, влияющих на результат;
c) проверка устойчивости метода посредством изменения управляемых параметров, таких как температура в термостате, дозируемый объем;
d) сравнение с результатами, полученными с помощью других валидированных методов;
e) межлабораторные сличения;
f) оценивание неопределенности измерений, связанной с результатами измерений, на основании понимания теоретических принципов метода и опыта его реализации при отборе образцов или проведении испытаний.
7.2.2.2 При внесении изменений в валидированный метод их влияние должно быть определено и, в случае если было установлено, что они оказывают влияние на первоначальную валидацию, должна быть выполнена новая валидация метода.
7.2.2.3 Характеристики валидированных методов, оцененные для предполагаемого использования, должны соответствовать потребностям заказчиков и установленным требованиям.
Примечание — Характеристики метода могут включать (но не ограничиваться) диапазон измерений, точность, неопределенность результатов измерений, предел обнаружения, предел количественного определения, избирательность метода, линейность, повторяемость или воспроизводимость, устойчивость к внешним воздействиям или эффектам влияния матрицы образца или испытываемого объекта и смещение.
7.2.2.4 Лаборатория должна сохранять следующие записи о валидации:
a) использованную процедуру валидации;
b) перечень требований;
c) определение характеристик метода;
d) полученные результаты;
e) заключение о пригодности метода вместе с подробным описанием его соответствия в отношении предполагаемого использования.
7.3 Отбор образцов
7.3.1 В случае когда лаборатория проводит отбор образцов веществ, материалов или продукции для последующих испытаний или калибровки, она должна иметь план и методы их отбора. Метод отбора образцов должен учитывать факторы, которые необходимо контролировать, чтобы обеспечить достоверность результатов последующих испытаний или калибровки. План и метод отбора образцов должны быть доступны на месте проведения отбора. Планы отбора образцов должны основываться, когда это целесообразно, на соответствующих статистических методах.
7.3.2 Методы отбора образцов должны описывать:
a) выбор образцов или точек отбора;
b) план отбора образцов;
c) подготовку или обработку образца(ов) вещества, материала или продукции с целью получения требуемого образца для последующего проведения испытаний или калибровки.
Примечание — После того как образец получен лабораторией, дальнейшее обращение, которое может потребоваться, описано в 7.4.
7.3.3 Лаборатория должна сохранять соответствующие записи об отборе образцов, который составляет часть проведенных испытаний или калибровки. Такие записи должны включать в себя (если применимо):
a) ссылку на примененный метод отбора образцов;
b) дату и время отбора образцов;
c) данные для идентификации и описания образца (например, номер, количество, наименование);
d) идентификацию лица, выполнившего отбор образцов;
e) идентификацию использованного оборудования;
f) условия окружающей среды и транспортировки;
g) схемы или другие эквивалентные способы идентификации места отбора образцов, если это необходимо;
h) отклонения, дополнения или исключения из метода и плана отбора образцов.
7.4 Обращение с объектами испытаний или калибровки
7.4.1 Лаборатория должна иметь процедуру для транспортировки, получения объектов испытаний или калибровки, обращения с объектами испытаний или калибровки, защиты, хранения, обеспечения сохранности, уничтожения или возврата объектов испытаний или калибровки, включая все условия, необходимые для защиты целостности объектов испытаний или калибровки и защиты интересов лаборатории и заказчика. Должны быть предприняты меры предосторожности, чтобы избежать ухудшения свойств, загрязнения, потери или повреждения объектов при обращении, транспортировке, хранении/ожидании и подготовке к испытаниям или калибровке. Инструкции по обращению с объектами, предоставленные вместе с ними, должны быть соблюдены.
7.4.2 Лаборатория должна иметь четкую систему идентификации объектов испытаний или калибровки. Идентификация должна сохраняться, пока объект находится под ответственностью лаборатории. Данная система должна обеспечивать, что объекты не будут перепутаны физически или при ссылке на них в записях или других документах. Система идентификации должна, если это необходимо, учитывать разделение объекта или группы объектов и их перемещение.
7.4.3 При получении объекта испытаний или калибровки отклонения от установленных условий должны быть зарегистрированы. Если есть сомнения относительно пригодности объекта для испытания или калибровки или если объект не соответствует представленному описанию, то лаборатория, перед тем как продолжить работу, должна обратиться к заказчику за дополнительными инструкциями и зарегистрировать результаты этого обсуждения. Если заказчик требует провести испытание или калибровку какого-либо объекта, признавая отклонение от установленных условий, лаборатория должна включить в отчет заключение о том, на какие результаты могут повлиять данные отклонения.
7.4.4 В случае если объекты необходимо хранить или кондиционировать при определенных условиях окружающей среды, эти условия должны поддерживаться, контролироваться и регистрироваться.
7.5 Технические записи
7.5.1 Лаборатория должна обеспечивать наличие в технических записях для каждого вида лабораторной деятельности результатов, отчета и достаточной информации, позволяющей, если это возможно, идентифицировать факторы, влияющие на результат измерения и связанную с ним неопределенность измерений, а также обеспечить возможность повторного проведения данной лабораторной деятельности в условиях, максимально близких к первоначальным. Технические записи должны включать дату и сведения о персонале лаборатории, который несет ответственность за каждый вид лабораторной деятельности и за проверку данных и результатов. Первичные наблюдения, данные и расчеты должны быть записаны в момент, когда они были получены, и должны отождествляться с конкретной работой.
7.5.2 Лаборатория должна обеспечивать прослеживаемость изменений, вносимых в технические записи, к предыдущим версиям либо к первичным наблюдениям. И первичные, и измененные данные и файлы должны сохраняться с указанием даты внесения изменений, сведений об аспектах, претерпевших изменения, и лицах, ответственных за данные изменения.
7.6 Оценивание неопределенности измерений
7.6.1 Лаборатории должны определять вклад(ы) в неопределенность измерений. При оценивании неопределенности измерений все существенные вклады, в том числе связанные с отбором образцов, должны учитываться с применением соответствующих методов анализа.
7.6.2 Лаборатория, выполняющая калибровки, в том числе собственного оборудования, должна оценивать неопределенность измерений для всех калибровок.
7.6.3 Лаборатория, выполняющая испытания, должна оценивать неопределенность измерений. В тех случаях, когда метод испытаний исключает строгую оценку неопределенности измерений, оценивание должно проводиться на основе понимания теоретических принципов или практического опыта выполнения метода.
Примечание 1 — В случае если хорошо известный метод испытаний устанавливает пределы значений основных источников неопределенности измерений и указывает форму представления результатов вычислений, считается, что лаборатория выполнила требования 7.6.3, следуя методу испытаний и инструкции по представлению результатов.
Примечание 2 — При использовании конкретного метода, для которого неопределенность результатов измерений уже была установлена и подтверждена, нет необходимости оценивать неопределенность измерений для каждого результата, если лаборатория может продемонстрировать, что выявленные критические факторы, оказывающие влияние, находятся под контролем.
Примечание 3 — Для подробной информации см. ISO/IEC Guide 98-3, ISO 21748 и стандарты серии ISO 5725.
7.7 Обеспечение достоверности результатов
7.7.1 Лаборатория должна иметь процедуру для мониторинга достоверности результатов своей деятельности. Полученные данные должны регистрироваться таким образом, чтобы можно было выявить тенденции, и там, где это практически возможно, должны применяться статистические методы для анализа результатов. Должен быть составлен план такого мониторинга и проводиться его анализ. Мониторинг должен включать (но не ограничиваться), где целесообразно, следующее:
a) использование стандартных образцов или образцов для контроля качества;
b) использование альтернативного оборудования, которое было калибровано, для обеспечения прослеживаемости результатов;
c) проверку(и) функционирования измерительного и испытательного оборудования;
d) использование контрольных или рабочих эталонов с ведением контрольных карт, где это применимо;
e) промежуточные проверки измерительного оборудования;
f) повторные испытания или калибровки с использованием одного и того же или различных методов;
g) повторные испытания или повторную калибровку хранящихся образцов;
h) корреляцию результатов для различных характеристик образца;
i) анализ полученных данных;
j) внутрилабораторные сличения;
k) испытания шифрованного(ых) образца(ов).
7.7.2 Лаборатория должна осуществлять мониторинг своей деятельности путем сравнения с результатами других лабораторий, если это возможно и применимо. Такой мониторинг должен планироваться, и его результаты должны анализироваться. Он должен включать (но не ограничиваться) следующие мероприятия или одно из них:
a) участие в проверках квалификации.
Примечание — В ISO/IEC 17043 приведена дополнительная информация о проверках квалификации и о провайдерах проверки квалификации. Провайдеры проверки квалификации, которые выполняют требования ISO/IEC 17043, считаются компетентными;
b) участие в межлабораторных сличениях, отличных от проверок квалификации.
7.7.3 Данные, полученные с помощью мониторинга, должны анализироваться и использоваться для управления лабораторной деятельностью, а также по возможности для внесения улучшений в работу лаборатории. Если было обнаружено, что результаты анализа данных, полученных при мониторинге, выходят за рамки установленных критериев, необходимо предпринять соответствующие действия с целью предотвращения включения в отчетную документацию неверных результатов.
7.8 Представление отчетов о результатах
7.8.1 Общие положения
7.8.1.1 Результаты должны быть рассмотрены и утверждены до их выдачи.
7.8.1.2 Результаты должны быть представлены точно, четко, недвусмысленно и объективно, как правило, в форме отчета (например, отчет об испытаниях, свидетельство (сертификат) о калибровке или акт отбора образцов) и должны включать в себя всю информацию, согласованную с заказчиком и необходимую для интерпретации результатов, а также всю информацию, требуемую в соответствии с применяемым методом. Все оформленные отчеты должны быть сохранены в качестве технических записей.
Примечание 1 — Для целей настоящего стандарта отчеты об испытаниях и свидетельства (сертификаты) о калибровке иногда могут называться протоколами испытаний и отчетами о калибровке соответственно.
Примечание 2 — Отчеты могут быть изданы на бумажном носителе или с помощью электронных средств при условии, что требования настоящего стандарта выполнены.
7.8.1.3 При согласовании с заказчиком результаты могут быть представлены в упрощенном виде. Любые сведения, указанные в 7.8.2-7.8.7, которые не были представлены заказчику, должны быть легкодоступными.
7.8.2 Общие требования к отчетам (об испытаниях, калибровке или отборе образцов)
7.8.2.1 В целях минимизации возможности неправильного понимания или неправильного использования информации каждый отчет должен включать как минимум следующую информацию, если у лаборатории нет обоснованных причин не выполнять это требование:
a) название (например, «Отчет об испытаниях», «Свидетельство (сертификат) о калибровке» или «Акт отбора образцов»);
b) наименование и адрес лаборатории;
c) место осуществления лабораторной деятельности, в том числе если она осуществлялась на площадях заказчика, либо на участках, удаленных от постоянных производственных площадей лаборатории, либо на соответствующих временно используемых или мобильных объектах;
d) уникальную идентификацию, для того чтобы все его составляющие воспринимались как часть общего отчета, и четкую идентификацию конца отчета;
e) наименование и контактные данные заказчика;
f) идентификацию применяемого метода;
g) описание, однозначную идентификацию и при необходимости состояние образца;
h) дату получения образца(ов) для испытаний или объекта калибровки и дату отбора образца(ов), когда это имеет важное значение для достоверности и применения результатов;
i) дату(ы) осуществления лабораторной деятельности;
j) дату выдачи отчета;
k) ссылку на план и метод отбора образцов, использованные лабораторией или другими органами, если это важно для достоверности или применения результатов;
I) заявление о том, что результаты относятся только к объектам, прошедшим испытания, калибровку или отбор;
m) результаты, где это применимо, с единицами измерения;
n) дополнения, отклонения или исключения из метода;
o) идентификацию лиц(а), утвердивших(его) отчет;
p) однозначную идентификацию результатов, полученных от внешних поставщиков.
Примечание — Включение заявления о том, что отчет не должен быть воспроизведен не в полном объеме без разрешения лаборатории, может обеспечить уверенность в том, что части отчета не интерпретируются вне контекста.
7.8.2.2 Лаборатория должна нести ответственность за всю информацию, представленную в отчете, за исключением случаев, когда информация предоставляется заказчиком. Данные, предоставленные заказчиком, должны быть четко идентифицированы. Кроме того, в случае если информация предоставлена заказчиком и она может повлиять на достоверность результатов, в отчет должно быть включено заявление об ограничении ответственности лаборатории. В случае если лаборатория не осуществляет и не несет ответственности за стадию отбора образцов (например, образец был предоставлен заказчиком), в отчете должно быть отражено, что полученные результаты относятся к предоставленному заказчиком образцу.
7.8.3 Специальные требования к отчетам об испытаниях
7.8.3.1 В дополнение к требованиям, перечисленным в 7.8.2, отчеты об испытаниях должны, если это необходимо для интерпретации результатов испытаний, включать в себя следующее:
a) информацию об особых условиях испытаний, таких как условия окружающей среды;
b) при необходимости заявление о соответствии требованиям или спецификациям (см. 7.8.6);
c) где это применимо, неопределенность измерений, представленную в тех же единицах, что и измеряемая величина, или в относительном по отношению к измеряемой величине виде (например, в процентах), когда:
— это имеет отношение к достоверности или применению результатов испытаний;
— этого требует заказчик; или
— неопределенность измерения влияет на соответствие установленному пределу;
d) мнения и интерпретации, где это применимо (см. 7.8.7);
e) дополнительную информацию, которая может потребоваться по конкретным методам, органам власти, заказчикам или группам заказчиков.
7.8.3.2 Если лаборатория несет ответственность за деятельность по отбору образцов, отчеты об испытаниях должны соответствовать требованиям, указанным в 7.8.5, если это необходимо для интерпретации результатов испытаний.
7.8.4 Специальные требования к свидетельствам (сертификатам) о калибровке
7.8.4.1 В дополнение к требованиям, перечисленным в 7.8.2, в свидетельства (сертификаты) о калибровке должны быть включены следующие сведения:
a) значение неопределенности измерений для результата измерений, представленное в тех же единицах, что и измеряемая величина, или в относительном по отношению к измеряемой величине виде (например, в процентах).
Примечание — В соответствии с ISO/IEC Guide 99 результат измерения, как правило, выражается одним измеренным значением величины с указанием единицы измерения и неопределенности измерений;
b) сведения об условиях (например, условиях окружающей среды), при которых выполнялись калибровки и которые могли оказать влияние на результаты измерений;
c) заявление о том, каким образом обеспечивается метрологическая прослеживаемость измерений (см. приложение А);
d) результаты, полученные до и после регулировки или ремонта, если таковые проводились;
e) заявление о соответствии требованиям или спецификациям при необходимости (см. 7.8.6);
f) мнения и интерпретации (см. 7.8.7) при необходимости.
7.8.4.2 Если лаборатория отвечает за деятельность по отбору образцов, свидетельства (сертификаты) о калибровке должны соответствовать требованиям, указанным в 7.8.5, если это необходимо для интерпретации результатов калибровки.
7.8.4.3 Свидетельство (сертификат) о калибровке или калибровочная этикетка не должны содержать никаких рекомендаций по выбору межкалибровочных интервалов, кроме тех случаев, когда это было согласовано с заказчиком.
7.8.5 Представление результатов по отбору образцов — специальные требования
Если лаборатория несет ответственность за деятельность по отбору образцов, в дополнение к требованиям, перечисленным в 7.8.2, когда это необходимо для интерпретации результатов, отчеты должны включать следующее:
a) дату отбора образцов;
b) уникальную идентификацию выбранного образца или материала (включая наименование производителя, обозначение модели или типа и серийные номера, когда это применимо);
c) место отбора образцов, включая любые диаграммы, эскизы или фотографии;
d) ссылку на план отбора и метод отбора;
e) сведения обо всех условиях окружающей среды во время отбора образцов, которые влияют на интерпретацию результатов;
f) информацию, необходимую для оценки неопределенности измерений для последующих испытаний или калибровки.
7.8.6 Представление заключений о соответствии
7.8.6.1 Если по результатам испытания или калибровки делается заключение о соответствии спецификации или стандарту, лаборатория должна документировать правило принятия решения, принимая во внимание уровень риска (например, ложноположительное или ложноотрицательное решение, статистические предположения), связанный с применяемым правилом принятия решения, и применить данное правило.
Примечание — Если правило принятия решения установлено заказчиком, правилами или нормативными документами, дальнейшее рассмотрение уровня риска не требуется.
7.8.6.2 Лаборатория должна представить заключение о соответствии, в котором четко определено:
a) к каким результатам применяется данное заключение;
b) каким спецификациям, стандартам или их частям соответствует или не соответствует объект;
c) правило принятия решения, которое было использовано (если оно не содержится в соответствующих спецификации или стандарте).
Примечание — Для получения дополнительной информации см. ISO/IEC Guide 98-4.
7.8.7 Представление мнений и интерпретаций
7.8.7.1 В случае представления мнений и интерпретаций лаборатория должна обеспечить, что только персонал, уполномоченный на представление мнений и интерпретаций, представляет соответствующие заявления. Лаборатория должна документировать основания, на которых базируются включенные в отчет мнения и интерпретации.
Примечание — Важно отличать мнения и интерпретации от выводов по результатам инспекций или сертификации продукции, как это установлено в ISO/IEC 17020 и ISO/IEC 17065, и от заявлений о соответствии, упомянутых в 7.8.6.
7.8.7.2 Мнения или интерпретации, содержащиеся в отчетах, должны быть основаны на результатах, полученных для объекта, проходившего испытания/калибровку, и должны быть четко обозначены как таковые.
7.8.7.3 Если мнения и интерпретации представляются путем непосредственного обсуждения результатов с заказчиком, необходимо сохранять соответствующие записи такого обсуждения.
7.8.8 Изменения к отчетам
7.8.8.1 Если необходимо внести изменения, дополнения в выданный отчет, переоформить его, любое изменение информации должно быть четко обозначено и, если необходимо, причина внесения изменения должна быть включена в отчет.
7.8.8.2 Изменения в отчет после его выдачи должны вноситься только в виде дополнительного документа или иного способа передачи данных, которые включают формулировку: «Изменение к отчету, порядковый номер… [или иная идентификация]» или другую подобную формулировку.
Такие изменения должны соответствовать всем требованиям настоящего стандарта.
7.8.8.3 Когда необходимо выдать новый отчет о результатах, он должен быть уникальным образом идентифицирован и содержать ссылку на отчет о результатах, который он заменяет.
7.9 Жалобы (претензии)
7.9.1 Лаборатория должна иметь документированный процесс для получения, рассмотрения жалоб (претензий) и принятия решений по жалобам (претензиям).
7.9.2 Описание процесса обращения с жалобами (претензиями) должно быть доступно всем заинтересованным сторонам по запросу. При получении жалобы (претензии) лаборатория должна удостовериться, относится ли жалоба (претензия) к лабораторной деятельности, за которую она несет ответственность, и, если это так, должна принять ее на рассмотрение. Лаборатория несет ответственность за принятие всех решений на всех стадиях процесса рассмотрения жалобы (претензии).
7.9.3 Процесс рассмотрения жалоб (претензий) должен включать по крайней мере следующие элементы и методы:
a) описание процесса получения, проверки, рассмотрения жалобы (претензии) и принятия решения об ответных действиях, которые должны быть предприняты;
b) отслеживание и регистрация жалоб (претензий), включая действия, предпринятые для их разрешения;
c) обеспечение того, что необходимые меры предпринимаются.
7.9.4 Лаборатория, получающая жалобу (претензию), должна нести ответственность за сбор и проверку всей необходимой информации с целью подтверждения обоснованности жалобы (претензии).
7.9.5 Когда это возможно, лаборатория должна подтвердить получение жалобы (претензии) и информировать заявителя о ходе и результатах рассмотрения.
7.9.6 Результаты рассмотрения жалобы (претензии), которые будут доведены до заявителя, должны быть подготовлены или рассмотрены и одобрены лицом(ами), которое(ые) не принимало(и) участия в деятельности лаборатории, по поводу которой поступила жалоба (претензия).
Примечание — Это может выполняться внешним персоналом.
7.9.7 Когда это возможно, лаборатория должна предоставить заявителю официальное уведомление о результатах рассмотрения жалобы (претензии).
7.10 Управление несоответствующей работой
7.10.1 Лаборатория должна иметь процедуру, которую применяют в случае, если какой-либо аспект лабораторной деятельности или результаты работы не соответствуют собственным процедурам лаборатории или требованиям, согласованным с заказчиком (например, оборудование или условия окружающей среды не соответствуют установленным пределам, результаты мониторинга не отвечают установленным критериям). Процедура должна обеспечивать, что:
a) определены ответственность и полномочия для управления несоответствующей работой;
b) действия (включая приостановку или повтор работы и приостановку выдачи отчетов, если необходимо) основаны на уровнях риска, установленных лабораторией;
c) проведено оценивание значимости несоответствующей установленным требованиям работы, в том числе анализ ее воздействия на предыдущие результаты;
d) принято решение о приемлемости несоответствующей работы;
e) когда это необходимо, уведомлен заказчик и аннулированы результаты работы;
f) определена ответственность за принятие решения о возобновлении работы.
7.10.2 Лаборатория должна вести записи в отношении несоответствующей работы и необходимых действий, как указано в 7.10.1, перечисления b)-f).
7.10.3 В тех случаях, когда оценка показывает, что несоответствующая работа может повториться, или есть сомнения в отношении соответствия деятельности лаборатории собственной системе менеджмента, лаборатория должна обеспечить выполнение корректирующих действий.
7.11 Управление данными и информацией
7.11.1 Лаборатория должна иметь доступ ко всем данным и информации, необходимым для выполнения лабораторной деятельности.
7.11.2 Правильность функционирования систем(ы) управления информацией лаборатории, используемых(ой) для сбора, обработки, записи, представления результатов, хранения или поиска данных, в том числе правильность функционирования интерфейсов систем(ы) управления информацией лаборатории, должна(ы) быть проверена лабораторией перед внедрением в работу. При любых изменениях, включая изменения конфигурации программного обеспечения лаборатории или модификации коммерческого программного обеспечения, они должны быть утверждены, документированы и валидированы до введения их в действие.
Примечание 1 — В настоящем стандарте «системы управления информацией лаборатории» включают в себя управление данными и информацией, содержащимися как в компьютеризированных, так и в некомпьютеризированных системах. Некоторые из требований могут быть в большей степени применимы к компьютеризированным системам, чем к некомпьютеризированным системам.
Примечание 2 — Доступное на рынке коммерческое программное обеспечение при обычном его использовании в области, для которой оно предназначено, может считаться в достаточной степени валидированным.
7.11.3 Система(ы) управления информацией лаборатории, должна(ы):
a) быть защищена(ы) от несанкционированного доступа;
b) быть защищена(ы) от искажения или потери данных;
c) функционировать в условиях окружающей среды, которые соответствуют спецификациям поставщика или лаборатории, или, в случае некомпьютеризированных систем, создавать условия, обеспечивающие неизменность выполненных от руки записей и расшифровки;
d) поддерживаться в таком состоянии, которое обеспечивает целостность данных и информации;
e) включать регистрацию системных сбоев и соответствующих оперативных и корректирующих действий.
7.11.4 В том случае, если управление данной системой и ее поддержание осуществляется дистанционно или через внешнего поставщика, лаборатория должна обеспечить соответствие поставщика или оператора системы всем применимым требованиям настоящего стандарта.
7.11.5 Лаборатория должна обеспечивать, чтобы инструкции, руководства и справочные данные, относящиеся к системе(ам) управления информацией лаборатории, были легкодоступными для персонала.
7.11.6 Расчеты и передачи данных должны подвергаться надлежащим систематическим проверкам.
8 Требования к системе менеджмента
8.1 Варианты
8.1.1 Общие положения
Лаборатория должна установить, документировать, внедрить и поддерживать систему менеджмента, которая способна обеспечивать и демонстрировать постоянное выполнение требований настоящего стандарта и обеспечивать качество выполненных лабораторией работ. В дополнение к соответствию требованиям разделов 4-7 лаборатория должна внедрить систему менеджмента в соответствии с вариантом А или вариантом В.
Примечание — См. приложение В для дополнительной информации.
8.1.2 Вариант А
Как минимум система менеджмента лаборатории должна предусматривать следующее:
— документацию системы менеджмента (см. 8.2);
— управление документами системы менеджмента (см. 8.3);
— управление записями (см. 8.4);
— действия, связанные с рисками и возможностями (см. 8.5);
— улучшения (см. 8.6);
— корректирующие действия (см. 8.7);
— внутренние аудиты (см. 8.8);
— анализ со стороны руководства (см. 8.9).
8.1.3 Вариант В
Лаборатория, которая установила и поддерживает систему менеджмента в соответствии с требованиями ISO 9001 и способна подтверждать и демонстрировать постоянное выполнение требований разделов 4-7, также демонстрирует как минимум готовность выполнять требования, содержащиеся в 8.2-8.9.
8.2 Документация системы менеджмента (вариант А)
8.2.1 Руководство лаборатории должно установить, документировать и поддерживать политики и цели для выполнения требований настоящего стандарта и должно обеспечить, чтобы эти политики и цели были признаны и внедрены на всех уровнях организации лаборатории.
8.2.2. Политики и цели должны быть направлены на обеспечение компетентности, беспристрастности и стабильности деятельности лаборатории.
8.2.3 Руководство лаборатории должно представить доказательства приверженности к разработке и внедрению системы менеджмента и постоянному повышению ее результативности.
8.2.4 Вся документация, процессы, системы, записи, относящиеся к выполнению требований настоящего стандарта, должны быть включены в систему менеджмента, соотнесены или связаны с ней.
8.2.5 Весь персонал, участвующий в лабораторной деятельности, должен иметь доступ к тем частям документации системы менеджмента и соответствующей информации, которые применяются в сфере его ответственности.
8.3 Управление документами системы менеджмента (вариант А)
8.3.1 Лаборатория должна управлять документами (внутренними и внешними), относящимися к выполнению требований настоящего стандарта.
Примечание — В данном контексте «документом» могут быть заявления о политике, процедуры, спецификации, инструкции производителя, калибровочные таблицы, схемы, пособия, плакаты, уведомления, памятки, чертежи, планы и т.д. Они могут быть представлены на различных носителях информации, например в печатном или цифровом формате.
8.3.2 Лаборатория должна обеспечить, что:
a) документы проверены на пригодность уполномоченным персоналом до их издания;
b) документы периодически анализируются и при необходимости пересматриваются;
c) идентифицируются изменения и статус текущей редакции документа;
d) актуальные версии применяемых документов доступны на всех рабочих местах и при необходимости их распространение управляется;
e) документы уникальным образом идентифицированы;
f) не допускается непреднамеренное использование устаревших документов и применяется соответствующая идентификация данных документов, в случае если они сохраняются с какой-либо целью.
8.4 Управление записями (вариант А)
8.4.1 Лаборатория должна вести и сохранять разборчивые записи с целью подтверждения соблюдения требований настоящего стандарта.
8.4.2 Лаборатория должна осуществлять управление, необходимое для идентификации, хранения, защиты, резервного копирования, архивирования, поиска, срока хранения и уничтожения своих записей. Лаборатория должна сохранять записи в течение периода, установленного договорными обязательствами. Доступ к данным записям должен соответствовать обязательствам в области конфиденциальности, и записи должны быть легкодоступными.
Примечание — В 7.5 приведены дополнительные требования, относящиеся к техническим записям.
8.5 Действия, связанные с рисками и возможностями (вариант А)
8.5.1 Лаборатория должна рассматривать риски и возможности, связанные с лабораторной деятельностью, для того чтобы:
a) обеспечивать, что система менеджмента достигает намеченных результатов;
b) наращивать возможности для достижения целей и задач лаборатории;
c) предотвращать или уменьшить нежелательные воздействия и возможные сбои в лабораторной деятельности;
d) добиваться улучшений.
8.5.2 Лаборатория должна планировать:
a) действия, связанные с данными рисками и возможностями;
b) каким образом:
1) интегрировать и внедрять данные действия в систему менеджмента;
2) оценивать результативность данных действий.
Примечание — Хотя в настоящем стандарте указывается, что лаборатория планирует действия по устранению рисков, требования к формальным методам управления рисками или документированному процессу управления рисками не установлены. Лаборатории могут решить, следует ли разрабатывать более обширную методологию управления рисками, чем это требуется в настоящем стандарте, например, посредством применения других руководств или стандартов.
8.5.3 Предпринимаемые действия, связанные с рисками и возможностями, должны быть соразмерны их потенциальному влиянию на достоверность лабораторных результатов.
Примечание 1 — Примерами действий, связанных с рисками, могут быть идентификация и предупреждение угроз, принятие рисков с целью реализации возможности, устранение источника риска, изменение вероятности риска или его последствий, разделение рисков или сохранение риска посредством обоснованного решения.
Примечание 2 — Возможности могут привести к расширению области лабораторной деятельности, привлечению новых заказчиков, использованию новых технологий или других возможностей с целью удовлетворения потребностей заказчиков.
8.6 Улучшения (вариант А)
8.6.1 Лаборатория должна идентифицировать и выбрать возможности для улучшений, а также предпринять необходимые действия.
Примечание — Возможности для улучшений могут быть идентифицированы по результатам анализа рабочих процедур, использования политик, основных целей, результатов аудитов, корректирующих действий, анализа со стороны руководства, предложений персонала, оценки риска, анализа данных и результатов проверок квалификации.
8.6.2 Лаборатория должна стремиться получать обратную связь от заказчиков, как положительную, так и отрицательную. Обратная связь должна анализироваться и применяться для улучшения системы менеджмента, лабораторной деятельности и обслуживания заказчиков.
Примечание — Примерами типов обратной связи являются опросы относительно удовлетворенности заказчиков, записи переговоров и обсуждение отчетов с заказчиками.
8.7 Корректирующие действия (вариант А)
8.7.1 При выявлении несоответствия лаборатория должна:
a) реагировать на несоответствие и при необходимости:
— предпринять действия для управления несоответствием и его устранения;
— отреагировать на последствия;
b) оценить необходимость действия для устранения причин(ы) несоответствия, для того чтобы предупредить его повторное или новое проявление, посредством:
— рассмотрения и анализа несоответствия;
— выявления причин несоответствия;
— выявления существования или потенциальной возможности возникновения подобных несоответствий;
c) предпринять необходимые действия;
d) оценить результативность предпринятых корректирующих действий;
e) повторно оценить риски и возможности, выявленные по итогам планирования, если это необходимо;
f) при необходимости внести изменения в систему менеджмента.
8.7.2 Корректирующие действия должны соответствовать масштабам и последствиям обнаруженного несоответствия.
8.7.3 Лаборатория должна сохранять записи в качестве свидетельств следующего:
a) сущности несоответствий, причин(ы) и любых предпринятых последующих действий;
b) результатов корректирующих действий.
8.8 Внутренние аудиты (вариант А)
8.8.1 Лаборатория должна проводить внутренние аудиты через запланированные интервалы для получения информации о том, является ли система менеджмента:
a) соответствующей:
— собственным требованиям лаборатории к своей системе менеджмента, в том числе лабораторной деятельности;
— требованиям настоящего стандарта;
b) результативно внедренной и реализуемой.
8.8.2 Лаборатория должна:
a) планировать, разрабатывать, внедрять и реализовывать программу аудита, в том числе в отношении периодичности, методов, сферы ответственности, планируемых требований и отчетности, которая должна учитывать важность соответствующей лабораторной деятельности, изменения, влияющие на лабораторию, а также результаты предыдущих аудитов;
b) определять критерии аудита и область проведения каждого аудита;
c) обеспечивать, что результаты аудита доведены до соответствующего руководства;
d) выполнять соответствующие коррекции и корректирующие действия без необоснованных задержек;
e) сохранять записи в качестве подтверждения реализации программы аудита и результатов аудитов.
Примечание — В ISO 19011 приведены руководящие указания для проведения внутренних аудитов.
8.9 Анализ со стороны руководства (вариант А)
8.9.1 Руководство лаборатории должно анализировать систему менеджмента с запланированной периодичностью, чтобы обеспечить ее постоянную пригодность, адекватность и результативность, включая заявленные политики и цели, связанные с выполнением требований настоящего стандарта.
8.9.2 Входные данные анализа со стороны руководства должны быть зарегистрированы и включать информацию относительно:
a) изменений во внутренних и внешних вопросах, имеющих отношение к лаборатории;
b) достижения поставленных целей;
c) пригодности политик и процедур;
d) статуса действий, запланированных после предыдущих анализов со стороны руководства;
e) результата(ов) последних внутренних аудитов;
f) корректирующих действий;
g) оценок, проводимых внешними органами;
h) изменений объема и вида работы или области деятельности лаборатории;
i) обратной связи от персонала и заказчиков;
j) жалоб (претензий);
k) результативности реализованных улучшений;
I) достаточности ресурсов;
m) результатов идентификации рисков;
n) итогов деятельности по обеспечению достоверности результатов; а также
o) других значимых факторов, такие как мониторинг деятельности и обучение.
8.9.3 Выходные данные анализа со стороны руководства должны включать записи обо всех решениях и действиях, относящихся по крайней мере к:
a) результативности системы менеджмента и ее процессов;
b) улучшению лабораторной деятельности, относящейся к выполнению требований настоящего стандарта;
c) предоставлению необходимых ресурсов;
d) любой необходимости изменений.
Приложение А
(справочное)
Метрологическая прослеживаемость
А.1 Общие сведения
В настоящем приложении приведена дополнительная информация о метрологической прослеживаемости, которая представляет собой важнейшую концепцию для обеспечения сопоставимости результатов измерений как на национальном, так и на международном уровне.
А.2 Установление метрологической прослеживаемости
А.2.1 Метрологическая прослеживаемость устанавливается с учетом и подтверждением:
a) определения измеряемой величины (величины, подлежащей измерению);
b) документированной непрерывной цепи калибровок, позволяющей установить связь с соответствующей основой для сравнения (в качестве такой соответствующей основы для сравнения могут выступать национальные или международные эталоны, а также внутренние (рабочие) эталоны);
c) оценивания неопределенности измерений на каждом этапе в цепи прослеживаемости с применением согласованных методов;
d) реализации каждого этапа в цепи прослеживаемости с применением соответствующих методов, с получением результатов измерений и связанных с ними зарегистрированных значений неопределенности измерений;
e) предоставления лабораториями, реализующими один или несколько этапов в цепи прослеживаемости, доказательств технической компетентности.
А.2.2 Систематическая погрешность (иногда называемая смещением) калиброванного оборудования учитывается при распространении метрологической прослеживаемости на результаты измерений, полученные в лаборатории. Существуют различные механизмы для учета систематической погрешности измерений при распространении метрологической прослеживаемости.
А.2.3 Для распространения метрологической прослеживаемости иногда применяют эталоны, информация о которых, предоставленная компетентной лабораторией, содержит только заявление о соответствии спецификации (без указания результатов измерений и значений неопределенности, связанных с ними). Реализация данного подхода, в соответствии с которым предельные значения, указанные в спецификациях, используются в качестве источника неопределенности, зависит от:
— применения соответствующего правила принятия решений для установления соответствия;
— последующего учета указанных в спецификациях предельных значений в бюджете неопределенности посредством технически обоснованного способа.
Техническое обоснование данного подхода заключается в том, что при заявлении о соответствии спецификации определяется интервал измеренных значений, в пределах которого при заданном уровне доверия предположительно находится истинное значение, и при этом рассматривается как любое смещение от истинного значения, так и неопределенность измерений.
Пример — Применение гирь с присвоенным согласно требованиям OIML R 111 классом точности для калибровки весов.
А.3 Демонстрация метрологической прослеживаемости
А.3.1 Лаборатории несут ответственность за установление метрологической прослеживаемости в соответствии с настоящим стандартом. Метрологическую прослеживаемость обеспечивают результаты калибровки, представленные лабораториями, соответствующими настоящему стандарту. Сертифицированные значения сертифицированных стандартных образцов, представленных поставщиками стандартных образцов, соответствующие требованиям ISO 17034, обеспечивают метрологическую прослеживаемость. Существуют различные способы демонстрации соответствия настоящему стандарту: признание третьей стороной (такой, как орган по аккредитации), внешняя оценка заказчиками или самооценка. К способам, принятым на международном уровне, относят (но не ограничиваются этим) следующие:
a) калибровочные и измерительные возможности, обеспечиваемые национальными метрологическими институтами и назначенными институтами, которые были подвергнуты соответствующим процессам паритетной оценки. Такая паритетная оценка проводится в рамках CIPM MRA (Соглашение о взаимном признании, подготовленное Международным комитетом мер и весов). С видами услуг, на которые распространяется действие CIPM MRA, можно ознакомиться в приложении С BIPM KCDB (базы данных по ключевым сличениям, сформированной Международным бюро мер и весов), где содержится подробная информация о диапазонах и неопределенности измерений для каждой из перечисленных в нем услуг;
b) калибровочные и измерительные возможности, которые подтверждены при аккредитации в органе по аккредитации, уполномоченном в рамках соглашения ILAC (Международное сотрудничество по аккредитации лабораторий) или региональных соглашений, признаваемых ILAC, обладают подтвержденной метрологической прослеживаемостью. Сведения об области аккредитации калибровочных лабораторий являются общедоступными и могут быть запрошены у соответствующих органов по аккредитации.
А.3.2 Совместная декларация BIPM, OIML (Международной организации по законодательной метрологии), ILAC и ISO о метрологической прослеживаемости содержит специальные указания по обеспечению международного признания цепи метрологической прослеживаемости.
Приложение В
(справочное)
Варианты системы менеджмента
В.1 Расширение применения систем менеджмента в целом увеличило потребность в подтверждении того, что лаборатории могут обеспечивать функционирование системы менеджмента, соответствующей и ISO 9001, и настоящему стандарту. Таким образом, настоящий стандарт обеспечивает два варианта требований, относящихся к реализации системы менеджмента.
В.2 Вариант А (см. 8.1.2) перечисляет минимальные требования к внедрению системы менеджмента в лаборатории. Внимание было уделено тому, чтобы включить все требования ISO 9001, имеющие отношение к областям лабораторной деятельности, на которые распространяется система менеджмента. Лаборатории, которые соответствуют требованиям ISO/IEC 17025 (разделы 4-7) и внедряют вариант А раздела 8, таким образом, также будут в целом действовать в соответствии с принципами ISO 9001.
В.3 Вариант В (см. 8.1.3) позволяет лабораториям создавать и поддерживать систему менеджмента в соответствии с требованиями ISO 9001 таким образом, чтобы обеспечивать и демонстрировать постоянное выполнение требований разделов 4-7. Лаборатории, которые применяют вариант В раздела 8, будут также работать в соответствии с ISO 9001. Соответствие требованиям ISO 9001 системы менеджмента, в рамках которой функционирует лаборатория, не является само по себе демонстрацией компетентности лаборатории в отношении предоставления технически достоверных данных и результатов. Это требование реализуется через соответствие разделам 4-7.
В.4 Оба варианта предполагают достижение того же результата в процессе функционирования системы менеджмента и соответствия требованиям разделов 4-7.
Примечание — Документы, данные и записи являются составными частями документированной информации в ISO 9001 и других стандартах по системам менеджмента. Управление документами рассматривается в 8.3. Управление записями рассматривается в 8.4 и 7.5. Управление данными, которые относятся к лабораторной деятельности, рассматривается в 7.11.
В.5 На рисунке В.1 показан пример варианта схематического представления рабочих процессов лаборатории, как описано в разделе 7.
|
|
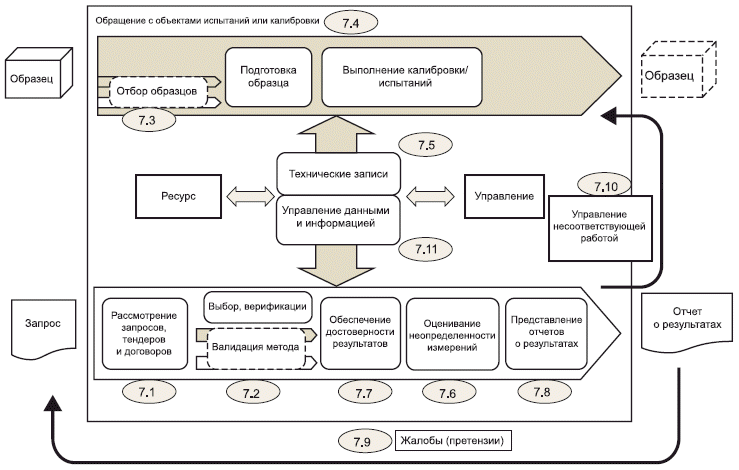
Рисунок В.1 — Вариант схематического представления рабочих процессов лаборатории
Приложение ДА
(справочное)
Сведения о соответствии ссылочных международных стандартов межгосударственным стандартам
Таблица ДА.1
|
Обозначение ссылочного международного стандарта |
Степень соответствия |
Обозначение и наименование соответствующего межгосударственного стандарта |
|
ISO/IEC Guide 99 |
— |
* |
|
ISO/IEC 17000 |
IDT |
ГОСТ ISO/IEC 17000-2012 «Оценка соответствия. Словарь и общие принципы» |
|
* Соответствующий межгосударственный стандарт отсутствует. До его принятия рекомендуется использовать перевод на русский язык данного международного стандарта. Примечание — В настоящей таблице использовано следующее условное обозначение степени соответствия стандарта: — IDT — идентичный стандарт. |
Библиография
|
ISO 5725-1 |
Accuracy (trueness and precision) of measurement methods and results — Part 1: General principles and definitions |
|
(Точность (правильность и прецизионность) методов и результатов измерений. Часть 1. Основные положения и определения) |
|
|
ISO 5725-2 |
Accuracy (trueness and precision) of measurement methods and results — Part 2: Basic method for the determination of repeatability and reproducibility of a standard measurement method |
|
(Точность (правильность и прецизионность) методов и результатов измерений. Часть 2. Основной метод определения повторяемости и воспроизводимости стандартного метода измерений) |
|
|
ISO 5725-3 |
Accuracy (trueness and precision) of measurement methods and results — Part 3: Intermediate measures of the precision of a standard measurement method |
|
(Точность (правильность и прецизионность) методов и результатов измерений. Часть 3. Промежуточные показатели прецизионности стандартного метода измерений) |
|
|
ISO 5725-4 |
Accuracy (trueness and precision) of measurement methods and results — Part 4: Basic methods for the determination of the trueness of a standard measurement method |
|
(Точность (правильность и прецизионность) методов и результатов измерений. Часть 4. Основные методы определения правильности стандартного метода измерений) |
|
|
ISO 5725-6 |
Accuracy (trueness and precision) of measurement methods and results — Part 6: Use in practice of accuracy values |
|
(Точность (правильность и прецизионность) методов и результатов измерений. Часть 6. Использование значений точности на практике) |
|
|
ISO 9000 |
Quality management systems — Fundamentals and vocabulary |
|
(Системы менеджмента качества. Основные положения и словарь) |
|
|
ISO 9001 |
Quality management systems — Requirements |
|
(Системы менеджмента качества. Требования) |
|
|
ISO 10012 |
Measurement management systems — Requirements for measurement processes and measuring equipment |
|
(Системы менеджмента измерений. Требования к процессам измерений и измерительному оборудованию) |
|
|
ISO/IEC 12207 |
Systems and software engineering — Software life cycle processes |
|
(Информационные технологии. Процессы жизненного цикла программных средств) |
|
|
ISO 15189 |
Medical laboratories — Requirements for quality and competence |
|
(Медицинские лаборатории. Специальные требования к качеству и компетентности) |
|
|
ISO 15194 |
In vitro diagnostic medical devices — Measurement of quantities in samples of biological origin — Requirements for certified reference materials and the content of supporting documentation |
|
(Медицинские изделия для in vitro-диагностики. Измерение величин в образцах биологического происхождения. Требования к аттестованным стандартным образцам и содержанию сопроводительной документации) |
|
|
ISO/IEC 17011 |
Conformity assessment — Requirements for accreditation bodies accrediting conformity assessment bodies |
|
(Оценка соответствия. Общие требования к органам по аккредитации органов по оценке соответствия) |
|
|
ISO/IEC 17020 |
Conformity assessment — Requirements for the operation of various types of bodies performing inspection |
|
(Оценка соответствия. Общие требования к работе различных типов органов, проводящих инспекции) |
|
|
ISO/IEC 17021-1 |
Conformity assessment — Requirements for bodies providing audit and certification of management systems — Part 1: Requirements |
|
(Оценка соответствия. Требования к органам, проводящим аудит и сертификацию систем менеджмента. Часть 1. Требования) |
|
|
ISO 17034 |
General requirements for the competence of reference material producers |
|
(Общие требования к компетенции производителей эталонных материалов) |
|
|
ISO/IEC 17043 |
Conformity assessment — General requirements for proficiency testing |
|
(Оценка соответствия. Основные требования к проведению проверки квалификации) |
|
|
ISO/IEC 17065 |
Conformity assessment — Requirements for bodies certifying products, processes and services |
|
(Оценка соответствия. Требования к органам по сертификации продукции, процессов и услуг) |
|
|
ISO 17511 |
In vitro diagnostic medical devices — Measurement of quantities in biological samples — Metrological traceability of values assigned to calibrators and control materials |
|
(Изделия медицинские для диагностики in vitro. Измерение величин в биологических пробах. Метрологическая прослеживаемость значений, приписанных калибраторам и контрольным материалам) |
|
|
ISO 19011 |
Guidelines for auditing management systems |
|
(Руководящие указания по аудиту систем менеджмента) |
|
|
ISO 21748 |
Guidance for the use of repeatability, reproducibility and trueness estimates in measurement uncertainty evaluation |
|
(Статистические методы. Руководство по использованию оценок повторяемости, воспроизводимости и правильности при оценке неопределенности измерений) |
|
|
ISO 31000 |
Risk management — Guidelines |
|
(Менеджмент рисков. Руководящие указания) |
|
|
ISO Guide 30 |
Reference materials — Selected terms and definitions |
|
(Стандартные образцы. Термины и определения, используемые в области стандартных образцов) |
|
|
ISO Guide 31 |
Reference materials — Contents of certificates, labels and accompanying documentation |
|
(Стандартные образцы. Содержание сертификатов (паспортов) и этикеток) |
|
|
ISO Guide 33 |
Reference materials — Good practice in using reference materials |
|
(Стандартные образцы. Установившаяся практика применения стандартных образцов) |
|
|
ISO Guide 35 |
Reference materials — Guidance for characterization and assessment of homogeneity and stability |
|
(Стандартные образцы. Общие и статистические принципы сертификации (аттестации)) |
|
|
ISO Guide 80 |
Guidance for the in-house preparation of quality control materials (QCMs) |
|
(Руководство по внутрилабораторному изготовлению материалов для контроля качества (МКК)) |
|
|
ISO/IEC Guide 98-3 |
Uncertainty of measurement — Part 3: Guide to the expression of uncertainty in measurement (GUM:1995) |
|
(Неопределенность измерений. Часть 3. Руководство по выражению неопределенности измерения) |
|
|
ISO/IEC Guide 98-4 |
Uncertainty of measurement — Part 4: Role of measurement uncertainty in conformity assessment |
|
(Неопределенность измерений. Часть 4. Роль неопределенности измерения в оценке соответствия) |
|
|
IEC Guide 115 |
Application of uncertainty of measurement to conformity assessment activities in the electrotechnical sector |
|
(Погрешность измерения при оценке согласованности деятельности в электротехническом секторе) |
|
|
Joint BIPM, OIML, ILAC and ISO declaration on metrological traceability, 2011 |
|
|
(Совместная декларация BIPM, OIML, ILAC и ISO о метрологической прослеживаемости, 2011) |
|
|
ILAC International Laboratory Accreditation Cooperation |
|
|
(Международная организация по аккредитации лабораторий) |
|
|
(VIML), OIML V1:2013 |
International vocabulary of terms in legal metrology, 2013 |
|
(Международный словарь терминов по законодательной метрологии, 2013) |
|
|
JCGM 106:2012 |
Evaluation of measurement data — The role of measurement uncertainty in conformity assessment |
|
(Оценивание данных измерений. Роль неопределенности измерения в оценке соответствия) |
|
|
EEE/RM/062rev3, Eurachem |
|
|
(Выбор и использование эталонных материалов) |
|
|
BIPM SI Brochure |
|
|
(Брошюра СИ: Международная система единиц (СИ), BIPM) |
_______________
![]() http://www.bipm.org/utils/common/pdf/BIPM-OIML-ILAC-ISO_joint_declaration_2011.pdf
http://www.bipm.org/utils/common/pdf/BIPM-OIML-ILAC-ISO_joint_declaration_2011.pdf
![]() http://ilac.org/
http://ilac.org/
![]() https://www.eurachem.org/images/stories/Guides/pdf/EEE-RM-062rev3.pdf
https://www.eurachem.org/images/stories/Guides/pdf/EEE-RM-062rev3.pdf
![]() http://www.bipm.org/en/publications/si-brochure/
http://www.bipm.org/en/publications/si-brochure/
(Нумерация сносок приведена по оригиналу.)
|
УДК 53.089.6+061.64(083.74)(476) |
МКС 03.120.20 |
IDT |
|
Ключевые слова: беспристрастность, прослеживаемость, компетентность, конфиденциальность, система менеджмента |
Электронный текст документа
и сверен по:
, 2021
ФГУП «СТАНДАРТИНФОРМ» зарегистрированы официальные переводы на русский язык ряда документов по стандартным образцам Международной организации по стандартизации:
- Руководство ИСО 80:2015 «Руководство по внутрилабораторному изготовлению материалов для контроля качества (МКК)» — ISO Guide 80:2014 «Guidance for in-house preparation of quality control materials (QCMs)»//Перевод ФГУП «УНИИМ» зарегистрирован ФГУП «СТАНДАРТИНФОРМ» (номер регистрации: 9262/ISO. Дата регистрации: 104.2017 г.). Краткий обзор документа приведен здесь.
- Технический отчет ИСО 79:2015 «Стандартные образцы. Примеры стандартных образцов качественных свойств» – ISO/TR 79:2015 «Reference materials – Examples of reference materials for qualitative properties»//Перевод ФГУП «УНИИМ» зарегистрирован ФГУП «СТАНДАРТИНФОРМ» (номер регистрации: 8965/ISO/TR. Дата регистрации: 11.2016 г.). Краткий обзор документа приведен здесь.
- Руководство ИСО 30:2015 Стандартные образцы – Некоторые термины и определения // Перевод ФГУП «УНИИМ» зарегистрирован ФГУП «СТАНДАРТИНФОРМ» (номер регистрации: 8924/ISO GUIDE. Дата регистрации: 01.11.2016)
- Руководство ИСО 31:2015 Стандартные образцы – Содержание сертификатов, этикеток и сопроводительной документации // Перевод ФГУП «УНИИМ» зарегистрирован ФГУП «СТАНДАРТИНФОРМ» (номер регистрации: 8917/ISO GUIDE. Дата регистрации: 28.10.2016)
- Руководство ИСО 33:2015 Стандартные образцы – Надлежащая практика применения стандартных образцов // Перевод ФГУП «УНИИМ» зарегистрирован ФГУП «СТАНДАРТИНФОРМ» (номер регистрации: 8923/ISO GUIDE. Дата регистрации: 01.11.2016)
- Технический отчет ИСО 16476:2016 Стандартные образцы – Установление и выражение метрологической прослеживаемости значений величин, приписанных стандартным образцам // Перевод ФГУП «УНИИМ» зарегистрирован ФГУП «СТАНДАРТИНФОРМ» (номер регистрации: 8937/ISO/TR. Дата регистрации: 08.11.2016)
Сведения о переводах других документов ИСО в области стандартных образцов приведены здесь.
Руководство ИСО 80:2015
Технический отчет ИСО 79:2015
Обзор официальных переводов на русский язык документов ИСО/РЕМКО
Полный текст:
- Аннотация
- Об авторе
Аннотация
ФГУП «УНИИМ», являясь Научным методическим центром ГССО, регулярно проводит работы, направленные на признание в РФ в качестве официально зарегистрированных переводов основополагающих документов ИСО/РЕМКО путем их переводов на русский язык и официальной регистрации в ФГУП «СТАНДАРТИНФОРМ» и, при необходимости, гармонизации действующих национальных нормативных документов со стандартами ИСО/РЕМКО. К числу зарегистрированных в последнее время официальных переводов относятся следующие документы. Технический отчет ИСО 79:2015 «Стандартные образцы. Примеры стандартных образцов качественных свойств» — ISO/TR 79:2015 «Reference materials — Examples of reference materials for qualitative properties» (Номер регистрации официального перевода: 8965/ISO/TR. Дата регистрации: 22.11.2016). — ИСО 80:2015 «Руководство по внутрилабораторному изготовлению материалов для контроля качества (МКК)» -ISO Guide 80:2014 «Guidance for in-house preparation of quality control materials (QCMs)» (Номер регистрации: 9262/ISO. Дата регистрации: 12.04.2017)
Об авторе
О. Н. Кремлёва
ФГУП «Уральский научно-исследовательский институт метрологии»
Россия
Рецензия
Для цитирования:
Кремлёва О.Н. Обзор официальных переводов на русский язык документов ИСО/РЕМКО. Эталоны. Стандартные образцы. 2017;13(1):61-62.
For citation:
. Overview of official translations of ISO/REMCO documents into Russian. Measurement Standards. Reference Materials. 2017;13(1):61-62.
(In Russ.)
Просмотров: 670
ГОСТ Р 52770-2020
НАЦИОНАЛЬНЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕРАЦИИ
Изделия медицинские
СИСТЕМА ОЦЕНКИ БИОЛОГИЧЕСКОГО ДЕЙСТВИЯ
Часть 1
Общие требования биологической безопасности
Medical devices. Evaluation system of biological effects. Part 1. General requirements for biological safety
ОКС 11.100.20
Дата введения 2021-03-01
Предисловие
1 РАЗРАБОТАН Автономной некоммерческой организацией «Институт медико-биологических исследований и технологий (АНО «ИМБИИТ»)
2 ВНЕСЕН Техническим комитетом по стандартизации ТК 422 «Оценка биологического действия медицинских изделий»
3 УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Приказом Федерального агентства по техническому регулированию и метрологии от 17 ноября 2020 г. N 1101-ст
4 ВЗАМЕН ГОСТ Р 52770-2016
Правила применения настоящего стандарта установлены в статье 26 Федерального закона от 29 июня 2015 г. N 162-ФЗ «О стандартизации в Российской Федерации». Информация об изменениях к настоящему стандарту публикуется в ежегодном (по состоянию на 1 января текущего года) информационном указателе «Национальные стандарты», а официальный текст изменений и поправок — в ежемесячном информационном указателе «Национальные стандарты». В случае пересмотра (замены) или отмены настоящего стандарта соответствующее уведомление будет опубликовано в ближайшем выпуске ежемесячного информационного указателя «Национальные стандарты». Соответствующая информация, уведомление и тексты размещаются также в информационной системе общего пользования — на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет (www.gost.ru)
Введение
Соблюдение положений стандартов серии «Система оценки биологического действия» позволит обеспечить системный подход к оценке биологического действия медицинских изделий (далее — МИ).
Настоящий стандарт является руководством по оценке биологического действия применения МИ и предназначен для применения исследователями (испытателями), разработчиками и изготовителями МИ при проведении токсикологических исследований, экспертизы, контроля качества, эффективности и безопасности МИ, при их регистрации, производстве, подтверждении соответствия и государственном контроле (надзоре).
Целью настоящего стандарта является установление общих требований биологической безопасности МИ с учетом потенциального риска для здоровья и подходов к исследованиям и испытаниям.
Настоящий стандарт является основополагающим. При применении стандартов на конкретные виды продукции следует учитывать требования настоящего стандарта.
Настоящий стандарт не заменяет стандарты серии ГОСТ ISO 10993, но разъясняет и дополняет некоторые положения, не учтенные в данных стандартах, содействует сотрудничеству между лабораториями и другими органами, обмену информацией и опытом, гармонизации стандартов и процедур.
В серию стандартов «Изделия медицинские. Система оценки биологического действия», входят следующие части:
— Часть 1. Общие требования биологической безопасности;
— Часть 2. Применение уровня предельно допустимого токсикологического воздействия для оценки биосовместимости компонентов медицинских изделий;
— Часть 3. Методы исследований (испытаний).
При формировании подхода к исследованиям следует учитывать новизну МИ (при выведении на рынок нового продукта) или опыт применения МИ в клинической практике (при изменении МИ с целью оценки рисков этих изменений).
Содержание в МИ лекарственного средства (ЛС) или другого биологически активного компонента является важным аспектом, который следует учитывать.
Для деградируемых/резорбируемых МИ важно учитывать время и степень деградации/резорбции, а также оценить степень опасности продуктов деградации/резорбции во временном аспекте.
МИ отнесены к социально значимой продукции, поэтому оценка риска их применения основана на критериях, отражающих непосредственное влияние компонентов конечного продукта на здоровье наиболее чувствительных групп населения, что требует обеспечения значительного запаса надежности допустимых показателей.
Ухудшение экологической среды привело к повышенному содержанию тяжелых металлов как в почве, так и в воде, что не могло не отразиться на биологической безопасности материалов природного происхождения, что потребовало введения дополнительных требований.
1 Область применения
Настоящий стандарт распространяется на медицинские изделия (далее — МИ) и материалы, применяемые для их изготовления, при использовании их по назначению в надлежащих условиях, а также на МИ, находящиеся в обращении.
Настоящий стандарт устанавливает общие требования биологической безопасности МИ и материалов, применяемых для их изготовления, контактирующих непосредственно или опосредованно с организмом человека при использовании их по назначению.
Настоящий стандарт является руководством для планирования исследований (испытаний) по показателям биологического действия.
Настоящий стандарт устанавливает общие требования к проведению исследований (испытаний) по определению санитарно-химических/физико-химических показателей, показателей биологического действия и микробиологических показателей с целью подтверждения безопасности МИ при использовании его по назначению в надлежащих условиях.
В настоящем стандарте приведены допустимые количества миграции химических веществ в экстракт некоторых потенциально опасных химических соединений для материалов и МИ.
В настоящем стандарте приведены подходы к исследованиям (испытаниям) материалов/МИ и установлены требования к выбору образца МИ для проведения исследований (испытаний), общие требования к приготовлению экстрактов из исследуемых образцов, к методам (методикам) определения показателей биологического действия, приведены значения допустимых количеств экстрагируемых/выщелачиваемых веществ из материалов/МИ.
2 Нормативные ссылки
В настоящем стандарте использованы нормативные ссылки на следующие стандарты:
ГОСТ 23932 Посуда и оборудование лабораторные стеклянные. Общие технические условия
ГОСТ 31209 Контейнеры для крови и ее компонентов. Требования химической и биологической безопасности и методы испытаний
ГОСТ 31214 Изделия медицинские. Требования к образцам и документации, представляемым на токсикологические, санитарно-химические испытания, испытания на стерильность и пирогенность
ГОСТ 31576 Оценка биологического действия медицинских стоматологических материалов и изделий. Классификация и приготовление проб
ГОСТ 31814 Оценка соответствия. Общие правила отбора образцов для испытаний продукции при подтверждении соответствия
ГОСТ EN 556-1 Стерилизация медицинских изделий. Требования к медицинским изделиям категории «стерильные». Часть 1. Требования к медицинским изделиям, подлежащим финишной стерилизации
ГОСТ ISO 10993-1 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 1. Оценка и исследования
ГОСТ ISO 10993-4 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 4. Исследования изделий, взаимодействующих с кровью
ГОСТ ISO 10993-5 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 5. Исследования на цитотоксичность: методы in vitro
ГОСТ ISO 10993-6 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 6. Исследования местного действия после имплантации
ГОСТ ISO 10993-7 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 7. Остаточное содержание этиленоксида после стерилизации
ГОСТ ISO 10993-9 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 9. Основные принципы идентификации и количественного определения потенциальных продуктов деструкции
ГОСТ ISO 10993-10 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 10. Исследование раздражающего и сенсибилизирующего действия
ГОСТ ISO 10993-11 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 11. Исследование общетоксического действия
ГОСТ ISO 10993-12 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 12. Приготовление проб и контрольные образцы
ГОСТ ISO 10993-13 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 13. Идентификация и количественное определение продуктов деградации полимерных медицинских изделий
ГОСТ ISO 10993-14 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 14. Идентификация и количественное определение продуктов деградации изделий из керамики
ГОСТ ISO 10993-15 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 15. Идентификация и количественное определение продуктов деградации из металлов и сплавов
ГОСТ ISO 10993-16 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 16. Концепция токсикокинетических исследований продуктов разложения и выщелачиваемых веществ
ГОСТ ISO 10993-17 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 17. Установление пороговых значений для вымываемых веществ
ГОСТ ISO 10993-18 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 18. Исследование химических свойств материалов
ГОСТ ISO/TS 10993-19 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 19. Исследования физико-химических, морфологических и топографических свойств материалов
ГОСТ ISO/TS 10993-20 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 20. Принципы и методы исследования иммунотоксичности медицинских изделий
ГОСТ ISO/TR 10993-22 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 22. Руководство по наноматериалам
ГОСТ ISO 14971 Изделия медицинские. Применение менеджмента риска к медицинским изделиям
ГОСТ ISO/IEC 17000 Оценка соответствия. Словарь и общие принципы
ГОСТ ISO/IEC 17025 Общие требования к компетентности испытательных и калибровочных лабораторий
Примечание — При пользовании настоящим стандартом целесообразно проверить действие ссылочных стандартов в информационной системе общего пользования — на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет или по ежегодному информационному указателю «Национальные стандарты», который опубликован по состоянию на 1 января текущего года, и по выпускам ежемесячного информационного указателя «Национальные стандарты» за текущий год. Если заменен ссылочный стандарт, на который дана недатированная ссылка, то рекомендуется использовать действующую версию этого стандарта с учетом всех внесенных в данную версию изменений. Если заменен ссылочный стандарт, на который дана датированная ссылка, то рекомендуется использовать версию этого стандарта с указанным выше годом утверждения (принятия). Если после утверждения настоящего стандарта в ссылочный стандарт, на который дана датированная ссылка, внесено изменение, затрагивающее положение, на которое дана ссылка, то это положение рекомендуется применять без учета данного изменения. Если ссылочный стандарт отменен без замены, то положение, в котором дана ссылка на него, рекомендуется применять в части, не затрагивающей эту ссылку.
3 Термины и определения
В настоящем стандарте применены термины по ГОСТ ISO/IEC 17000, ГОСТ ISO 14971 (см. также [1]*), а также следующие термины с соответствующими определениями:
3.1 безопасность МИ: Состояние, при котором отсутствует недопустимый риск причинения вреда жизни или здоровью граждан при использовании МИ в условиях, предусмотренных изготовителем.
Примечание — Безопасность МИ должна обеспечиваться сохранением его эксплуатационных свойств в течение установленного изготовителем срока службы или срока годности.
3.2 биологическая безопасность МИ: Состояние МИ/материала при использовании по назначению в надлежащих условиях, при котором МИ/материал не оказывает вредного воздействия на организм человека по санитарно-химическим/физико-химическим показателям, показателям биологического действия, микробиологическим показателям.
3.3 оценка биологического действия МИ: Анализ и оценка существующих данных биологической безопасности применения МИ, а также исследования/испытания, проводимые в целях определения соответствия МИ общим требованиям безопасности.
3.4 исследуемый материал: Материал, изделие, часть изделия или его компонент или его типичный образец, изготовленный и обработанный эквивалентными методами, пробу или экстракт которых отбирают для биологического или химического исследования или оценки.
3.5 интегральные показатели: Обобщенные показатели качества материала/МИ (восстановительные примеси, ультрафиолетовое поглощение, изменение величины рН).
3.6 пирогенность: Способность химического агента или другого вещества вызывать лихорадочное состояние организма.
3.7 допустимое количество миграции химических веществ; ДКМ: Допустимое количество миграции химических веществ в экстракт.
3.8 санитарно-химические/физико-химические испытания: Испытания, проводимые с целью определения интегральных показателей, количественного определения компонентов, мигрирующих в экстракт из МИ, и материалов, применяемых для их изготовления, а также индивидуальных потенциально опасных соединений (составляющих веществ, технологических добавок, стерилизующих агентов, примесей и т.п.).
3.9 скрининговые исследования (испытания): Исследования (испытания), проводимые с целью предварительной характеристики возможных источников и уровней рисков.
3.10 экстракт: Раствор, полученный в результате экстракции исследуемого материала или контрольного материала.
3.11 экстрагирующая жидкость: Модельная среда, применяемая для приготовления экстракта.
3.12 компонент: Химическое вещество или соединение, присутствующее в готовом МИ или материалах изготовления.
Примечания
1 Компоненты могут присутствовать преднамеренно (например, добавка, такая как антиоксидант) или непреднамеренно (например, примесь).
2 При применении уровня предельно допустимого токсикологического воздействия к экстрагируемому или выщелачиваемому веществу сущность экстрагируемого/выщелачиваемого вещества представляет собой компонент, который может оказывать потенциальное воздействие на человека при использовании медицинского изделия.
3.13 экстрагируемое вещество: Компонент, высвобождаемый при экстрагировании или из материала изготовления при использовании условий лабораторной экстракции и модельный* среды.
Примечание — При применении уровня предельно допустимого токсикологического воздействия к экстрагируемому веществу предполагается, что экстрагированное количество потенциально контактирует с пользователями МИ во время клинического применения, см. ГОСТ ISO 10993-17.
3.14 выщелачиваемое вещество: Компонент, высвобождаемый из МИ и потенциально контактирующий с организмом при клиническом применении.
Примечание — При применении уровня предельно допустимого токсикологического воздействия к выщелачиваемому веществу предполагается, что выщелоченное количество потенциально контактирует с пользователями МИ во время его клинического применения, см. стандарты серии ГОСТ ISO 10993.
3.15 уровень предельно допустимого токсикологического воздействия/порог токсикологической опасности: Уровень воздействия для компонентов, ниже которого не существует значимого риска для здоровья человека.
4 Общие требования
4.1 Оценка биологического действия МИ включает в себя проведение комплекса исследований (испытаний) с целью качественного и количественного определения биологической безопасности, в т.ч. санитарно-химических/физико-химических показателей (интегральных и специфических) и микробиологических показателей в рамках менеджмента риска.
4.2 Для проведения исследований (испытаний) представляют образцы МИ, контактирующие непосредственно или опосредованно с организмом человека при использовании его в соответствии с назначением (образцы готового изделия, единообразные по характеру и качеству, изготовленные в определенном цикле производства, прошедшие все производственные этапы, установленные изготовителем).
4.3 При выборе материалов, применяемых для изготовления МИ, необходимо оценивать их биологическую безопасность. Для проведения исследований (испытаний) могут быть представлены как опытные образцы МИ, так и материалы, из которых они изготовлены.
Выбор и оценка любого материала для МИ, предназначенного для применения в медицинской практике, требуют системного подхода к оценке преимуществ/недостатков физических и химических характеристик в рамках анализа и оценки риска (см. ГОСТ ISO 14971).
4.4 МИ должны соответствовать требованиям биологической безопасности по санитарно-химическим/физико-химическим показателям.
4.5 МИ должны соответствовать требованиям биологической безопасности по показателям биологического действия, см. стандарты серии ГОСТ ISO 10993, [2].
4.6 МИ, поставляемые в стерильном состоянии, должны сохранять стерильность до вскрытия защитной упаковки в течение срока годности.
4.7 Программу исследования составляют с учетом назначения МИ, вида и продолжительности контакта с организмом человека, свойств материалов, из которых оно изготовлено, современных методов исследования (испытания), а также информации из литературных источников о применяемых материалах, их рецептурном составе, способах предстерилизационной очистки, дезинфекции и стерилизации.
4.8 Программа исследований (испытаний) МИ должна учитывать требования стандартов и нормативных документов (далее — НД) изготовителя.
4.9 Во избежание недостаточных или избыточных требований внешних экспертов, программу исследований (испытаний) составляют и документально оформляют компетентные специалисты (в соответствии с требованиями ГОСТ ISO/IEC 17025), имеющие необходимый опыт работы в области оценки биологического действия МИ.
4.10 Для МИ, в состав которых входит вещество, представляющее собой лекарственный препарат или другое биологически активное средство и воздействующее на человеческий организм в дополнение к воздействию МИ, оценку биологического действия МИ проводят с учетом действия (свойств) данного вещества.
4.11 Исследование (испытание) МИ, содержащего активные источники излучения, проводят на неактивных («холодных») исследуемых образцах.
4.12 При оценке биологического действия МИ, содержащего биологические ткани и их компоненты, учитывают требования соответствующих НД.
4.13 МИ, содержащие ткани животного происхождения, оценивают с учетом требований соответствующих стандартов (см. также [3]).
4.14 МИ, содержащие ткани человеческого происхождения, оценивают с учетом требований соответствующих стандартов (см. также [4]).
4.15 МИ, контактирующие с системой кровообращения, кровью и ее компонентами, препаратами для внутрисосудистого введения, подвергают испытаниям на гемосовместимость по ГОСТ ISO 10993-4.
4.16 МИ, контактирующие с системой кровообращения, кровью и ее компонентами, препаратами для внутрисосудистого введения, испытывают на пирогенность.
4.17 МИ из материалов природного происхождения испытывают на наличие свободного белка.
4.18 Скрининговые исследования (испытания) проводят с целью предварительного установления показателей (характеристик) биологической безопасности при контрольных испытаниях.
4.19 При контрольных испытаниях МИ, находящихся в обращении (изъятых из обращения), за исключением случаев, повлекших угрозу нанесения вреда здоровью и жизни человека, допустимо применение реперных точек оценки их биологического действия, руководствуясь составом материала и назначением МИ, установленным изготовителем. Реперными точками в санитарно-химических/физико-химических испытаниях являются интегральные показатели в экстрактах из материала/МИ: восстановительные примеси, ультрафиолетовое поглощение, изменение значения рН (если в НД на изделие не предусмотрено иное); содержание формальдегида, мономера исходного полимерного материала. Определение остаточных количеств растворителей, стерилизующих агентов, тяжелых металлов проводят с учетом анализа риска. При необходимости рекомендуется руководствоваться соответствующими стандартами (см. стандарты серии ГОСТ ISO 10993, [2], [5]).
4.20 Повторные исследования (испытания) по показателям биологического действия проводят при изменении:
— источника поступления или любой характеристики материалов, применяемых при изготовлении МИ;
— упаковки (барьерная система для стерилизации);
— способа или условия проведения стерилизации МИ;
— назначения МИ, предусмотренного инструкций изготовителя.
5 Общие требования биологической безопасности по санитарно-химическим/физико-химическим показателям
5.1 Общие положения
Санитарно-химические/физико-химические испытания проводят в целях качественного и количественного определения мигрирующих в экстракт компонентов композиции, остаточных количеств химических соединений, используемых в технологическом процессе синтеза некоторых композиций и изготовления МИ остаточных количеств стерилизующих агентов исследуемого (испытуемого) МИ.
Санитарно-химические/физико-химические испытания являются обязательными для всех групп МИ и предваряют исследования (испытания) на биологических тест-объектах. Испытаниям подлежат как экстракты из материалов/МИ, так и МИ жидких, в том числе в форме геля. Условия приготовления экстрактов (виды экстрагирующей жидкости (среды)), температура и продолжительность экстракции, соотношение поверхности или массы исследуемого материала к объему экстрагирующей жидкости (среды) выбирают в зависимости от категории МИ (см. приложения А, Б, В).
В ходе санитарно-химических/физико-химических испытаний с помощью интегральных и специфических (селективных) методов анализа экстрактов из исследуемого материала оценивают химическую стабильность материала и характеристику мигрирующих в экстракт веществ, что дает представление о природе высвобождающихся из изделий химических соединений и их потенциальной опасности. С этой целью используют такие высокочувствительные, эффективные и селективные методы анализа, как спектрофотометрия, высокоэффективная жидкостная хроматография, газовая и жидкостная хроматографии, масс-спектрометрия, атомно-абсорбционная спектрометрия и др. (см. ГОСТ ISO 10993-9).
Результаты санитарно-химических/физико-химических испытаний могут служить основанием для прекращения дальнейших исследований (испытаний) материала/МИ в случае обнаружения миграции веществ с известной токсикологической характеристикой в количествах, превышающих допустимые значения (см. приложение Г), осуществления коррекции в подборе материалов и оптимизации технологического процесса изготовления МИ, включая способ и режим стерилизации, что позволит создать продукт, отвечающий требованиям биологической безопасности. Остановить испытания или продолжить решает компетентный специалист, руководствуясь оценкой полученных результатов и анализом риска, который должен быть обоснован и подкреплен научными данными.
Если для материалов/МИ с биологически активными добавками (ЛС, антисептики, аппретирующие вещества и др.) интегральные показатели: восстановительные примеси, ультрафиолетовое поглощение, изменение значения рН, превышают допустимые (см. приложение Д, таблица Д.1), то решение об их соответствии требованиям биологической безопасности принимают по результатам исследований (испытаний) на биологических тест-объектах.
Наименование и допустимые значения некоторых санитарно-химических/физико-химических показателей с учетом природы и назначения материалов/МИ, вида и степени опасности приведены в приложениях Г, Д.
5.2 Общие требования к выбору образца для исследования (испытания)
Правила отбора образцов приведены в ГОСТ 31214, ГОСТ 31814 и других НД на конкретный вид МИ или метод испытаний.
5.3 Общие требования к приготовлению экстрактов из исследуемых материалов
Основные требования к приготовлению экстрактов установлены в ГОСТ ISO 10993-12.
При приготовлении экстрактов следует учитывать требования, установленные в стандартах на конкретные МИ. Следует выбирать более «жесткие» условия экстракции.
В процедуре пробоподготовки должны быть учтены все процедуры обращения с образцом, предусмотренные инструкцией по применению МИ. Выбор экстрагирующей жидкости следует проводить с учетом методики испытаний. Как правило, санитарно-химические/физико-химические испытания МИ, изготовленных из полимерных материалов, экстракцию проводят на дистиллированной воде. Экстрагирующая жидкость для МИ из металлов и керамики — 0,9%-ный раствор хлорида натрия, если в НД изготовителя не установлены другие требования (см. приложение А).
Примечание — Водные экстракты готовят с использованием дистиллированной воды или воды для лабораторного анализа (см. [6]).
Принципы экстракции исследуемого материала приведены в ГОСТ ISO 10993-12.
Жидкие формы (растворы) МИ испытывают «в чистом виде».
Если значение рН экстракта из испытуемого материала или самой жидкой формы менее либо равно 2,0, либо равно или более 11,5, то материал признают потенциальным раздражителем и дальнейшие исследования его не проводят (см. ГОСТ ISO 10993-10).
Посуда и оборудование лабораторное стеклянное должны соответствовать требованиям ГОСТ 23932.
При выборе материалов для имплантатов, разработке новых МИ, отсутствии информации о характеристиках в отношении безопасности экстракты готовят в соотношении между массой испытуемого материала и объемом модельной среды по формуле
, (1)
где М — максимально возможное количество имплантируемого материала (в граммах);
V — объем крови в организме человека (5 л);
K — коэффициент аггравации, равный 10.
Сроки экспозиции экстрактов определяются природой материала/МИ, продолжительностью контакта с организмом человека, применяемостью методик. При применении динамического режима приготовления экстракта необходимо регламентировать сроки обновления модельной среды и проводить анализ полученных экстрактов на каждом этапе. При контрольных исследованиях режимы экстракции могут быть изменены в сторону уменьшения времени и увеличения температуры, если данные изменения обоснованы.
5.4 Общие требования к методам (методикам) определения санитарно-химических/ физико-химических показателей
Средства измерений и оборудование, применяемое для исследований (испытаний), должны быть откалиброваны и/или аттестованы и/или поверены в установленном порядке с учетом требований НД на методы исследований (испытаний).
Методики выполнения измерений, применяемые при исследованиях (испытаниях), должны быть аттестованы или стандартизованы в соответствии с установленными требованиями.
Для надлежащего осуществления лабораторной деятельности испытательная лаборатория должна иметь эталоны, стандартные образцы согласно ГОСТ ISO 10993-12 (см. также [7]-[9]).
6 Общие требования биологической безопасности по показателям биологического действия
6.1 Общие положения
Оценка биологического действия МИ — это исследования, проводимые для выявления биологического действия материала/МИ in vitro и in vivo с целью дальнейшей экстраполяции полученных результатов на человека.
Основные подходы к исследованиям биологического действия приведены в соответствующих стандартах серии ГОСТ ISO 10993, [2].
6.2 Общие требования к выбору образца для исследования (испытания)
Общие требования к выбору образцов для исследований (испытаний) установлены в ГОСТ ISO 10993-12.
Число образцов, предоставляемых на исследования (испытания), должно быть таким, чтобы можно было провести статистическую обработку результатов и провести повторные исследования для исключения возможной ошибки или проверки атипичных результатов.
Исследуемые материалы и контрольные образцы следует обрабатывать в условиях, исключающих контаминацию.
Изучению подлежит такое количество материала, которое рассчитано по формуле
, (2)
где М — максимальное количество материала (в граммах), имплантируемое человеку;
Р — масса тела человека (70 кг);
K — коэффициент аггравации, равный 20.
Образцы для исследований (испытаний) имплантируемых МИ отбирают в соответствии с ГОСТ ISO 10993-6.
6.3 Общие требования к приготовлению экстрактов из исследуемых материалов
Общие требования к приготовлению экстрактов, используемых для оценки биологического действия материалов/МИ, установлены в ГОСТ ISO 10993-12. Условия приготовления экстрактов для конкретных групп материалов/МИ приведены в приложении А.
Примечание — Экстракты для исследования на лабораторных животных, в том числе и для испытания на пирогенность, готовят на 0,9%-ном стерильном апирогенном растворе хлорида натрия.
6.4 Общие требования к методам (методикам) оценки биологического действия
Методики исследования биологического действия изложены в соответствующих стандартах серии ГОСТ ISO 10993.
Специалист, планирующий исследования и составляющий их программу по испытаниям, вправе выбрать альтернативный метод испытаний, соответствующий применению МИ при условии его валидации.
6.5 Испытания на пирогенность, вызванную материалом
Испытание на пирогенность, вызванную материалом/МИ, на кроликах основано на измерении температуры тела до и после внутривенной инъекции растворов, полученных в результате экстракции исследуемого материала и контрольного материала.
Испытание на пирогенность следует проводить для материалов/МИ, контактирующих с системой кровообращения в одной точке и служащих для входа в кровеносную систему (см. ГОСТ ISO 10993-1, ГОСТ ISO 10993-11), контактирующих с циркулирующей кровью (см. ГОСТ ISO 10993-1, ГОСТ ISO 10993-11), контактирующих с кровью, ее компонентами, кровозаменяющими растворами (см. ГОСТ ISO 10993-11, ГОСТ 31209).
Примечание — Экстракты для испытания на пирогенность готовят на 0,9%-ном стерильном апирогенном растворе хлорида натрия с использованием апирогенной посуды, в асептических условиях.
7 Общие требования биологической безопасности по микробиологическим показателям
7.1 Общие положения
Целью микробиологических исследований является обнаружение микроорганизмов, их идентификация и изучение свойств (при необходимости) (см. [10]).
7.2 Испытания на стерильность
Одним из критериев биологической безопасности МИ является их стерильность.
Требования к МИ, прошедшим финишную стерилизацию, которые будут отнесены к категории «СТЕРИЛЬНЫЕ», установлены в ГОСТ EN 556-1.
Стерильность МИ подтверждают:
а) предварительной валидацией процесса стерилизации и последующими валидациями, показывающими, что данный процесс приемлем (см. [11]-[13]);
б) информацией, собранной во время текущего контроля стерильности и мониторинга процесса, показывающей, что валидированный процесс эффективно применен.
Требования к разработке, валидации и текущему контролю процесса стерилизации МИ изложены в соответствующих стандартах.
7.3 Микробиологическая чистота
Испытаниям на микробиологическую чистоту подвергают нестерилизуемые МИ с определяемыми микробиологическими характеристиками, например: МИ в форме исполнения крем или гель, руководствуясь НД изготовителя.
7.4 Антимикробные свойства
Антимикробные свойства МИ исследуют, оценивая антибактериальную способность (активность) на стандартных штаммах микроорганизмов.
Данные исследования проводят, руководствуясь соответствующими стандартами или НД на конкретный материал/МИ.
Методы (методики) исследования антимикробных свойств приведены в соответствующих стандартах.
7.5 Испытания на пирогенность, вызванную эндотоксинами
Описание теста лизата амебоцитов мечехвоста (ЛАЛ-тест) для определения эндотоксина in vitro в образцах материалов/МИ при использовании клеточных биологических систем см. в [14]. Испытание на бактериальные эндотоксины основано на специфической реакции реактива, представляющего собой лизат клеток крови (амебоцитов) мечехвоста с бактериальными эндотоксинами. В результате ферментативной реакции происходит изменение реакционной смеси, пропорциональное концентрации эндотоксина.
Следует учитывать, что использование ЛАЛ-реактива имеет ограничение и применимо не для всех МИ (например, биополимерные гели).
Другим методом in vitro является исследование активации моноцитов, количественный метод, который использует естественный человеческий механизм повышения температуры и может быть применен для теста на эндотоксины, связанные с материалами/МИ, а также на другие биологические пирогены (например, дрожжевые, паразитические и вирусные пирогены) (см. ГОСТ ISO 10993-11).
Так как результаты исследований могут значительно отличаться в зависимости от применяемого метода, то изготовитель МИ должен в документации на изделие привести валидированную методику определения концентрации бактериального эндотоксина.
8 Представление отчетов о результатах
Результаты исследований (испытаний) должны быть представлены точно, четко, недвусмысленно и объективно (см. ГОСТ ISO/IEC 17025).
Выводы, содержащиеся в протоколах, должны быть однозначными и понятными.
Протоколы с результатами исследований (испытаний) МИ могут сопровождаться экспертными заключениями, отчетами, пояснительными записками в зависимости от требований организации, в которую предоставляются результаты.
Приложение А
(рекомендуемое)
Условия приготовления экстрактов для определения санитарно-химических/физико-химических показателей
Таблица А.1 — Условия приготовления экстрактов для групп изделий из полимеров (экстрагирующая жидкость — вода дистиллированная)
|
Категория изделия |
Соотно- шение площади поверх- ности к объему экстраги- рующей жидкости (S/V), см /см
|
Соотно- шение массы изделия к объему экстраги- рующей жидкости (M/V), г/см
|
Соотно- шение количест- ва образцов к объему экстраги- рующей жидкости ( N/V ), шт./см
|
Темпе- ратура экстрак- ции, °С |
Продол- житель- ность экстрак- ции, ч |
Приме- чание |
|
Изделия поверхностного контакта |
||||||
|
Средства перевязочные, контактирующие с неповрежденной кожей: — повязки — пленки — бинты — пластыри фиксирующие — гигиенические салфетки |
1/1 |
— |
— |
37±1 |
24±2 |
— |
|
— вата медицинская гигроскопическая |
— |
1/500 |
— |
37±1 |
24±2 |
— |
|
— гипсовые бинты — бандажи — ортезы — шины — корсеты — ремни-фиксаторы |
1/1 |
— |
— |
37±1 |
24±2 |
— |
|
Средства перевязочные, контактирующие с поврежденными кожей и слизистыми оболочками: — противоожоговые повязки, гемостатические рассасывающиеся и др. — ватно-марлевые средства (салфетки, бинты, перевязочные пакеты) |
1,6/1 |
— |
— |
37±1 |
24±2 |
— |
|
— клеи, сорбенты, гемостатические порошки |
— |
М/250 |
— |
37±1 |
24±2 |
М — максимальная разовая доза |
|
— средства перевязочные пластырного типа |
1,6/1 |
— |
— |
37±1 |
24±2 |
— |
|
Гели для УЗИ |
— |
5/100 |
— |
37±1 |
2,0±0,1 |
— |
|
Линзы очковые для коррекции зрения |
1/1 |
— |
— |
37±1 |
24±2 |
— |
|
Оправы очковые |
— |
— |
3/500 |
37±1 |
24±2 |
— |
|
Протезы глазные |
— |
— |
1/50 |
37±1 |
24±2 |
— |
|
Одежда и белье для пациентов, персонала |
1/1 |
— |
— |
37±1 |
8±1 |
— |
|
Хирургическая одежда и белье, применяемые как МИ для пациентов, хирургического персонала |
1/1 |
— |
— |
37±1 |
24±2 |
— |
|
Изделия рентгенозащитные (фартуки, нагрудники, перчатки, бахилы) |
1/10 |
— |
— |
37±1 |
2,0±0,1 |
— |
|
Медицинские гигиенические прокладки, подгузники, пеленки |
1/1 |
— |
— |
37±1 |
24±2 |
После полного водопогло- щения |
|
Презервативы |
— |
— |
1/50 |
37±1 |
8±1 |
— |
|
Перчатки медицинские |
1/1 |
— |
— |
37±1 |
8±1 |
— |
|
Перчатки медицинские из латекса натурального каучука |
1/1 |
— |
— |
37±1 |
2,0±0,1 |
— |
|
Спринцовки, кружки Эсмарха, клизменные наконечники |
— |
— |
1/V |
37±1 |
2,0±0,1 |
Заполняют номинальным объемом |
|
Молокоотсосы |
— |
— |
1/V |
37±1 |
24±2 |
Заполняют номинальным объемом |
|
Моче- и калоприемники, подкладные судна, грелки, пузыри для льда |
1/5 |
— |
— |
37±1 |
2,0±0,1 |
— |
|
Экзопротезы грудной железы, изделия протезно-ортопедические и полуфабрикаты к ним |
1/2 |
— |
— |
37±1 |
24±2 |
— |
|
Внутриматочные контрацептивы, кольца |
— |
— |
1/50 |
37± 1 |
72±2 |
— |
|
Линзы контактные |
1/1 |
— |
— |
37±1 |
24±2 |
— |
|
Гинекологические инструменты: |
||||||
|
— зеркало по Куско |
— |
— |
1/500 |
37±1 |
2,0±0,1 |
— |
|
— шпатель, ложка Фолькмана, цервикальная щетка |
— |
— |
1/100 |
37±1 |
2,0±0,1 |
— |
|
Датчики, электроды и др. устройства, контактирующие с кожей |
— |
— |
1/250 |
37±1 |
2,0±0,1 |
— |
|
Камеры неонатальных инкубаторов |
1/1 |
— |
— |
37±1 |
24±2 |
— |
|
Ингаляторы |
— |
— |
1/V |
37±1 |
24±2 |
Заполняют номинальным объемом |
|
Слуховые аппараты |
— |
— |
1/50 |
37±1 |
24±2 |
— |
|
Криопакеты |
— |
— |
1/250 |
37±1 |
2,0±0,1 |
— |
|
Микросферы для противоожоговых кроватей |
— |
0,1/1 |
— |
37±1 |
72±2 |
— |
|
Изделия, присоединяемые извне |
||||||
|
Устройства комплектные эксфузионные, инфузионные и трансфузионные |
— |
— |
3/250 |
37±1 |
2,0±0,1 |
По приложению Б |
|
Аппараты и устройства для замещения функций органов и систем организма: |
||||||
|
— аппараты искусственного кровообращения, искусственной почки, для гемосорбции в комплекте с магистралями или без них и их функциональные элементы Магистрали кровопроводящие |
— |
— |
1/500 |
37±1 |
2,0±0,1 |
По приложению Б |
|
Катетеры внутрисосудистые, трубки медицинские для переливания крови, препаратов из крови, кровезаменителей и инфузионных растворов |
— |
— |
1/250 |
37±1 |
24±2 |
— |
|
Шприцы инъекционные однократного применения |
— |
— |
1/V |
37±1 |
24±2 |
Заполняют через иглу номинальным объемом |
|
Контейнеры для крови, препаратов из крови, кровезаменителей и инфузионных растворов: |
||||||
|
— 500 мл |
— |
— |
1/100 |
70±2 |
24±2 |
По ГОСТ 31209 |
|
— 300 мл |
— |
— |
1/50 |
70±2 |
24±2 |
По ГОСТ 31209 |
|
Катетеры, зонды, дренажи, бужи различных типов |
— |
— |
1/250 |
37±1 |
24±2 |
— |
|
Имплантируемые изделия |
||||||
|
Клапаны сердца, перикарда, опорные кольца, кардио- и нервно-мышечные стимуляторы; протезы кровеносных сосудов, грудной железы, гортани, яичка, фаллопротезы; имплантаты для челюстно-лицевой хирургии, протезы мягких тканей, имплантируемые датчики и устройства для постоянного дозируемого введения лекарственных веществ |
— |
— |
1/500 |
37±1 |
72±2 |
— |
|
Костные цементы, имплантируемые гели |
— |
М/500 |
— |
37±1 |
72±2 |
М — максимальная разовая доза |
|
Нити хирургические |
— |
— |
— |
37±1 |
24±2 |
Отношение длины нити (см) к объему дистиллирован- ной воды (мл) 0,4/1 |
|
Имплантаты для офтальмохирургии |
— |
1/1 |
— |
37±1 |
72±2 |
М — максимальная разовая доза |
|
Интраокулярные линзы |
— |
— |
1,0/0,3 |
37±1 |
72±2 |
— |
|
Изделия офтальмологические вискохирургические |
— |
1/1 |
— |
37± 1 |
8±1 |
М — максимальная разовая доза |
|
Упаковка |
||||||
|
Пробки для укупоривания сосудов с кровью, препаратов из крови, кровезаменителей и инфузионных растворов |
1/2 |
— |
— |
121±1 |
1,0±0,1 |
Автоклавирова- ние (P=1 атм) |
|
Емкости для хранения ЛС |
1/V |
— |
— |
37±1 |
24±2 |
Заполняют номинальным объемом |
|
Материалы для упаковки ЛС |
1/1 |
— |
— |
37±1 |
24±2 |
— |
|
Материалы для упаковки МИ |
1/10 |
— |
— |
37±1 |
24±2 |
— |
|
Медицинские стоматологические изделия и материалы |
||||||
|
Медицинские стоматологические изделия и материалы |
— |
— |
— |
— |
— |
По ГОСТ 31576 |
Таблица А.2 — Условия приготовления экстрактов для групп изделий из металлов, сплавов, керамики и стекла
|
Категория изделия |
Экстрагирующая жидкость (среда) |
Соотно- шение площади поверх- ности к объему экстраги- рующей жидкости (S/V), см /см
|
Соотно- шение массы изделия к объему экстраги- рующей жидкости (M/V), г/см
|
Соотно- шение количест- ва образцов к объему экстраги- рующей жидкости ( N/V ), шт./см
|
Темпе- ратура экстрак- ции, °С |
Продол- житель- ность экстрак- ции, ч |
Приме- чание |
|
Изделия поверхностного контакта |
|||||||
|
Оправы очковые |
Вода дистиллированная |
— |
— |
3/500 |
37±1 |
24±2 |
— |
|
Линзы очковые |
Вода дистиллированная |
— |
— |
3/500 |
37±1 |
24±2 |
— |
|
Пипетки глазные |
Вода дистиллированная |
— |
— |
1/V |
37±1 |
2,0±0,1 |
Заполняют номиналь- ным объемом |
|
Датчики, электроды, контактирующие с кожей, слизистыми |
0,9%-ный раствор хлорида натрия |
— |
— |
1/150 |
37±1 |
2,0±0,1 |
— |
|
Изделия, присоединяемые извне |
|||||||
|
Шприцы медицинские инъекционные многократного применения |
Вода дистиллированная |
— |
— |
1/V |
37±1 |
24±2 |
Заполняют номиналь- ным объемом |
|
Иглы инъекционные, скарификаторы |
0,9%-ный раствор хлорида натрия |
— |
— |
1/5 |
37±1 |
2,0±0,1 |
— |
|
Автономные желудочно- кишечные электростимуля- торы |
0,9%-ный раствор хлорида натрия |
— |
— |
1/100 |
37±1 |
24±2 |
— |
|
Иглы атравматические без нитей |
0,9%-ный раствор хлорида натрия |
— |
— |
1/5 |
37±1 |
2,0±0,1 |
— |
|
Хирургические, гинекологические и др. инструменты: |
|||||||
|
— многократного применения |
0,9%-ный раствор хлорида натрия |
— |
— |
1/150 |
37±1 |
24±2 |
— |
|
— однократного применения |
0,9%-ный раствор хлорида натрия |
— |
— |
1/150 |
37±1 |
2,0±0,1 |
— |
|
Кардиостимулято- ры, изделия для ортопедии (пластины для остеосинтеза, спицы и др. приспособления), протезы суставов, внутрисосудистые стенты |
0,9%-ный раствор хлорида натрия |
— |
— |
1/500 |
37±1 |
336±2 |
— |
|
Имплантируемые изделия |
|||||||
|
Нейрохирурги- ческие и сосудистые клипсы, скобы, сшивающие |
0,9%-ный раствор хлорида натрия |
— |
— |
1/50 |
37±1 |
336±2 |
— |
|
Материалы хирургические шовные (нити металлические) |
0,9%-ный раствор хлорида натрия |
— |
— |
— |
37±1 |
336±2 |
Отношение длины нити (см) к объему экстрагиру- ющей жидкости (мл) 0,4/1 |
|
Автономные желудочно- кишечные электростимуля- торы |
0,9%-ный раствор хлорида натрия |
— |
— |
1/100 |
37±1 |
24±2 |
— |
|
Упаковка |
|||||||
|
Тара для ЛС, контактирующих с внутренними средами организма, а также ЛС наружного применения |
Вода дистиллированная |
— |
— |
1/V |
37±1 |
72±2 |
Заполняют номиналь- ным объемом |
|
Медицинские стоматологические изделия и материалы |
|||||||
|
Медицинские стоматологи- ческие изделия и материалы |
— |
— |
— |
— |
— |
— |
По ГОСТ 31576 |
|
Примечания 1 Режимы экстракции, приведенные в приложении А, могут быть изменены, если эти изменения обоснованы (например, контрольные испытания). 2 Если МИ не вошли в настоящее приложение, то экстракцию проводят в соответствии с ГОСТ ISO 10993-12. |
Приложение Б
(обязательное)
Методика приготовления экстракта с использованием стенда
Б.1 Назначение стенда
Стенд предназначен для приготовления экстракта, применяемого при санитарно-химических/физико-химических испытаниях и оценке биологического действия МИ: устройств комплектных эксфузионных, инфузионных и трансфузионных, аппаратов и устройств для замещения функций органов и систем организма (аппаратов искусственного кровообращения, искусственной почки, аппаратов для гемосорбции в комплекте с магистралями или без них и их функциональных элементов, а также магистралей к перечисленным аппаратам).
Б.2 Описание стенда
Функциональная схема стенда должна соответствовать схеме, приведенной на рисунке Б.1.
|
|
1 — испытуемое МИ (при испытании эксфузионных, инфузионных и трансфузионных устройств последовательно соединяют три МИ); 2 — стеклянная емкость, заполненная 500 мл дистиллированной воды для всех видов МИ (для эксфузионных, инфузионных и трансфузионных устройств — 250 мл, для оксигенаторов — 1000 мл); 3 — термостат, поддерживающий температуру дистиллированной воды в испытательном контуре (37±1)°С; 4 — перистальтический насос, подключенный к испытательному контуру через инертную силиконовую трубку минимально возможной длины и обеспечивающий циркуляцию дистиллированной воды в контуре со скоростью потока 1 л/ч для эксфузионных, инфузионных и трансфузионных устройств, для остальных МИ скорость потока указана в инструкции по применению
Рисунок Б.1 — Функциональная схема стенда для приготовления экстракта
Б.3 Приготовление экстракта
Через замкнутый контур в режиме рециркуляции в течение 2 ч с указанной скоростью потока пропускают соответствующий объем дистиллированной воды при температуре (37±1)°С. Через 2 ч, не нарушая герметичности контура, извлекают колбу из термостата, охлаждают до комнатной температуры. В первую очередь непосредственно из контура проводят отбор проб для определения этиленоксида, циклогексанона и тетрагидрофурана методами хроматографии путем прокола иглой шприца полимерной трубки устройства. Остальную часть вытяжки сливают в стеклянную емкость со шлифом.
В качестве контрольного раствора используют соответствующий объем дистиллированной воды, пропущенной через замкнутый контур в тех же условиях, но без присоединения испытуемых МИ.
Приложение В
(рекомендуемое)
Показатели (характеристики), определяемые в экстрактах из исследуемых материалов в зависимости от их природы
Таблица В.1
|
Наименование исследуемых материалов в зависимости от их природы |
Наименование показателя (характеристики) |
|
1 Материалы полимерные синтетические, в зависимости от состава |
|
|
Материалы полимерные синтетические, общие показатели |
Восстановительные примеси Изменение значения рН Ультрафиолетовое поглощение в диапазоне длин волн от 220 до 360 нм включ. Наименования компонентов: — железо (Fe), — кадмий (Cd) , — медь (Cu), — олово (Sn), — свинец (Pb) , — хром (Cr) Формальдегид |
|
Полиэтилен, полипропилен и другие материалы на основе полиолефинов |
Ацетальдегид Ацетон Изопропанол Метанол Пропанол |
|
Полистирол и сополимеры на основе стирола |
Стирол Метанол |
|
Поливинилхлорид |
Ацетальдегид Ацетон Винилхлорид Изопропанол Метанол Пропанол Диоктилфталат (ДОФ) |
|
Полиорганосилоксаны (силиконы) |
Ацетальдегид Метанол Фенол |
|
Полиакрилаты |
Акрилонитрил Бутилакрилат Метилакрилат Метилметакрилат |
|
Полиамиды: |
|
|
— капрон (полиамид 6, поликапроамид); |
е-капролактам Бензол Фенол |
|
— нейлон (полиамид 66, полигексаметиленадипамид); — полиакриламид |
Гексаметилендиамин Метанол |
|
Акриламид |
|
|
Полиуретан |
Ацетальдегид Ацетон Изопропанол Метанол Пропанол Этиленгликоль |
|
Полиэфиры: — полиэтилентерефталат и сополимеры на основе терефталетовой кислоты; |
Ацетальдегид Ацетон Изопропанол Метанол Пропанол Этиленгликоль |
|
— поликарбонат; |
Фенол Дифенилолпропан |
|
— полисульфон |
Дифенилолпропан |
|
— эпоксидные смолы |
Дифенилолпропан Фенол Эпихлоргидрин |
|
Фторопласт (фторопласт-3, фторопласт-4, тефлон) |
Формальдегид |
|
2 Резины и латексы синтетические, в зависимости от состава |
|
|
Восстановительные примеси Изменение значения pH, ед. pH Ультрафиолетовое поглощение в диапазоне длин волн от 220 до 360 нм включ. Кадмий (Cd), Свинец (Pb), Цинк (Zn), Формальдегид Акрилонитрил |
|
|
Ацетальдегид Агидол-2 (2,2-метилен-бис (4-метил-6-трет-бутил-фенол) Изопрен Тиурам Д (тетраметилтиурамдисульфид) Тиурам Е (тетраэтилтиурамдисульфид) Дифенилгуанидин Альтакс (2,2-дибензтиазолдисульфид) Каптакс (2-меркаптобензтиазол) Сульфенамид Ц (циклогексил-2-бензтиазол-сульфенамид) Неозон Д (нафтам-2,- фенилнафтиламин) |
|
|
3 Материалы природного происхождения, в зависимости от состава |
|
|
Биологические ткани и их производные человеческого происхождения в нежизнеспособном состоянии, в зависимости от способа обработки, состава. Биологические ткани и их производные животного происхождения в нежизнеспособном состоянии, в зависимости от способа обработки, состава. Растительное сырье в зависимости от способа обработки, состава, в том числе: — латекс натурального каучука; — целлюлоза |
Восстановительные примеси Изменение значения рН Ультрафиолетовое поглощение в диапазоне длин волн от 220 до 360 нм включ. Наименования компонентов: — ванадий (V), — висмут (Bi), — железо (Fe), — кадмий (Cd), — кобальт (Co), — марганец (Mn), — медь (Cu), — мышьяк (As), — никель (Ni), — олово (Sn), |
|
— ртуть (Hg), — свинец (Pb), — хром (Cr), — цинк (Zn) Ацетальдегид Ацетон Изопропанол Метанол Пропанол Формальдегид |
|
|
Агидол-2 (2,2-метилен-бис (4-метил-6-трет-бутил-фенол) Изопрен Тиурам Д (тетраметилтиурамдисульфид) Дифенилгуанидин Альтакс (2,2-дибензтиазолдисульфид) Каптакс (2-меркаптобензтиазол) Сульфенамид Ц (циклогексил-2-бензтиазол-сульфенамид) Неозон Д (нафтам-2,- фенилнафтиламин) Этилацетат |
|
|
4 Металлы и сплавы, в зависимости от состава |
|
|
Изменение значения pH Наименования компонентов: — алюминий (Al), — ванадий (V), — вольфрам (W), — железо (Fe), — кобальт (Co), — марганец (Mn), — медь (Cu), — молибден (Mo), — никель (Ni), — титан (Ti), — хром (Cr), — цинк (Zn) |
|
|
5 Стекло и керамика, в зависимости от состава |
|
|
Изменение значения рН Наименования компонентов: — алюминий (Al), — бор (B), — кадмий (Cd), — кобальт (Co), — марганец (Mn), — медь (Cu), — мышьяк (As), — олово (Sn), — ртуть (Hg), — серебро (Ag), — свинец (Pb), — титан (Ti), — хром (Cr), — цинк (Zn) |
|
|
6 Агенты, стерилизующие и консервирующие, пластификаторы, рентгеноконтрастные вещества |
|
|
Барий (Ва) Глутаровый альдегид Формальдегид Этиленоксид Фталаты |
|
|
7 Растворители для склеивания деталей и узлов изделий |
|
|
Ацетон Бутан-2-он (метилэтилкетон) Тетрагидрофуран Циклогексанон |
|
|
________________ Для имплантируемых МИ. Примечание — В таблице приведены вещества, наиболее часто встречающиеся в МИ. В МИ могут находиться вещества, не встречающиеся в таблице. Определение веществ в конкретном МИ осуществляют с учетом результатов анализа риска. |
Приложение Г
(обязательное)
Допустимое количество экстрагируемых/выщелачиваемых веществ в экстрактах из исследуемых материалов с учетом анализа биологических рисков
Таблица Г.1
|
Определяемый показатель |
Массовая концентрация ДКМ, мг/дм , не более |
|
Тяжелые металлы: |
|
|
— ванадий (V) |
0,10 0,001
|
|
— висмут (Bi) |
0,10 |
|
— железо (Fe) |
0,30 |
|
— кадмий (Cd) |
0,001 0,0001
|
|
— кобальт (Co) |
0,10 |
|
— марганец (Mn) |
0,10 |
|
— медь (Cu) |
0,10 |
|
— мышьяк (As) |
0,05 |
|
— никель (Ni) |
0,10 0,01
|
|
— олово (Sn) |
1,0 |
|
— ртуть (Hg) |
0,005 0,0001
|
|
— свинец (Pb) |
0,030 |
|
— хром (Cr) |
0,10 0,050
|
|
Органические вещества |
— |
|
Ацетон |
0,10 |
|
Глутаровый альдегид |
0,05 |
|
Диоктилфталат (ДОФ) |
0,05 |
|
Бутан-2-он (метилэтилкетон) |
1,0 |
|
Тетрагидрофуран |
20,0 |
|
Формальдегид |
0,10 |
|
Циклогексанон |
2,50 |
|
Этиленоксид |
—
|
|
Для имплантируемых МИ. Допустимые уровни установлены в ГОСТ ISO 10993-7. Примечание — Если в МИ содержание вышеперечисленных веществ (компонентов) превышает ДКМ, то их количество должно быть обосновано. |
Приложение Д
(рекомендуемое)
Санитарно-химические/физико-химические показатели
Таблица Д.1 — Интегральные показатели
|
Определяемый показатель |
Допустимое значение, не более |
|
Восстановительные примеси, мл 0,02 Н раствора |
1,0 |
|
Изменение значения рН, ед. рН |
±1,0 |
|
Ультрафиолетовое поглощение, ед. О.П. в диапазоне длин волн, нм: |
|
|
— от 220 до 360 включ. |
0,30 |
|
— от 230 до 360 включ. |
0,20
|
|
— от 250 до 320 включ. |
0,10
|
|
Для контейнеров для крови и ее компонентов. Для укупорочных пробок и устройств комплектных эксфузионных, инфузионных и трансфузионных однократного применения. |
Таблица Д.2 — Допустимое количество миграции химических веществ (ДКМ)
|
Определяемый показатель |
Массовая концентрация ДКМ, мг/дм , не более |
|
Алюминий (Al) |
0,50 |
|
Барий (Ba) |
0,10
|
|
Бор (B) |
0,50 |
|
Вольфрам (W) |
0,050 |
|
Молибден (Mo) |
0,25 |
|
Олово (Sn) |
2,0 |
|
Серебро (Ag) |
0,050 |
|
Титан (Ti) |
0,10 |
|
Цинк (Zn) |
0,01
|
|
Агидол-2 (2,2-метилен-бис (4-метил-6-трет-бутил-фенол) |
2,0 |
|
Акрилонитрил |
0,050 |
|
Альтакс (2,2-дибензтиазолдисульфид) |
0,5
|
|
0,4 |
|
|
Ацетальдегид |
0,20 |
|
Бензол |
0,010 |
|
Бутилакрилат |
0,010 |
|
Винилхлорид |
0,010 |
|
Гексаметилендиамин |
0,010 |
|
Дифенилгуанидин |
0,5
|
|
0,15 |
|
|
Дифенилолпропан |
0,010 |
|
е-капролактам |
0,01 |
|
Каптакс (2-меркаптобензтиазол) |
0,4 |
|
Метилакрилат |
0,020 |
|
Метилметакрилат |
0,250 |
|
Неозон Д (нафтам-2, фенилнафтиламин) |
0,2 |
|
Изопропанол |
0,10 |
|
Метанол |
0,20 |
|
Пропанол |
0,10 |
|
Стирол |
0,010 |
|
Сульфенамид Ц (циклогексил-2-бензтиазолсульфенамид) |
0,4 |
|
Тиурам Д (тетраметилтиурамдисульфид) |
0,05
|
|
Фенол |
0,050 |
|
Эпихлоргидрин |
0,10 |
|
Этиленгликоль |
1,0 |
|
Этилацетат |
0,10 |
|
Этилцимат (диэтилдитиокарбамат цинка) |
0,05 0,6 |
|
Для МИ, контактирующих с кровью. Для имплантируемых МИ. Для МИ, контактирующих с кожей. Для укупорочных пробок и устройств комплектных эксфузионных, инфузионных и трансфузионных однократного применения. Примечание — Если в НД заявителя или по результатам исследований (испытаний) значения показателей превышают значения, указанные в таблице, то обоснование их безопасности проводят в соответствии с ГОСТ ISO 10993-17 (см. также [5]). |
Библиография
|
[1] |
Руководство ИСО/МЭК 51:2014* |
Аспекты безопасности. Руководящие указания по их включению в стандарты (Safety aspects — Guidelines for their inclusion in standards) |
|
[2] |
ИСО 10993 (все части) |
Оценка биологическая медицинских изделий (Biological evaluation of medical devices) |
|
[3] |
ИСО 22442 (все части) |
Изделия медицинские, использующие ткани животных и их производные (Medical devices utilizing animal tissues and their derivatives) |
|
[4] |
ИСО 13022:2012 |
Медицинская продукция, содержащая жизнеспособные человеческие клетки. Применение менеджмента риска и требования к методикам обработки (Medical products containing viable human cells — Application of risk management and requirements for processing practices) |
|
[5] |
ISO/TS 21726:2019 |
Оценка биологического действия медицинских изделий. Применение уровня предельно допустимого токсикологического воздействия (TTC) для оценки биосовместимости компонентов медицинских изделий [Biological evaluation of medical devices — Application of the threshold of toxicological concern (TTC) for assessing biocompatibility of medical device constituents] |
|
[6] |
ИСО 3696:1987 |
Вода для лабораторного анализа. Технические требования и методы испытаний (Water for analytical laboratory use; Specification and test methods) |
|
[7] |
ИСО 17034:2016 |
Общие требования к компетентности производителей стандартных образцов (General requirements for the competence of reference material producers) |
|
[8] |
Руководство ИСО 33:2015 |
Стандартные образцы. Надлежащая практика применения стандартных образцов (Reference materials — Good practice in using reference materials) |
|
[9] |
Руководство ИСО 80:2014 |
Руководство по внутрилабораторному изготовлению материалов для контроля качества (МКК) [Guidance for the in-house preparation of quality control materials (QCMs)] |
|
[10] |
ИСО 11737-2:2019 |
Стерилизация медицинских изделий. Микробиологические методы. Часть 1. Оценка популяции микроорганизмов на продукции (Sterilization of medical devices — Microbiological methods — Part 2: Tests of sterility performed in the definition, validation and maintenance of a sterilization process) |
|
[11] |
ИСО 17665-1:2006 |
Стерилизация медицинской продукции. Стерилизация паром. Часть 1. Требования к разработке, валидации и текущему контролю процесса стерилизации медицинских изделий (Sterilization of health care products — Moist heat — Part 1: Requirements for the development, validation and routine control of a sterilization process for medical devices) |
|
[12] |
ИСО 11135:2014 |
Стерилизация медицинской продукции. Этиленоксид. Требования к разработке, валидации и текущему контролю процесса стерилизации медицинских изделий (Sterilization of health-care products — Ethylene oxide — Requirements for the development, validation and routine control of a sterilization process for medical devices) |
|
[13] |
ИСО 11137-1:2006 |
Стерилизация медицинской продукции. Облучение. Часть 1. Требования к разработке, валидации и текущему контролю процесса стерилизации медицинских приборов (Sterilization of health care products — Radiation — Part 1: Requirements For development, validation and routine control of a sterilization process for medical devices) |
|
[14] |
ИСО 29701:2010 |
Нанотехнологии. Испытания эндотоксинов на образцах наноматериалов для систем in vitro. Испытание Limulus amebocyte lysate (LAL) [Nanotechnologies — Endotoxin test on nanomaterial samples for in vitro systems — Limulus amebocyte lysate (LAL) test] |
|
УДК 615.46:006.354 |
ОКС 11.100.20 |
|
Ключевые слова: медицинские изделия, система оценки соответствия в области безопасности, общие требования биологической безопасности, физико-химические исследования, санитарно-химические испытания, токсикологические испытания |
1 сообщение в этой теме
Рекомендуемые сообщения

inara
0
Опубликовано
-
- Жалоба
- Поделиться
Опубликовано
Ищу ISO Guide 80:2014 «Руководство по внутрилабораторному изготовлению материалов для контроля качества» на русском языке. Пожалуйста, может кто-то имеет перевод любого качества! Спасибо!
Ссылка на комментарий
Поделиться на других сайтах
В архиве
Эта тема в настоящий момент находится в архиве и закрыта для публикации сообщений.