12.09.2022

Описание препарата Алеценза® (капсулы, 150 мг) основано на официальной инструкции, утверждено компанией-производителем в 2022 году
Дата согласования: 12.09.2022
Особые отметки:
![]()
![]()
Содержание
- Действующее вещество
- ATX
- Фармакологическая группа
- Нозологическая классификация (МКБ-10)
- Состав
- Описание лекарственной формы
- Фармакологическое действие
- Фармакодинамика
- Фармакокинетика
- Показания
- Противопоказания
- Применение при беременности и кормлении грудью
- Способ применения и дозы
- Побочные действия
- Взаимодействие
- Передозировка
- Особые указания
- Форма выпуска
- Производитель
- Условия отпуска из аптек
- Условия хранения
- Срок годности
- Заказ в аптеках Москвы
Действующее вещество
ATX
Фармакологическая группа
Состав
| Капсулы | 1 капс. |
| действующее вещество: | |
| алектиниб | 150 мг |
| (в виде алектиниба гидрохлорида – 161,33 мг) | |
| вспомогательные вещества: лактозы моногидрат — 33,67 мг; гидроксипропилцеллюлоза (гипролоза) — 15 мг; натрия лаурилсульфат — 75 мг; кальция карбоксиметилцеллюлоза (кальция кармеллоза) — 43,35 мг; магния стеарат — 1,65 мг | |
| оболочка капсулы: каррагинан — 0,25 мг; калия хлорид — 0,42 мг; титана диоксид (Е171) — 4,2 мг; воск карнаубский — <0,110; крахмал кукурузный — <0,110; гипромеллоза — 61,63 мг | |
| чернила для нанесения надписи на капсуле: краситель железа оксид красный (Е172); краситель железа оксид желтый (Е172); алюминиевый лак FD&C Blue No.2 (Е132); воск карнаубский; шеллак белый; глицерил моноолеат; 1-бутанол; спирт этиловый безводный |
Описание лекарственной формы
Твердые капсулы, размер 1, корпус и крышечка капсулы от белого до желтовато-белого цвета.
На корпусе капсулы нанесена надпись «150 mg» черного цвета. На крышечке капсулы нанесена надпись «ALE» черного цвета.
Фармакологическое действие
Фармакологическое действие
—
противоопухолевое.
Фармакодинамика
Механизм действия. Алектиниб является мощным высокоселективным ингибитором тирозинкиназ ALK (anaplastic lymphoma kinase; киназа анапластической лимфомы) и RET (REarranged during Transfection). В доклинических исследованиях ингибирование активности тирозинкиназы ALK приводило к блокаде нисходящих сигнальных путей, включая STAT 3 (переносчик сигнала и активатор транскрипции 3) и PI3K/AKT (фосфоинозитол-3-киназу/протеинкиназу В), и вызывало апоптоз опухолевых клеток.
В условиях in vitro и in vivo алектиниб проявлял активность в отношении мутантных форм фермента ALK, в т.ч. с мутациями, обуславливающими резистентность к кризотинибу.
Основной метаболит алектиниба — М4 — в условиях in vitro показал эффективность и активность, сопоставимую с алектинибом.
Согласно данным доклинических исследований, алектиниб не является субстратом Р-gp или BCRP, выполняющих функцию белков-переносчиков в ГЭБ, и таким образом обладает способностью проникать в ЦНС и задерживаться в ней.
Алектиниб индуцирует регрессию опухоли на моделях ксенотрансплантатов опухоли у мышей, в т.ч. демонстрирует противоопухолевую активность в мозге, и увеличивает выживаемость на моделях внутричерепных опухолей у животных.
Фармакокинетика
Фармакокинетические показатели алектиниба и М4 оценивали у пациентов с ALK-положительным немелкоклеточным раком легкого (НМРЛ) и здоровых добровольцев. Средние геометрические (коэффициент вариации, %) показатели Cmax, Cmin и AUC0–12 в равновесном состоянии для алектиниба составляли ~665 нг/мл (44,3%), 572 нг/мл (47,8%) и 7430 нг·ч/мл (45,7%) соответственно. Средние геометрические показатели Cmax, Cmin и AUC0–12 в равновесном состоянии для М4 составляли ~246 нг/мл (45,4%), 222 нг/мл (46,6%) и 2810 нг·ч/мл (45,9%) соответственно.
Всасывание. После перорального приема алектиниба у пациентов с ALK-положительным НМРЛ в дозе 600 мг 2 раза в сутки после приема пищи отмечалось быстрое всасывание препарата. Tmax составляло ~ от 4 до 6 ч.
При приеме алектиниба в дозе 600 мг 2 раза в сутки равновесное состояние алектиниба достигается в течение 7 дней и остается неизменным, при этом согласно данным популяционного фармакокинетического анализа средний геометрический коэффициент накопления равен 5,6. Популяционный фармакокинетический анализ показал пропорциональность доз алектиниба в диапазоне от 300 до 900 мг после приема пищи.
У здоровых добровольцев абсолютная биодоступность при применении алектиниба после приема пищи составляла 36,9% (33,9–40,3%).
При однократном пероральном применении алектиниба в дозе 600 мг прием высококалорийной пищи с большим содержанием жиров увеличивал экспозицию препарата в 3 раза по сравнению с приемом натощак (среднее геометрическое соотношение комбинированных показателей для алектиниба и М4: Cmax — 3,31 (2,79–3,93), AUCinf — 3,11 (2,73–3,55).
Распределение. Алектиниб и M4 имеют высокий уровень связывания с белками плазмы крови человека (>99%) независимо от концентрации действующего вещества. В условиях in vitro среднее соотношение концентраций в крови и плазме человека составляет 2,64 и 2,5 для алектиниба и М4 соответственно (при клинически значимых концентрациях).
Средний геометрический Vss алектиниба после в/в введения составлял 475 л, указывая на широкое распределение алектиниба в тканях.
Метаболизм. Исследования метаболизма в условиях in vitro показали, что изофермент CYP3A4 является основным изоферментом CYP, который опосредует метаболизм алектиниба и М4, при этом определено, что данный изофермент осуществляет 40–50% метаболизма в гепатоцитах человека. Согласно результатам исследования массового баланса алектиниб и М4 являлись основными циркулирующими веществами в плазме крови, их общее содержание составляло 76% от общей радиоактивности в плазме крови. Среднее геометрическое соотношение метаболит/препарат в равновесном состоянии — 0,399.
Выведение. У здоровых добровольцев после однократного перорального применения 14С-меченого алектиниба бóльшая часть радиоактивности была выявлена в кале (средняя степень извлечения 97,8%, диапазон 95,6–100%), минимальная — в моче (средняя степень извлечения 0,46%, диапазон 0,3–0,6%). Доля неизмененного алектиниба в кале составляла 84% от введенной дозы, а доля М4 составила 5,8%.
Согласно данным популяционного фармакокинетического анализа, CL/F алектиниба составлял 81,9 л/ч, М4 — 217 л/ч. Индивидуальный расчетный средний геометрический T1/2 алектиниба составлял 32,5 ч, M4 — 30,7 ч.
Особые группы пациентов
Пациенты детского возраста. Исследования фармакокинетики алектиниба у пациентов детского возраста (<18 лет) не проводились.
Пациенты пожилого возраста. Возраст не оказывает влияния на экспозицию алектиниба.
Пациенты с нарушением функции почек. Алектиниб и М4 выводятся почками в незначительных количествах в неизмененном виде (<0,2% дозы). Данные, полученные у пациентов с нарушением функции почек легкой и средней степени тяжести, показывают, что нарушение функции почек не оказывает значительного влияния на фармакокинетику алектиниба. Специальные исследования фармакокинетики не проводились, данные популяционной фармакокинетики для пациентов с нарушением функции почек тяжелой степени отсутствуют, однако коррекции дозы у таких пациентов не требуется, поскольку алектиниб выводится почками в незначительной степени.
Пациенты с нарушением функции печени. Основным путем выведения алектиниба является печеночный метаболизм. У пациентов с нарушением функции печени возможно увеличение концентрации алектиниба и/или М4 в плазме крови. Согласно данным популяционного фармакокинетического анализа экспозиции алектиниба и М4 у пациентов с нарушением функции печени легкой степени тяжести (общий билирубин (ОБ) на исходном уровне ≤ВГН и активность АСТ >ВГН или ОБ на исходном уровне >1–1,5 × ВГН и любая активность АСТ на исходном уровне) соответствовали таковым у пациентов с нормальной функцией печени (ОБ ≤ВГН и активность АСТ ≤ВГН).
После однократного перорального приема алектиниба в дозе 300 мг пациентами с нарушением функции печени тяжелой степени (класс С по Чайлд-Пью) Сmax алектиниба была такой же как и у здоровых добровольцев; AUCinf у пациентов с нарушением функции печени тяжелой степени (класс С по Чайлд-Пью) была в 2,2 раза выше, чем у здоровых добровольцев, а Сmax и AUCinf для М4 были на 39 и 34% ниже соответственно. Таким образом, общая комбинированная экспозиция для алектиниба и М4 (AUCinf) была в 1,8 раза выше у пациентов с нарушением функции печени тяжелой степени по сравнению со здоровыми добровольцами.
В ходе изучения применения препарата у пациентов с нарушением функции печени средней степени тяжести (класс B по Чайлд-Пью) наблюдалась умеренно повышенная экспозиция алектиниба по сравнению со здоровыми добровольцами. Однако у пациентов с нарушением функции печени средней степени тяжести (класс B по Чайлд-Пью) в целом не отмечалось отклонений в показателях билирубина, альбумина или ПВ, что указывает на их возможное неполное соответствие пациентам с нарушением функции печени умеренной степени тяжести со сниженной метаболической способностью.
Показания
Монотерапия местно-распространенного или метастатического немелкоклеточного рака легкого с опухолевой экспрессией киназы анапластической лимфомы (ALK-положительный).
Противопоказания
- повышенная чувствительность к алектинибу или другим вспомогательным веществам препарата в анамнезе;
- беременность;
- период грудного вскармливания;
- детский возраст до 18 лет (эффективность и безопасность применения не изучались).
С осторожностью: нарушение функции почек тяжелой степени; редкая наследственная непереносимость галактозы, врожденная недостаточность лактазы или глюкозо-галактозная мальабсорбция.
Применение при беременности и кормлении грудью
Контрацепция. Пациентки или половые партнерши пациентов, обладающие детородным потенциалом, должны использовать высокоэффективные методы контрацепции во время терапии препаратом Алеценза® и в течение не менее 3 мес после приема последней дозы препарата.
Беременность. Женщинам детородного потенциала необходимо избегать беременности при приеме препарата Алеценза®. Клинические исследования препарата Алеценза® у беременных женщин не проводились. В силу своего механизма действия препарат Алеценза® может оказывать повреждающее воздействие на плод.
Доклинические данные. В исследованиях на животных алектиниб вызывал развитие эмбриофетальной токсичности.
Период грудного вскармливания. Неизвестно, выводится ли препарат Алеценза® с грудным молоком. Исследований по оценке влияния препарата Алеценза® на выработку грудного молока или наличию препарата Алеценза® в грудном молоке не проводилось. Поскольку большинство препаратов проникает в грудное молоко и риск для грудных детей не может быть исключен, пациенткам следует воздержаться от грудного вскармливания на фоне приема препарата Алеценза®.
Способ применения и дозы
Реклама: ООО «РЛС-Патент», ИНН 5044031277, erid=4CQwVszH9pUkpHxmQQo
Внутрь. Капсулы препарата Алеценза® следует принимать одновременно с приемом пищи и проглатывать целиком. Открывать или растворять капсулы нельзя.
Перед применением препарата Алеценза® в первой линии терапии НМРЛ необходимо установить наличие опухолевой экспрессии ALK. Для выявления пациентов с ALK-положительным НМРЛ необходимо использовать валидированный метод определения ALK.
Рекомендуемая доза препарата Алеценза® составляет 600 мг (4 капс. по 150 мг) 2 раза в сутки (суточная доза составляет 1200 мг).
Пациенты с исходным нарушением функции печени тяжелой степени (класс С по Чайлд-Пью) должны получать препарат Алеценза® в начальной дозе 450 мг 2 раза в сутки (суточная доза составляет 900 мг).
Продолжительность лечения. Лечение препаратом Алеценза® рекомендуется проводить до прогрессирования заболевания или до развития непереносимой токсичности.
Задержка приема или пропуск дозы. Если прием очередной дозы пропущен, ее можно принять позднее, при условии, что интервал между приемом пропущенной дозы и приемом следующей запланированной дозы составляет не менее 6 ч. Несмотря на возникновение рвоты после приема препарата Алеценза®, следующую дозу необходимо принять в запланированное время.
Коррекция дозы. Для контроля нежелательных явлений может потребоваться временное прерывание лечения, уменьшение дозы или полное прекращение применения препарата Алеценза®. Дозу препарата необходимо уменьшать пошагово, на 150 мг 2 раза в сутки, в зависимости от переносимости. Если применение препарата в дозе 300 мг 2 раза в сутки невозможно из-за непереносимости, терапию препаратом Алеценза® следует полностью прекратить.
В таблице 1 ниже приведены общие рекомендации по коррекции дозы препарата Алеценза®.
Таблица 1
Схема снижения дозы
| Схема снижения дозы | Уровень дозы |
| Доза | 600 мг 2 раза в сутки |
| Первое снижение дозы | 450 мг 2 раза в сутки |
| Второе снижение дозы | 300 мг 2 раза в сутки |
Таблица 2
Коррекция дозы при развитии отдельных нежелательных реакций
| Нежелательная реакция, степень тяжести | Терапия препаратом Алеценза® |
| Интерстициальная болезнь легких (ИБЛ)/пневмонит (все степени тяжести) | Следует немедленно прервать лечение и полностью прекратить терапию, если иные потенциальные причины возникновения ИБЛ/пневмонита не установлены |
| Повышение активности АЛТ или АСТ до ≥3-й степени тяжести (>5 × ВГН), с сопутствующим повышением концентрации ОБ ≤2 × ВГН | Временно приостановить терапию; дождаться восстановления показателей до исходного уровня или до ≤1-й степени тяжести (≤3 × ВГН), затем возобновить применение препарата в сниженной дозе (см. таблицу 1) |
| Повышение активности АЛТ или АСТ до ≥2-й степени тяжести (>3 × ВГН), с сопутствующим повышением концентрации ОБ >2 × ВГН (в отсутствие холестаза или гемолиза) | Следует полностью прекратить терапию препаратом Алеценза® |
| Брадикардияа 2-й или 3-й степени тяжести (симптоматическая, возможно тяжелая и клинически значимая, требующая медицинского вмешательства) | Следует временно приостановить терапию препаратом; дождаться уменьшения степени тяжести брадикардии до ≤1-й степени (без симптомов) или восстановления ЧСС до 60 уд./мин. Необходимо оценить действие сопутствующих препаратов с известной способностью вызывать брадикардию, а также действие антигипертензивных препаратов. Если способствующий одновременно применяемый препарат был выявлен и отменен или была произведена коррекция его дозы, следует возобновить терапию препаратом Алеценза® в исходной дозе после уменьшения степени тяжести брадикардии до ≤1-й степени (без симптомов) или увеличения ЧСС до ≥60 уд./мин. Если такой препарат не был выявлен или не был отменен, или не произведена коррекция его дозы, следует возобновить терапию препаратом Алеценза® в сниженной дозе (см. таблицу 1) после уменьшения степени тяжести брадикардии до ≤1-й степени (без симптомов) или увеличения ЧСС до ≥60 уд./мин |
| Брадикардияа 4-й степени тяжести (с жизнеугрожающими последствиями, требующая незамедлительного медицинского вмешательства) | Если способствующий одновременно применяемый препарат не был выявлен, следует полностью прекратить терапию препаратом Алеценза®. Если такой препарат выявлен и отменен или произведена коррекция его дозы, следует возобновить терапию препаратом Алеценза® в сниженной дозе (см. таблицу 1) после уменьшения степени тяжести брадикардии до ≤1-й степени (без симптомов) или увеличения ЧСС до ≥60 уд./мин при условии постоянного наблюдения в соответствии с клиническими показаниями. При повторном возникновении брадикардии применение препарата Алеценза® следует полностью прекратить |
| Повышение активности КФК >5 × ВГН | Следует временно приостановить терапию препаратом и дождаться восстановления активности КФК до исходного уровня или до ≤2,5 × ВГН, затем возобновить применение препарата в исходной дозе |
| Повышение активности КФК >10 х ВГН или повторное повышение активности КФК >5 х ВГН | Следует временно приостановить терапию препаратом и дождаться восстановления активности КФК до исходного уровня или до ≤2,5 × ВГН, затем возобновить применение препарата в сниженной дозе (см. таблицу 1) |
аЧСС <60 уд./мин.
Дозирование в особых случаях
Пациенты детского возраста. Безопасность и эффективность препарата Алеценза® у детей и подростков (<18 лет) не изучены.
Пациенты пожилого/старческого возраста. Коррекции дозы препарата Алеценза® у пациентов ≥65 лет не требуется.
Пациенты с нарушением функции почек. Коррекции дозы препарата Алеценза® у пациентов с нарушением функции почек легкой и средней степени тяжести не требуется. Применение препарата Алеценза® у пациентов с нарушением функции почек тяжелой степени не изучено, однако поскольку алектиниб выводится почками в незначительной степени, коррекции дозы у таких пациентов не требуется.
Пациенты с нарушением функции печени. Пациентам с исходным нарушением функции печени легкой (класс А по Чайлд-Пью) или средней степени тяжести (класс В по Чайлд-Пью) коррекции начальной дозы не требуется. Пациенты с исходным нарушением функции печени тяжелой степени (класс С по Чайлд-Пью) должны получать препарат Алеценза® в начальной дозе 450 мг 2 раза в сутки (суточная доза составляет 900 мг). Рекомендуется соответствующее наблюдение (контроль за показателями функции печени) для всех пациентов с нарушением функции печени.
Побочные действия
Безопасность препарата Алеценза® оценивали в клинических исследованиях (КИ) у пациентов с ALK-положительным НМРЛ, ранее получавших терапию кризотинибом, при применении в рекомендованной дозе 600 мг 2 раза в сутки; медиана экспозиции составила 11 мес (0–35 мес). Безопасность препарата Алеценза® также оценивали в КИ у пациентов с ALK-положительным НМРЛ, ранее не получавших лечение, при применении в рекомендованной дозе 600 мг 2 раза в сутки; медиана экспозиции составила 17,9 мес.
Наиболее частыми (≥20%) нежелательными реакциями были запор (36%), отеки (34%, включая периферический, генерализованный, отек век и периорбитальный отек); миалгия (31%, включая миалгию и скелетно-мышечную боль), тошнота (22%), повышение концентрации билирубина (21%, включая повышение концентрации билирубина в крови, гипербилирубинемию и повышение концентрации конъюгированного билирубина), анемия (20%, включая анемию и снижение уровня Hb) и сыпь (20%, включая сыпь, макуло-папулезную сыпь, акнеиформный дерматит, эритему, генерализованную сыпь, папулезную сыпь, зудящую сыпь и макулярную сыпь).
Ниже представлен перечень нежелательных реакций, отмечавшихся в КИ у пациентов с ALK-положительным НМРЛ, ранее получавших терапию кризотинибом, и у пациентов с ALK-положительным НМРЛ, ранее не получавших лечение. Нежелательные реакции сгруппированы в соответствии с классами систем органов MedDRA.
Для описания частоты нежелательных реакций используется следующая классификация: очень часто (≥10%); часто (≥1% и <10%); нечасто (≥0,1% и <1%); редко (≥0,01% и <0,1%); очень редко (<0,01%).
Со стороны крови и лимфатической системы: очень часто — анемия1.
Со стороны нервной системы: часто — дисгевзия (нарушение вкусовых восприятий)#, 2.
Со стороны органа зрения: очень часто — расстройства зрения3.
Со стороны сердца: очень часто — брадикардия4.
Со стороны дыхательной системы, органов грудной клетки и средостения: часто — ИБЛ/пневмонит.
Со стороны ЖКТ: очень часто — запор, тошнота, диарея, рвота; часто — стоматит#, 5.
Со стороны печени и желчевыводящих путей: очень часто — повышение концентрации билирубина6, повышение активности АСТ, повышение активности АЛТ; нечасто — лекарственно-индуцированное поражение печени7.
Со стороны кожи и подкожных тканей: очень часто — сыпь8, реакция фоточувствительности.
Со стороны скелетно-мышечной системы и соединительной ткани: очень часто — миалгия9, повышение активности КФК в крови.
Со стороны почек и мочевыводящих путей: часто — повышение концентрации креатинина в крови*, острая почечная недостаточность*, #.
Общие расстройства и нарушения в месте введения: очень часто — отеки10.
Лабораторные и инструментальные данные: часто — увеличение массы тела#.
*Включая одно явление с летальным исходом.
#Явление, отмечено в КИ у пациентов с ALK-положительным НМРЛ, ранее не получавших лечение.
1Включая случаи анемии и снижения уровня Hb.
2Включая случаи дисгевзии и гипогевзии (снижение вкусовых восприятий).
3Включая случаи нечеткости зрения, нарушения зрения, плавающих помутнений в стекловидном теле, снижения остроты зрения, астенопии и диплопии.
4Включая случаи брадикардии и синусовой брадикардии.
5Включая случаи стоматита и язвенного стоматита.
6Включая случаи повышения концентрации билирубина в крови, гипербилирубинемии, повышения концентрации конъюгированного билирубина.
7Включая одного пациента с лекарственно-индуцированным поражением печени (термин MedDRA) и одного пациента с повышением активности АСТ и АЛТ 4-й степени и задокументированным лекарственно-индуцированным поражением печени, подтвержденным биопсией.
8Включая случаи сыпи, макуло-папулезной сыпи, акнеиформного дерматита, эритемы, генерализованной сыпи, папулезной сыпи, зудящей сыпи и макулярной сыпи.
9Включая случаи миалгии и скелетно-мышечной боли.
10Включая случаи отека, отека век и периферического, генерализованного, периорбитального отека.
Описание отдельных нежелательных реакций
Профиль безопасности препарата Алеценза® в целом был сходным в КИ у пациентов с ALK-положительным НМРЛ, ранее получавших терапию кризотинибом, и у пациентов, ранее не получавших лечение.
ИБЛ/пневмонит
При применении препарата Алеценза® отмечались тяжелые случаи ИБЛ/пневмонита. В КИ у 0,4% пациентов с ALK-положительным НМРЛ, ранее получавших терапию кризотинибом, развивалась ИБЛ 3-й степени тяжести, что привело к прекращению терапии препаратом Алеценза®. Явлений ИБЛ с летальным исходом ни в одном КИ отмечено не было.
Гепатотоксичность
Повышение активности АСТ/АЛT 3–4 степени тяжести отмечалось в КИ у двух пациентов с ALK-положительным НМРЛ, ранее получавших терапию кризотинибом; при этом было задокументировано лекарственно-индуцированное поражение печени, подтвержденное биопсией.
Повышение активности АСТ и АЛТ отмечалось у 16 и 14% пациентов в КИ соответственно. Большинство явлений были 1-й и 2-й степеней тяжести; явления ≥3-й степени тяжести наблюдались у 2,8 и 3,2% пациентов соответственно. Данные явления обычно развивались в течение первых 3 мес терапии, были, как правило, преходящими и разрешались после временной приостановки лечения (у 1,2 и 3,2% пациентов соответственно) или снижения дозы препарата Алеценза® (у 1,6 и 0,8% соответственно). Повышение активности АСТ и АЛТ приводило к прекращению терапии препаратом Алеценза® у 1,2 и 1,6% пациентов соответственно.
Повышение концентрации ОБ отмечалось в КИ у 17% пациентов с ALK-положительным НМРЛ, ранее получавших терапию кризотинибом. Большинство явлений были 1-й и 2-й степеней тяжести; явления 3-й степени тяжести наблюдались у 3,2% пациентов. Данные явления обычно отмечались в течение первых 3 мес терапии, были преходящими и разрешались после временной приостановки лечения (у 4,7% пациентов) или снижения дозы препарата Алеценза® (у 2,8% пациентов). Повышение концентрации билирубина приводило к прекращению терапии препаратом Алеценза® у 1,6% пациентов.
Одновременное повышение активности АЛТ или АСТ ≥3 × ВГН и повышение концентрации ОБ ≥2 × ВГН при нормальной активности ЩФ отмечалось у одного пациента в КИ препарата Алеценза®.
Брадикардия
Случаи брадикардии отмечались в КИ у 7,9% пациентов с ALK-положительным НМРЛ, ранее получавших терапию кризотинибом. Все явления были 1-й или 2-й степени тяжести. У 20% пациентов после приема препарата Алеценза® ЧСС составляла <50 уд./мин.
Тяжелая миалгия и повышение активности КФК
Сообщения о миалгии, включавшие миалгию (25%) и скелетно-мышечную боль (7,5%), регистрировались в КИ у пациентов с ALK-положительным НМРЛ, ранее получавших терапию кризотинибом, с общей частотой 31%. Большинство явлений были 1-й или 2-й степени тяжести, у 1,2% пациентов наблюдались явления 3-й степени тяжести. Только 0,8% пациентов потребовалась коррекция дозы в связи с данными явлениями.
Повышение активности КФК отмечалось в КИ у 46% пациентов с доступными лабораторными данными. Частота повышения активности КФК 3-й степени тяжести составила 5%; при этом медиана времени до возникновения явления составила 14 дней у пациентов с ALK-положительным НМРЛ, ранее получавших терапию кризотинибом, и 27,5 дней у пациентов с ALK-положительным НМРЛ, ранее не получавших лечение. Коррекция дозы препарата Алеценза® потребовалась у 4% пациентов с повышением активности КФК.
Отклонения со стороны лабораторных показателей
У пациентов с ALK-положительным НМРЛ, ранее получавших терапию кризотинибом, и у пациентов, ранее не получавших лечение, на фоне терапии препаратом Алеценза® наблюдались отклонения со стороны лабораторных показателей, представленные в таблице 3.
Таблица 3
| Показатель | Алектиниб | |
| Все степени (%) | 3–4-я степень тяжести (%)° | |
| Биохимия | ||
| Повышение концентрации креатинина в крови** | 38# | 3,4# |
| Повышение активности АСТ | 53* | 6,2# |
| Повышение активности АЛТ | 40# | 6,1# |
| Повышение активности КФК в крови | 46* | 5* |
| Повышение концентрации билирубина в крови | 53# | 5,5# |
| Гематология | ||
| Снижение уровня Hb | 62# | 6,8# |
Примечание: лабораторные отклонения определялись с учетом нормальных значений по классификации NCI CTCAE (Национального института рака, National Cancer Institute Common Terminology Criteria of Adverse Events).
*Частота, отмечавшаяся в КИ у пациентов с ALK-положительным НМРЛ, ранее получавших терапию кризотинибом.
**Только пациенты с повышением концентрации креатинина на основании определения ВГН по классификации нежелательных явлений NCI СТСAE.
#Частота, отмечавшаяся в КИ у пациентов с ALK-положительным НМРЛ, ранее не получавших лечение.
°Отклонений лабораторных показателей 5-й степени тяжести не наблюдалось.
Пострегистрационный опыт применения
При пострегистрационном применении препарата Алеценза® отмечалось повышение активности ЩФ; при этом в КИ данное явление наблюдалось у 7,5% пациентов с ALK-положительным НМРЛ, ранее получавших терапию кризотинибом.
Взаимодействие
Реклама: ООО «ВЕДАНТА», ИНН 7714886235, erid 4CQwVszH9pUkKJ7jUDd
Реклама: ООО «РЛС-Библиомед» ИНН 7714758963
Реклама: ООО «ВЕДАНТА», ИНН 7714886235, erid 4CQwVszH9pUkKJ7jUDd
Реклама: ООО «РЛС-Библиомед» ИНН 7714758963
Влияние алектиниба на сопутствующие лекарственные препараты
Субстраты изоферментов цитохрома P450
В условиях in vitro ни алектиниб, ни его основной активный метаболит — М4 — не оказывали ингибирующего действия на изоферменты CYP1A2, CYP2B6, CYP2C9, CYP2C19 или CYP2D6 в клинически значимых концентрациях. Алектиниб и M4 оказывают слабое, зависящее от времени, ингибирующее действие на CYP3A4. Кроме того, алектиниб является слабым индуктором изоферментов CYP3A4 и CYP2B6 в клинических концентрациях in vitro.
Согласно результатам исследования межлекарственного взаимодействия у пациентов с ALK-положительным НМРЛ, многократное применение алектиниба не оказывает влияния на экспозицию мидазолама, который является чувствительным субстратом CYP3A. Таким образом, при одновременном применении с субстратами CYP3A коррекции дозы не требуется.
Несмотря на то что исследования в условиях in vitro показывают, что алектиниб является ингибитором изофермента CYP2C8, данные фармакокинетического моделирования, учитывающего физиологические процессы организма человека, свидетельствуют о том, что алектиниб в клинически значимых концентрациях не будет увеличивать плазменные концентрации одновременно применяемых препаратов-субстратов изофермента CYP2C8.
Субстраты P-gp и BCRP
В условиях in vitro алектиниб и М4 являются ингибиторами P-gp и BCRP. Таким образом, алектиниб и М4 могут способствовать увеличению плазменных концентраций субстратов Р-gp или BCRP при их одновременном применении (не ожидается, что увеличение экспозиции будет более чем двукратным). Рекомендуется соответствующее наблюдение пациентов, которые одновременно принимают алектиниб и субстраты Р-gp или BCRP с узким терапевтическим индексом (например, дигоксин, дабигатран, метотрексат).
Влияние сопутствующих лекарственных препаратов на алектиниб
Согласно данным, полученным in vitro, изофермент CYP3A4 является основным ферментом, ответственным за метаболизм алектиниба и М4, при этом предполагается, что посредством CYP3A осуществляется 40–50% общего печеночного метаболизма. М4 и алектиниб обладают сходной эффективностью и активностью в отношении ALK в условиях in vitro.
Индукторы изофермента CYP3A
При многократном пероральном применении рифампицина (мощного индуктора изофермента CYP3A) в дозе 600 мг 1 раз в сутки одновременно с однократным пероральным приемом алектиниба в дозе 600 мг отмечалось небольшое влияние на общую экспозицию алектиниба и М4 (среднее геометрическое соотношение с/без рифампицина: Cmax — 0,96 (0,88–1,05), AUCinf — 0,82 (0,74–0,9). Таким образом, при одновременном приеме препарата Алеценза® с индукторами изофермента CYP3A коррекции дозы не требуется.
Ингибиторы изофермента CYP3A
При многократном пероральном применении позаконазола (мощного ингибитора изофермента CYP3A) в дозе 400 мг 2 раза в сутки одновременно с однократным пероральным приемом алектиниба в дозе 300 мг отмечалось небольшое влияние на общую экспозицию алектиниба и М4 (среднее геометрическое соотношение с/без позаконазола: Cmax — 0,93 (0,81–1,08), AUCinf — 1,36 (1,24–1,49). Таким образом, при одновременном приеме препарата Алеценза® с ингибиторами изофермента CYP3A коррекции дозы не требуется.
Лекарственные препараты, повышающие рН желудка
Несмотря на то что растворимость алектиниба в воде в условиях in vitro зависит от рН, специальное клиническое исследование межлекарственного взаимодействия показало, что применение эзомепразола (ингибитора протонного насоса) в дозе 40 мг 1 раз в сутки не оказывает клинически значимого влияния на общую экспозицию алектиниба и М4. Таким образом, при одновременном применении препарата Алеценза® с ингибиторами протонного насоса или другими препаратами, повышающими рН желудка (например, антагонистами Н2-рецепторов или антацидами), коррекции дозы не требуется.
Влияние белков-переносчиков на распределение алектиниба
Алектиниб не является субстратом P-gp in vitro. Ни алектиниб, ни М4 не являются субстратами для BCRP или OATP 1B1/B3. При этом М4 является субстратом Р-gp. Алектиниб ингибирует Р-gp, таким образом, не ожидается, что одновременное применение ингибиторов Р-gp будет оказывать значимое влияние на экспозицию М4.
Передозировка
В клинических исследованиях не наблюдалось случаев передозировки.
Лечение: необходимо тщательно наблюдать за состоянием пациентов и проводить поддерживающую терапию. Специфического антидота, который можно было бы использовать при передозировке препарата Алеценза®, нет.
Особые указания
ИБЛ/пневмонит
В клинических исследованиях препарата Алеценза® отмечались случаи ИБЛ/пневмонита. Необходимо наблюдать за пациентами на предмет развития легочных симптомов пневмонита.
Cледует незамедлительно прервать лечение препаратом Алеценза® у пациентов с диагностированным ИБЛ/пневмонитом и полностью прекратить терапию препаратом Алеценза®, если иные потенциальные причины возникновения ИБЛ/пневмонита не установлены.
Гепатотоксичность
В клинических исследованиях препарата Алеценза® отмечалось повышение активности АЛТ и АСТ >5 × ВГН, а также повышение концентрации билирубина >3 × ВГН. Большинство явлений возникало в ходе первых 3 мес терапии.
У трех пациентов с повышением активности АСТ/АЛT 3–4-й степени тяжести наблюдалось лекарственно-индуцированное поражение печени. У одного пациента зарегистрировано одновременное повышение активности АЛТ или АСТ ≥3 × ВГН и повышение концентрации ОБ ≥2 × ВГН при нормальной активности ЩФ.
Следует проводить мониторинг показателей функции печении, в т.ч. АЛТ, АСТ и ОБ, на исходном уровне и затем каждые 2 нед в течение первых 3 мес лечения. Далее необходимо продолжать периодический мониторинг, поскольку отклонения могут возникать и после 3 мес лечения. Пациентам, у которых отмечалось повышение активности трансаминаз и концентрации билирубина, необходим более частый мониторинг. В зависимости от степени тяжести нежелательной реакции, следует или приостановить лечение с последующим возобновлением приема препарата в сниженной дозе, или полностью прекратить терапию препаратом Алеценза® (см. таблицу 2).
Тяжелая миалгия и повышение активности КФК
В клинических исследованиях препарата Алеценза® отмечались миалгия или скелетно-мышечная боль, в т.ч. 3-й степени тяжести.
В клинических исследованиях препарата Алеценза® отмечалось повышение активности КФК, в т.ч. 3-й степени тяжести. Медиана времени до возникновения явлений 3-й степени тяжести составила 14 дней у пациентов с ALK-положительным НМРЛ, ранее получавших терапию кризотинибом, и 27,5 дня у пациентов с ALK-положительным НМРЛ, ранее не получавших лечение.
Следует рекомендовать пациентам сообщать о любых случаях мышечной боли, болезненности или слабости неясной этиологии. Необходимо оценивать активность КФК каждые 2 нед в первый месяц терапии и в соответствии с клиническими показаниями у тех пациентов, которые сообщают о симптомах.
В зависимости от степени тяжести повышения активности КФК, следует приостановить лечение препаратом Алеценза® с последующим возобновлением приема препарата в прежней дозе или снизить дозу (см. таблицу 2).
Брадикардия
При применении препарата Алеценза® возможно возникновение симптоматической брадикардии.
Необходим мониторинг ЧСС и АД в соответствии с клиническими показаниями. В случае возникновения бессимптомной брадикардии коррекции дозы не требуется.
При развитии симптоматической брадикардии или жизнеугрожающих состояний следует оценить действие сопутствующих препаратов с известной способностью вызывать брадикардию, а также действие антигипертензивных препаратов. Дозу препарата Алеценза® корректируют (см. таблицу 2).
Фоточувствительность
При применении препарата Алеценза® развивалась фоточувствительность к воздействию солнечных лучей. Необходимо рекомендовать пациентам избегать длительного пребывания на солнце во время приема препарата Алеценза® и в течение не менее 7 дней после прекращения терапии. Также следует использовать солнцезащитные средства и бальзам для губ с защитой от УФ-лучей широкого спектра (от воздействия УФ-диапазона А/УФ- излучения диапазона В) с SPF ≥50 для защиты от возможных солнечных ожогов.
Эмбриофетальная токсичность
Применение препарата при беременности может оказывать повреждающее воздействие на плод. У беременных крыс и кроликов применение алектиниба сопровождалось эмбриофетальной токсичностью. Пациентки или половые партнерши пациентов, обладающие детородным потенциалом, должны использовать высокоэффективные методы контрацепции во время терапии препаратом Алеценза®и в течение не менее 3 мес после приема последней дозы препарата.
Непереносимость лактозы
Препарат Алеценза®содержит лактозу. Следует соблюдать осторожность при применении препарата Алеценза® у пациентов с редкой наследственной непереносимостью галактозы, врожденной недостаточностью лактазы или глюкозо-галактозной мальабсорбцией.
Инструкции по уничтожению неиспользованного препарата или препарата с истекшим сроком годности
Попадание лекарственного препарата в окружающую среду должно быть сведено к минимуму. Не следует утилизировать препарат с помощью сточных вод или вместе с бытовыми отходами. Уничтожение неиспользованного препарата или препарата с истекшим сроком годности должно проводиться в соответствии с локальными требованиями.
Влияние на способность управлять транспортными средствами и механизмами. Влияние препарата Алеценза® на способность управлять автомобилем и работать с механизмами не изучалось.
Форма выпуска
Капсулы, 150 мг. По 8 капс. в контурной ячейковой упаковке (блистере) из композиционного материала (полиамид/алюминий/ПВХ) и фольги алюминиевой композиционной (алюминий/сополимер винилхлорида/винилацетата полибутилметакрилат). По 7 контурных ячейковых упаковок (блистеров) вместе с инструкцией по применению помещают в картонную пачку (промежуточная упаковка), на которую с целью контроля первого вскрытия наклеивают голограммы. По 4 промежуточных упаковки помещают в картонную пачку (потребительская упаковка) с контролем первого вскрытия (в виде перфорации).
Производитель
Экселла ГмбХ и Ко. КГ, Германия/Excella GmbH & Co. KG, Nurnberger Strasse 12, 90537 Feucht, Germany.
Фасовщик (первичная упаковка): Делфарм Милано С.р.Л., Италия/Delpharm Milano S.r.l., Via Carnevale 1, 20090 Segrate, Milano, Italy.
Упаковщик (вторичная/потребительская упаковка): Делфарм Милано С.р.Л., Италия/Delpharm Milano S.r.l., Via Carnevale 1, 20090 Segrate, Milano, Italy.
Выпускающий контроль качества:
1. Делфарм Милано С.р.Л., Италия/Delpharm Milano S.r.l., Via Carnevale 1, 20090 Segrate, Milano, Italy.
2. Ф. Хоффманн-Ля Рош Лтд., Швейцария/F. Hoffmann-La Roche Ltd, Viaduktstrasse 33, 4051 Basel, Switzerland.
Владелец регистрационного удостоверения: Ф. Хоффманн-Ля Рош Лтд., Швейцария/F. Hoffmann-La Roche Ltd, Grenzacherstrasse 124, 4070 Basel, Switzerland.
Претензии потребителей направлять в компанию ЗАО «Рош-Москва» по адресу: 107031, Россия, Москва, Трубная пл., 2, пом. 1, эт. 1-й, комн. 42.
Тел.: (495) 229-29-99; факс: (495) 229-79-99.
Или через форму обратной связи на сайте: www.roche.ru.
Условия отпуска из аптек
По рецепту.
Условия хранения
При температуре не выше 30 °C, в оригинальной упаковке для защиты от влаги и света.
Хранить в недоступном для детей месте.
Срок годности
5 лет.
Не применять по истечении срока годности, указанного на упаковке.
Представленная информация о ценах на препараты не является предложением о продаже или покупке товара.
Информация предназначена исключительно для сравнения цен в стационарных аптеках, осуществляющих деятельность в
соответствии со статьей 55 Федерального закона «Об обращении лекарственных средств» от 12.04.2010 № 61-ФЗ.
Алеценза — инструкция по применению
Синонимы, аналоги
Статьи
Регистрационный номер
ЛП-005109
Торговое наименование
Алеценза®
Международное непатентованное наименование
Алектиниб
Лекарственная форма
Капсулы
Состав
Одна капсула содержит:
действующее вещество: алектиниб – 150 мг (в виде алектиниба гидрохлорида – 161.33 мг);
вспомогательные вещества: лактозы моногидрат – 33.67 мг, гидроксипропилцеллюлоза (гипролоза) – 15.00 мг, натрия лаурилсульфат – 75.00 мг, кальция карбоксиметилцеллюлоза (кальция кармеллоза) – 43.35 мг, магния стеарат – 1.65 мг;
оболочка капсулы: каррагинан – 0.25 мг, калия хлорид – 0.42 мг, титана диоксид (Е171) – 4.20 мг, воск карнаубский <0.1010, крахмал кукурузный <0.1010, гипромеллоза – 61.63 мг;
чернила для нанесения надписи на капсуле: краситель железа оксид красный (Е172), краситель железа оксид желтый (Е172), алюминиевый лак FD&C Blue No.2 (E132), воск карнаубский, шеллак белый, глицерил моноолеат, 1-бутанол, спирт этиловый безводный.
Описание
Твердые капсулы, размер 1, корпус и крышечка капсулы от белого до желтовато-белого цвета.
На корпусе капсулы нанесена надпись «150 mg» черного цвета. На крышечке капсулы нанесена надпись «ALE» черного цвета.
Содержимое капсулы: порошок или скомковавшийся порошок от белого до светло-желтого цвета.
Фармакотерапевтическая группа
Противоопухолевое средство – протеинкиназы ингибитор.
Код АТХ
L01XE36
Фармакологические свойства
Фармакодинамика
Механизм действия
Алектиниб является мощным высокоселективным ингибитором тирозинкиназ ALK (anaplastic lymphoma kinase) и RET (REarranged during Transfection). В доклинических исследованиях ингибирование активности тирозинкиназы ALK приводило к блокаде нисходящих сигнальных путей, включая STAT 3 (переносчик сигнала и активатор транскрипции 3) и PI3K/AKT (фосфоинозитол-3-киназу/протеинкиназу В), и вызывало апоптоз опухолевых клеток.
В условиях in vitro и in vivo алектиниб проявлял активность в отношении мутантных форм фермента ALK, в том числе с мутациями, обуславливающими резистентность к кризотинибу.
Основной метаболит алектиниба – М4 – в условиях in vitro показал эффективность и активность, сопоставимую с алектинибом.
Согласно данным доклинических исследований алектиниб не является субстратом Р-гликопротеина или BCRP (белка устойчивости рака молочной железы), выполняющих функцию белков-переносчиков в гематоэнцефалическом барьере, и, таким образом, обладает способностью проникать в центральную нервную систему и задерживаться в ней. Алектиниб индуцирует регрессию опухоли на моделях ксенотрансплантатов опухоли у мышей, в том числе демонстрирует противоопухолевую активность в мозге и увеличивает выживаемость на моделях внутричерепных опухолей у животных.
Фармакокинетика
Фармакокинетические показатели алектиниба и М4 оценивали у пациентов с ALK-положительным НМРЛ (немелкоклеточным раком легкого) и у здоровых добровольцев.
Средние геометрические (коэффициент вариации,%) показатели Сmax (максимальной концентрации), Cmin (минимальной концентрации) и AUC0-12 ч (площади под кривой «концентрация – время» от 0 до 12 ч) в равновесном состоянии для алектиниба составляли ~665 нг/мл (44.3%), 572 нг/мл (47.8%) и 7430 нг*ч/мл (45.7%), соответственно. Средние геометрические показатели Сmax, Сmin и AUC0-12 ч В равновесном состоянии для М4 составляли ~246 нг/мл (45.4%), 222 нг/мл (46.6%) и 2810 нг*ч/мл (45.9%), соответственно.
Всасывание
После перорального приема алектиниба у пациентов с ALK-положительным НМРЛ в дозе 600 мг два раза в сутки после приема пищи отмечалось быстрое всасывание препарата. Тmax (время до достижения максимальной концентрации) составляло ~ от 4 до 6 ч.
При приеме алектиниба в дозе 600 мг два раза в сутки равновесное состояние алектиниба достигается в течение 7 дней и остается неизменным, при этом согласно данным популяционного фармакокинетического анализа средний геометрический коэффициент накопления равен 5.6. Популяционный фармакокинетический анализ показал пропорциональность доз алектиниба в диапазоне от 300 мг до 900 мг после приема пищи.
У здоровых добровольцев абсолютная биодоступность при применении алектиниба после приема пищи составляла 36.9% (33.9% – 40.3%).
При однократном пероральном применении алектиниба в дозе 600 мг прием высококалорийной пищи с большим содержанием жиров увеличивал экспозицию препарата в 3 раза по сравнению с приемом натощак (среднее геометрическое соотношение комбинированных показателей для алектиниба и М4 – Сmax: 3.31 [2.79-3.93], AUCinf: 3.11 [2.73-3.55]).
Распределение
Алектиниб и М4 имеют высокий уровень связывания с белками плазмы крови человека (>99%) независимо от концентрации действующего вещества. В условиях in vitro среднее соотношение концентраций в крови и в плазме человека составляет 2.64 и 2.50 для алектиниба и М4, соответственно (при клинически значимых концентрациях).
Средний геометрический VSS (объем распределения в равновесном состоянии) алектиниба после внутривенного введения составлял 475 л, указывая на широкое распределение алектиниба в тканях.
Метаболизм
Исследования метаболизма в условиях in vitro показали, что изофермент CYP3A4 является основным изоферментом CYP (цитохрома Р450), который опосредует метаболизм алектиниба и М4, при этом определено, что данный изофермент осуществляет 40-50% метаболизма в гепатоцитах человека. Согласно результатам исследования массового баланса алектиниб и М4 являлись основными циркулирующими веществами в плазме крови, их общее содержание составляло 76% от общей радиоактивности в плазме крови. Среднее геометрическое соотношение метаболит/препарат в равновесном состоянии – 0.399.
Выведение
У здоровых добровольцев после однократного перорального применения 14С-меченого алектиниба большая часть радиоактивности была выявлена в кале (средняя степень извлечения 97.8%, диапазон 95.6% – 100%), минимальная – в моче (средняя степень извлечения 0.46%, диапазон 0.30% – 0.60%). Доля неизмененного алектиниба в кале составляла 84% от введенной дозы, а доля М4 составила 5.8%.
Согласно данным популяционного фармакокинетического анализа CL/F (кажущийся клиренс) алектиниба составлял 81.9 л/ч, М4 – 217 л/ч. Индивидуальный расчетный средний геометрический период полувыведения алектиниба составлял 32.5 ч, М4 – 30.7 ч.
Особые группы пациентов
Пациенты детского возраста
Исследования фармакокинетики алектиниба у пациентов детского возраста (<18 лет) не проводились.
Пациенты пожилого возраста
Возраст не оказывает влияния на экспозицию алектиниба.
Пациенты с нарушением функции почек
Алектиниб и М4 выводятся почками в незначительных количествах в неизмененном виде (<0.2% дозы). Данные, полученные у пациентов с нарушением функции почек легкой и средней степени тяжести, показывают, что нарушение функции почек не оказывает значительного влияния на фармакокинетику алектиниба. Специальные исследования фармакокинетики не проводились, данные популяционной фармакокинетики для пациентов с нарушением функции почек тяжелой степени тяжести отсутствуют, однако коррекции дозы у таких пациентов не требуется, поскольку алектиниб выводится почками в незначительной степени.
Пациенты с нарушением функции печени
Основным путем выведения алектиниба является печеночный метаболизм. У пациентов с нарушением функции печени возможно увеличение концентрации алектиниба и/или М4 в плазме крови. Согласно данным популяционного фармакокинетического анализа экспозиции алектиниба и М4 у пациентов с нарушением функции печени легкой степени тяжести (ОБ (общий билирубин) на исходном уровне ≤ВГН (верхняя граница нормы) и активность ACT (аспартатаминотрансферазы) >ВГН или ОБ на исходном уровне >1.0-1.5 х ВГН и любая активность ACT на исходном уровне) соответствовали таковым у пациентов с нормальной функцией печени (ОБ ≤ВГН и активность ACT ≤ВГН).
После однократного перорального приема алектиниба в дозе 300 мг пациентами с нарушением функции печени тяжелой степени тяжести (класс С по Чайлд-Пью) Сmax алектиниба была такой же как и у здоровых добровольцев; AUCinf у пациентов с нарушением функции печени тяжелой степени тяжести (класс С по Чайлд-Пью) была в 2.2 раза выше, чем у здоровых добровольцев, а Сmax и AUCinf для М4 были на 39% и 34% ниже, соответственно. Таким образом, общая комбинированная экспозиция для алектиниба и М4 (AUCinf) была в 1.8 раз выше у пациентов с нарушением функции печени тяжелой степени тяжести по сравнению со здоровыми добровольцами.
В ходе изучения применения препарата у пациентов с нарушением функции печени средней степени тяжести (класс В по Чайлд-Пью) наблюдалась умеренно повышенная экспозиция алектиниба по сравнению со здоровыми добровольцами. Однако, у пациентов с нарушением функции печени средней степени тяжести (класс В по Чайлд-Пью), в целом, не отмечалось отклонений в показателях билирубина, альбумина или протромбинового времени, что указывает на их возможное неполное соответствие пациентам с нарушением функции печени умеренной степени тяжести со сниженной метаболической способностью.
Показания к применению
Монотерапия местно-распространенного или метастатического немелкоклеточного рака легкого с опухолевой экспрессией киназы анапластической лимфомы (ALK-положительный):
- в первой линии терапии;
- при прогрессировании заболевания на фоне терапии кризотинибом или при ее непереносимости.
Противопоказания
Повышенная чувствительность к алектинибу или к другим вспомогательным веществам препарата в анамнезе.
Беременность и период грудного вскармливания.
Детский возраст до 18 лет (эффективность и безопасность применения не изучались).
С осторожностью
Нарушение функции почек тяжелой степени тяжести.
Редкая наследственная непереносимость галактозы, врожденная недостаточность лактазы или глюкозо-галактозная мальабсорбция.
Применение при беременности и в период грудного вскармливания
Контрацепция
Пациентки или половые партнерши пациентов, обладающие детородным потенциалом, должны использовать высокоэффективные методы контрацепции во время терапии препаратом Алеценза® и в течение не менее 3 месяцев после приема последней дозы препарата.
Беременность
Женщинам детородного потенциала необходимо избегать беременности при приеме препарата Алеценза®. Клинические исследования препарата Алеценза® у беременных женщин не проводились. В силу своего механизма действия препарат Алеценза® может оказывать повреждающее воздействие на плод.
Доклинические данные
В исследованиях на животных алектиниб вызывал развитие эмбриофетальной токсичности.
Период грудного вскармливания
Неизвестно, выводится ли препарат Алеценза® с грудным молоком. Исследований по оценке влияния препарата Алеценза® на выработку грудного молока или наличию препарата Алеценза® в грудном молоке не проводилось. Поскольку большинство препаратов проникает в грудное молоко, и риск для грудных детей не может быть исключен, пациенткам следует воздержаться от грудного вскармливания на фоне приема препарата Алеценза®.
Способ применения и дозы
Перед применением препарата Алеценза® в первой линии терапии НМРЛ необходимо установить наличие опухолевой экспрессии киназы анапластической лимфомы (ALK). Для выявления пациентов с ALK-положительным НМРЛ необходимо использовать валидированный метод определения ALK.
Капсулы препарата Алеценза® следует принимать одновременно с приемом пищи и проглатывать целиком. Открывать или растворять капсулы нельзя.
Рекомендуемая доза препарата Алеценза® составляет 600 мг (четыре капсулы по 150 мг) два раза в сутки (суточная доза составляет 1200 мг), внутрь
Пациенты с исходным нарушением функции печени тяжелой степени тяжести (класс С по Чайлд-Пью) должны получать препарат Алеценза® в начальной дозе 450 мг 2 раза в сутки (суточная доза составляет 900 мг), внутрь.
Продолжительность лечения
Лечение препаратом Алеценза® рекомендуется проводить до прогрессирования заболевания или до развития непереносимой токсичности.
Задержка приема или пропуск дозы
Если прием очередной дозы пропущен, ее можно принять позднее, при условии, что интервал между приемом пропущенной дозы и приемом следующей запланированной дозы составляет не менее 6 часов. Несмотря на возникновение рвоты после приема препарата Алеценза®, следующую дозу необходимо принять в запланированное время.
Коррекция дозы
Для контроля нежелательных явлений может потребоваться временное прерывание лечения, уменьшение дозы или полное прекращение применения препарата Алеценза®.
Дозу препарата необходимо уменьшать пошагово, на 150 мг 2 раза в сутки, в зависимости от переносимости.
Если применение препарата в дозе 300 мг 2 раза в сутки невозможно из-за непереносимости, терапию препаратом Алеценза® следует полностью прекратить.
В таблице 1 ниже приведены общие рекомендации по коррекции дозы препарата Алеценза®.
Таблица 1. Схема снижения дозы.
| Схема снижения дозы | Уровень дозы |
| Доза | 600 мг 2 раза в сутки |
| Первое снижение дозы | 450 мг 2 раза в сутки |
| Второе снижение дозы | 300 мг 2 раза в сутки |
Таблица 2. Коррекция дозы при развитии отдельных нежелательных реакций.
| Нежелательная реакция, степень тяжести | Терапия препаратом Алеценза® |
| Интерстициальная болезнь легких (ИБЛ)/пневмонит (все степени тяжести) | Следует немедленно прервать лечение и полностью прекратить терапию, если иные потенциальные причины возникновения ИБЛ/пневмонита не установлены. |
| Повышение активности АЛТ (аланинаминотрансферазы) или ACT (аспартатаминотрансферазы) до ≥3 степени тяжести (>5 х ВГН), с сопутствующим повышением концентрации общего билирубина ≤2 х ВГН | Временно приостановить терапию; дождаться восстановления показателей до исходного уровня или до ≤1 степени тяжести (≤3 х ВГН), затем возобновить применение препарата в сниженной дозе (см. Таблицу 1). |
| Повышение активности АЛТ или ACT до ≥2 степени тяжести (>3 х ВГН), с сопутствующим повышением концентрации общего билирубина >2 х ВГН (в отсутствие холестаза или гемолиза) | Следует полностью прекратить терапию препаратом Алеценза®. |
| Брадикардияа 2 или 3 степени тяжести (симптоматическая, возможно тяжелая и клинически значимая, требующая медицинского вмешательства) | Следует временно приостановить терапию препаратом; дождаться уменьшения степени тяжести брадикардии до ≤1 степени (без симптомов) или восстановления частоты сердечных сокращений до ≥60 уд/мин. Необходимо оценить действие сопутствующих препаратов с известной способностью вызывать брадикардию, а также действие антигипертензивных препаратов. Если способствующий одновременно применяемый препарат был выявлен и отменен или была произведена коррекция его дозы, следует возобновить терапию препаратом Алеценза® в исходной дозе после уменьшения степени тяжести брадикардии до ≤1 степени (без симптомов) или увеличения частоты сердечных сокращений до ≥60 уд/мин. Если такой препарат не был выявлен или не был отменен, или не произведена коррекция его дозы, следует возобновить терапию препаратом Алеценза® в сниженной дозе (см. Таблицу 1) после уменьшения степени тяжести брадикардии до ≤1 степени (без симптомов) или увеличения частоты сердечных сокращений до ≥60 уд/мин. |
| Брадикардияа 4 степени тяжести (с жизнеугрожающими последствиями, требующая незамедлительного медицинского вмешательства) | Если способствующий одновременно применяемый препарат не был выявлен, следует полностью прекратить терапию препаратом Алеценза®. Если такой препарат выявлен и отменен или произведена коррекция его дозы, следует возобновить терапию препаратом Алеценза® в сниженной дозе (см. Таблицу 1) после уменьшения степени тяжести брадикардии до ≤1 степени (без симптомов) или увеличения частоты сердечных сокращений до ≥60 уд/мин при условии постоянного наблюдения в соответствии с клиническими показаниями. При повторном возникновении брадикардии применение препарата Алеценза® следует полностью прекратить. |
| Повышение активности КФК >5 х ВГН | Следует временно приостановить терапию препаратом и дождаться восстановления активности КФК до исходного уровня или до ≤2.5 х ВГН, затем возобновить применение препарата в исходной дозе. |
| Повышение активности КФК >10 х ВГН или повторное повышение активности КФК >5 х ВГН | Следует временно приостановить терапию препаратом и дождаться восстановления активности КФК до исходного уровня или до ≤2.5 х ВГН, затем возобновить применение препарата в сниженной дозе (см. Таблицу 1). |
а Частота сердечных сокращений менее 60 ударов в минуту
Дозирование в особых случаях
Пациенты детского возраста
Безопасность и эффективность препарата Алеценза® у детей и подростков (<18 лет) не изучены.
Пациенты пожилого/старческого возраста
Коррекции дозы препарата Алеценза® у пациентов ≥65 лет не требуется.
Пациенты с нарушением функции почек
Коррекции дозы препарата Алеценза® у пациентов с нарушением функции почек легкой и средней степени тяжести не требуется. Применение препарата Алеценза® у пациентов с нарушением функции почек тяжелой степени тяжести не изучено, однако поскольку алектиниб выводится почками в незначительной степени, коррекции дозы у таких пациентов не требуется.
Пациенты с нарушением функции печени
Пациентам с исходным нарушением функции печени легкой (класс А по Чайлд-Пью) или средней степени тяжести (класс В по Чайлд-Пью) коррекции начальной дозы не требуется. Пациенты с исходным нарушением функции печени тяжелой степени тяжести (класс С по Чайлд-Пью) должны получать препарат Алеценза® в начальной дозе 450 мг 2 раза в сутки (суточная доза составляет 900 мг), внутрь. Рекомендуется соответствующее наблюдение (контроль за показателями функции печени) для всех пациентов с нарушением функции печени.
Побочное действие
Безопасность препарата Алеценза® оценивали в клинических исследованиях (КИ) у пациентов с ALK-положительным НМРЛ, ранее получавших терапию кризотинибом, при применении в рекомендованной дозе 600 мг два раза в сутки; медиана экспозиции составила 11 месяцев (0-35 месяцев). Безопасность препарата Алеценза® также оценивали в КИ у пациентов с ALK-положительным НМРЛ, ранее не получавших лечение, при применении в рекомендованной дозе 600 мг два раза в сутки; медиана экспозиции составила 17.9 месяцев.
Наиболее частыми (≥20%) нежелательными реакциями были запор (36%), отеки (34%, включая периферический, генерализованный, отек век и периорбитальный отек); миалгия (31%, включая миалгию и скелетно-мышечную боль), тошнота (22%), повышение концентрации билирубина (21%, включая повышение концентрации билирубина в крови, гипербилирубинемии) и повышение концентрации конъюгированного билирубина), анемия (20%, включая анемию и снижение уровня гемоглобина) и сыпь (20%, включая сыпь, макуло-папулезную сыпь, акнеиформный дерматит, эритему, генерализованную сыпь, папулезную сыпь, зудящую сыпь и макулярную сыпь).
Ниже представлен перечень нежелательных реакций, отмечавшихся в КИ у пациентов с ALK-положительным НМРЛ. ранее получавших терапию кризотинибом, и у пациентов с ALK-положительным НМРЛ, ранее не получавших лечение. Нежелательные реакции сгруппированы в соответствии с классами систем органов медицинского словаря для нормативно-правовой деятельности MedDRA.
Для описания частоты нежелательных реакций используется следующая классификация: очень часто (≥10%), часто (≥1% и <10%), нечасто (≥0.1% и <1%), редко (≥0.01% и <0.1%), очень редко (<0.01%).
Нарушения со стороны крови и лимфатической системы: очень часто – анемия1. Нарушения со стороны нервной системы: часто – дисгевзия (нарушение вкусовых восприятий)#2.
Нарушения со стороны органа зрения: очень часто – расстройства зрения3.
Нарушения со стороны сердца: очень часто – брадикардия4.
Нарушения со стороны дыхательной системы, органов грудной клетки и средостения: часто – интерстициальная болезнь легких/пневмонит.
Нарушения со стороны желудочно-кишечного тракта: очень часто – запор, тошнота, диарея, рвота; часто – стоматит#5.
Нарушения со стороны печени и желчевыводящих путей: очень часто – повышение концентрации билирубина6, повышение активности ACT, повышение активности АЛТ; нечасто – лекарственно-индуцированное поражение печени7.
Нарушения со стороны кожи и подкожных тканей: очень часто – сыпь8, реакция фоточувствительности.
Нарушения со стороны скелетно-мышечной и соединительной ткани: очень часто – миалгия9, повышение активности КФК в крови.
Нарушение со стороны почек и мочевыводящих путей: часто – повышение концентрации креатинина в крови*, острая почечная недостаточность*#.
Общие расстройства и нарушения в месте введения: очень часто – отеки10.
Лабораторные и инструментальные данные: часто – увеличение массы тела#.
Список обозначений (ссылок):
* включая одно явление с летальным исходом.
# явление, отмечено в клиническом исследовании у пациентов с ALK-положительным НМРЛ, ранее не получавших лечение.
1 включая случаи анемии и снижения уровня гемоглобина.
2 включая случаи дисгевзии и гипогевзии (снижение вкусовых восприятий).
3 включая случаи нечеткости зрения, нарушения зрения, плавающих помутнений в стекловидном теле, снижения остроты зрения, астенопии и диплопии.
4 включая случаи брадикардии и синусовой брадикардии.
5 включая случаи стоматита и язвенного стоматита.
6 включая случаи повышения концентрации билирубина в крови, гипербилирубинемии, повышения концентрации конъюгированного билирубина.
7 включая одного пациента с лекарственно-индуцированным поражением печени (термин медицинского словаря для нормативно-правовой деятельности MedDRA) и одного пациента с повышением активности ACT и АЛТ 4 степени и задокументированным лекарственно-индуцированным поражением печени, подтвержденным биопсией.
8 включая случаи сыпи, макуло-папулезной сыпи, акнеиформного дерматита, эритемы, генерализованной сыпи, папулезной сыпи, зудящей сыпи и макулярной сыпи.
9 включая случаи миалгии и скелетно-мышечной боли.
10 включая случаи отека, отека век и периферического, генерализованного, периорбитального отека.
Описание отдельных нежелательных реакций
Профиль безопасности препарата Алеценза® в целом был сходным в КИ у пациентов с ALK-положительным НМРЛ, ранее получавших терапию кризотинибом, и у пациентов, ранее не получавших лечение.
Интерстициальная болезнь легких (ИБЛ)/пневмонит
При применении препарата Алеценза® отмечались тяжелые случаи интерстициальной болезни легких/пневмонита. В КИ у 0.4% пациентов с ALK-положительным НМРЛ, ранее получавших терапию кризотинибом, развивалась ИБЛ 3 степени тяжести, что привело к прекращению терапии препаратом Алеценза®. Явлений ИБЛ с летальным исходом ни в одном КИ отмечено не было.
Гепатотоксичность
Повышение активности АСТ/АЛТ 3-4 степени тяжести отмечалось в КИ у двух пациентов с ALK-положительным НМРЛ, ранее получавших терапию кризотинибом; при этом было задокументировано лекарственно-индуцированное поражение печени, подтвержденное биопсией.
Повышение активности ACT и АЛТ отмечалось у 16% и 14% пациентов в КИ. соответственно. Большинство явлений были 1 и 2 степеней тяжести; явления ≥3 степени тяжести наблюдались у 2.8% и 3.2% пациентов, соответственно. Данные явления обычно развивались в течение первых трех месяцев терапии, были как правило преходящими и разрешались после временной приостановки лечения (у 1.2% и 3.2% пациентов, соответственно) или снижения дозы препарата Алеценза® (у 1.6% и 0.8%, соответственно). Повышение активности ACT и АЛТ приводило к прекращению терапии препаратом Алеценза® у 1.2% и 1.6% пациентов, соответственно.
Повышение концентрации общего билирубина отмечалось в КИ у 17% пациентов с ALK-положительным НМРЛ, ранее получавших терапию кризотинибом. Большинство явлений были 1 и 2 степеней тяжести; явления 3 степени тяжести наблюдались у 3.2% пациентов. Данные явления обычно отмечались в течение первых трех месяцев терапии, были преходящими и разрешались после временной приостановки лечения (у 4.7% пациентов) или снижения дозы препарата Алеценза® (у 2.8% пациентов). Повышение концентрации билирубина приводило к прекращению терапии препаратом Алеценза® у 1.6% пациентов. Одновременное повышение активности АЛТ или ACT ≥3 х ВГН и повышение концентрации общего билирубина ≥2 х ВГН при нормальной активности ЩФ отмечалось у одного пациента в КИ препарата Алеценза®.
Брадикардия
Случаи брадикардии отмечались в КИ у 7.9% пациентов с ALK-положительным НМРЛ, ранее получавших терапию кризотинибом. Все явления были 1 или 2 степени тяжести. У 20% пациентов после приема препарата Алеценза® частота сердечных сокращений составляла <50 у д/мин.
Тяжелая миалгия и повышение активности КФК
Сообщения о миалгии, включавшие миалгию (25%) и скелетно-мышечную боль (7.5%) регистрировались в КИ у пациентов с ALK-положительным НМРЛ, ранее получавших терапию кризотинибом, с общей частотой 31%. Большинство явлений были 1 или 2 степени тяжести, у 1.2% пациентов наблюдались явления 3 степени тяжести. Только 0.8% пациентов потребовалась коррекция дозы в связи с данными явлениями.
Повышение активности КФК отмечалось в КИ у 46% пациентов с доступными лабораторными данными. Частота повышения активности КФК 3 степени тяжести составила 5.0%; при этом медиана времени до возникновения явления составила 14 дней у пациентов с ALK-положительным НМРЛ, ранее получавших терапию кризотинибом, и 27.5 дней у пациентов с ALK-положительным НМРЛ, ранее не получавших лечение. Коррекция дозы препарата Алеценза® потребовалась у 4.0% пациентов с повышением активности КФК.
Отклонения со стороны лабораторных показателей
У пациентов с ALK-положительным НМРЛ, ранее получавших терапию кризотинибом, и у пациентов, ранее не получавших лечение, на фоне терапии препаратом Алеценза® наблюдались следующие отклонения со стороны лабораторных показателей:
| Показатель | Алектиниб | |
| Все степени (%) | 3-4 степень тяжести (%)° | |
| Биохимия | ||
| Повышение концентрации креатинина в крови** | 38# | 3.4#; |
| Повышение активности ACT | 53* | 6.2# |
| Повышение активности АЛТ | 40# | 6.1# |
| Повышение активности КФК в крови | 46* | 5.0* |
| Повышение концентрации билирубина в крови | 46* | 5.5* |
| Гематология | ||
| Снижение уровня гемоглобина | 62# | 6.8# |
Примечание: лабораторные отклонения определялись с учетом нормальных значений по классификации NCI СТСАЕ (Национального института рака National Cancer Institute Common Terminology Criteria of Adverse Events).
* частота, отмечавшаяся в КИ у пациентов с ALK-положительным НМРЛ, ранее получавших терапию кризотинибом.
** только пациенты с повышением концентрации креатинина на основании определения ВГН по классификации нежелательных явлений СТСАЕ.
# частота, отмечавшаяся в КИ у пациентов с ALK-положительным НМРЛ, ранее не получавших лечение.
° отклонений лабораторных показателей 5 степени тяжести не наблюдалось.
Пострегистрационный опыт применения
При пострегистрационном применении препарата Алеценза® отмечалось повышение активности ЩФ; при этом в КИ данное явление наблюдалось у 7.5% пациентов с ALK-положительным НМРЛ, ранее получавших терапию кризотинибом.
Передозировка
В клинических исследованиях не наблюдалось случаев передозировки. При передозировке необходимо тщательно наблюдать за состоянием пациентов и проводить поддерживающую терапию. Специфического антидота, который можно было бы использовать при передозировке препарата Алеценза®, нет.
Взаимодействие с другими лекарственными средствами
Влияние алектиниба на сопутствующие лекарственные препараты
Субстраты изоферментов цитохрома Р450
В условиях in vitro ни алектиниб, ни его основной активный метаболит – М4 – не оказывали ингибирующего действия на изоферменты CYP1А2, CYP2B6, CYP2C9, CYP2C19 или CYP2D6 в клинически значимых концентрациях. Алектиниб и М4 оказывают слабое, зависящее от времени, ингибирующее действие на CYP3A4. Кроме того, алектиниб является слабым индуктором изоферментов CYP3A4 и CYP2B6 в клинических концентрациях in vitro.
Согласно результатам исследования межлекарственного взаимодействия у пациентов с ALK-положительным НМРЛ, многократное применение алектиниба не оказывает влияния на экспозицию мидазолама, который является чувствительным субстратом CYP3A. Таким образом, при одновременном применении с субстратами CYP3A коррекции дозы не требуется.
Несмотря на то, что исследования в условиях in vitro показывают, что алектиниб является ингибитором изофермента CYP2C8, данные фармакокинетического моделирования, учитывающего физиологические процессы организма человека, свидетельствуют о том, что алектиниб в клинически значимых концентрациях не будет увеличивать плазменные концентрации одновременно применяемых препаратов-субстратов изофермента CYP2C8.
Субстраты Р-гликопротеина и BCRP
В условиях in vitro алектиниб и М4 являются ингибиторами Р-гликопротеина и BCRP. Таким образом, алектиниб и М4 могут способствовать увеличению плазменных концентраций субстратов Р-гликопротеина или BCRP при их одновременном применении (не ожидается, что увеличение экспозиции будет более чем двукратным). Рекомендуется соответствующее наблюдение пациентов, которые одновременно принимают алектиниб и субстраты Р-гликопротеина или BCRP с узким терапевтическим индексом (например, дигоксин, дабигатран, метотрексат).
Влияние сопутствующих лекарственных препаратов на алектиниб
Согласно данным, полученным in vitro, изофермент CYP3A4 является основным ферментом, ответственным за метаболизм алектиниба и М4, при этом предполагается, что посредством CYP3A осуществляется 40-50% общего печеночного метаболизма. М4 и алектиниб обладают сходной эффективностью и активностью в отношении ALK в условиях in vitro.
Индукторы изофермента CYP3A
При многократном пероральном применении рифампицина (мощного индуктора изофермента CYP3A) в дозе 600 мг один раз в сутки одновременно с однократным пероральным приемом алектиниба в дозе 600 мг отмечалось небольшое влияние на общую экспозицию алектиниба и М4 (среднее геометрическое соотношение с/без рифампицина – Сmax: 0.96 [0.88-1.05], AUCinf: 0.82 [0.74-0.90]). Таким образом, при одновременном приеме препарата Алеценза® с индукторами изофермента CYP3A коррекции дозы не требуется.
Ингибиторы изофермента CYP3A
При многократном пероральном применении позаконазола (мощного ингибитора изофермента CYP3A) в дозе 400 мг два раза в сутки одновременно с однократным пероральным приемом алектиниба в дозе 300 мг отмечалось небольшое влияние на общую экспозицию алектиниба и М4 (среднее геометрическое соотношение с/без позаконазола – Сmax: 0.93 [0.81-1.08], AUCinf: 1.36 [1.24-1.49]). Таким образом, при одновременном приеме препарата Алеценза® с ингибиторами изофермента CYP3A коррекции дозы не требуется.
Лекарственные препараты, повышающие pH желудка
Несмотря на то, что растворимость алектиниба в воде в условиях in vitro зависит от pH, специальное клиническое исследование межлекарственного взаимодействия показало, что применение эзомепразола (ингибитора протонного насоса) в дозе 40 мг один раз в сутки не оказывает клинически значимого влияния на общую экспозицию алектиниба и М4. Таким образом, при одновременном применении препарата Алеценза® с ингибиторами протонного насоса или другими препаратами, повышающими pH желудка (например, антагонистами Н2-рецепторов или антацидами), коррекции дозы не требуется.
Влияние белков-переносчиков на распределение алектиниба
Алектиниб не является субстратом Р-гликопротеина in vitro. Ни алектиниб, ни М4 не являются субстратами для BCRP или ОАТР 1В1/В3. При этом М4 является субстратом Р-гликопротеина. Алектиниб ингибирует Р-гликопротеин, таким образом, не ожидается, что одновременное применение ингибиторов Р-гликопротеина будет оказывать значимое влияние на экспозицию М4.
Особые указания
Интерстициальная болезнь легких (ИБЛ)/пневмонит
В клинических исследованиях препарата Алеценза® отмечались случаи ИБЛ/пневмонита. Необходимо наблюдать за пациентами на предмет развития легочных симптомов пневмонита.
Следует незамедлительно прервать лечение препаратом Алеценза® у пациентов с диагностированным ИБЛ/пневмонитом и полностью прекратить терапию препаратом Алеценза®, если иные потенциальные причины возникновения ИБЛ/пневмонита не установлены.
Гепатотоксичность
В клинических исследованиях препарата Алеценза® отмечалось повышение активности АЛТ и ACT >5 х ВГН, а также повышение концентрации билирубина >3 х ВГН. Большинство явлений возникало в ходе первых 3 месяцев терапии.
У трех пациентов с повышением активности АСТ/АЛТ 3-4 степени тяжести наблюдалось лекарственно-индуцированное поражение печени. У одного пациента зарегистрировано одновременное повышение активности АЛТ или АСТ≥3 х ВГН и повышение концентрации общего билирубина ≥2 х ВГН при нормальной активности ЩФ.
Следует проводить мониторинг показателей функции печении, в том числе АЛТ, ACT и общего билирубина, на исходном уровне и затем каждые 2 недели в течение первых 3 месяцев лечения. Далее необходимо продолжать периодический мониторинг, поскольку отклонения могут возникать и после 3 месяцев лечения. Пациентам, у которых отмечалось повышение активности трансаминаз и концентрации билирубина, необходим более частый мониторинг. В зависимости от степени тяжести нежелательной реакции следует или приостановить лечение с последующим возобновлением приема препарата в сниженной дозе, или полностью прекратить терапию препаратом Алеценза® (см. таблицу 2, раздел «Способ применения и дозы»).
Тяжелая миалгия и повышение активности КФК
В клинических исследованиях препарата Алеценза® отмечались миалгия или скелетно-мышечная боль, в том числе 3 степени тяжести.
В клинических исследованиях препарата Алеценза® отмечалось повышение активности КФК, в том числе 3 степени тяжести. Медиана времени до возникновения явлений 3 степени тяжести составила 14 дней у пациентов с ALK-положительным НМРЛ, ранее получавших терапию кризотинибом, и 27.5 дней у пациентов с ALK-положительным НМРЛ, ранее не получавших лечение.
Следует рекомендовать пациентам сообщать о любых случаях мышечной боли, болезненности или слабости неясной этиологии. Необходимо оценивать активность КФК каждые 2 недели в первый месяц терапии и в соответствии с клиническими показаниями у тех пациентов, которые сообщают о симптомах.
В зависимости от степени тяжести повышения активности КФК следует приостановить лечение препаратом Алеценза® с последующим возобновлением приема препарата в прежней дозе или снизить дозу (см. таблицу 2, раздел «Способ применения и дозы»).
Брадикардия
При применении препарата Алеценза® возможно возникновение симптоматической брадикардии.
Необходим мониторинг частоты сердечных сокращений и артериального давления в соответствии с клиническими показаниями. В случае возникновения бессимптомной брадикардии коррекции дозы не требуется.
При развитии симптоматической брадикардии или жизнеугрожающих состояний следует оценить действие сопутствующих препаратов с известной способностью вызывать брадикардию, а также действие антигипертензивных препаратов. Дозу препарата Алеценза® корректируют (см. таблицу 2. раздел «Способ применения и дозы»).
Фоточувствительность
При применении препарата Алеценза® развивалась фоточувствительность к воздействию солнечных лучей. Необходимо рекомендовать пациентам избегать длительного пребывания на солнце во время приема препарата Алеценза® и в течение не менее 7 дней после прекращения терапии. Также следует использовать солнцезащитные средства и бальзам для губ с защитой от УФ лучей широкого спектра (от воздействия УФ А (ультрафиолетового излучения диапазона А)/УФ В (ультрафиолетового излучения диапазона В)) с SPF (солнцезащитным фактором) ≥50 для защиты от возможных солнечных ожогов.
Эмбриофетальная токсичность
Применение препарата при беременности может оказывать повреждающее воздействие на плод. У беременных крыс и кроликов применение алектиниба сопровождалось эмбриофетальной токсичностью. Пациентки или половые партнерши пациентов, обладающие детородным потенциалом, должны использовать высокоэффективные методы контрацепции во время терапии препаратом Алеценза® и в течение не менее 3 месяцев после приема последней дозы препарата.
Непереносимость лактозы
Препарат Алеценза® содержит лактозу. Следует соблюдать осторожность при применении препарата Алеценза® у пациентов с редкой наследственной непереносимостью галактозы, врожденной недостаточностью лактазы или глюкозо-галактозной мальабсорбцией.
Инструкции по уничтожению неиспользованного препарата или препарата с истекшим сроком годности
Попадание лекарственного препарата в окружающую среду должно быть сведено к минимуму. Не следует утилизировать препарат с помощью сточных вод или вместе с бытовыми отходами. Уничтожение неиспользованного препарата или препарата с истекшим сроком годности должно проводиться в соответствии с локальными требованиями.
Влияние на способность управлять транспортными средствами и механизмами
Влияние препарата Алеценза® на способность управлять автомобилем и работать с механизмами не изучалось.
Форма выпуска
Капсулы 150 мг
По 8 капсул в контурную ячейковую упаковку (блистер) из композиционного материала (полиамид/алюминий/поливинилхлорид) и фольги алюминиевой композиционной (алюминий/сополимер винилхлорида/винилацетата полибутилметакрилат).
7 контурных ячейковых упаковок (блистеров) вместе с инструкцией по применению помещают в картонную пачку (промежуточная упаковка), на которую с целью контроля первого вскрытия наклеивают голограммы.
4 промежуточных упаковки помещают в картонную пачку (потребительская упаковка) с контролем первого вскрытия (в виде перфорации).
Срок годности
3 года
Не использовать по истечении срока годности, указанного на упаковке.
Условия хранения
При температуре не выше 30°С в оригинальной упаковке для защиты от света и влаги.
Условия отпуска
Отпускают по рецепту.
Владелец Регистрационного удостоверения
Ф. Хоффманн-Ля Рош Лтд., Швейцария
F. Hoffmann-La Roche Ltd, Grenzacherstrasse 124, 4070 Basel, Switzerland
Производитель
Экселла ГмбХ и Ко. КГ, Германия
Excella GmbH & Со. KG, Nurnberger Strasse 12, 90537 Feucht, Germany
Фасовщик (первичная упаковка)
Рош С.п.А., Италия
Roche S.p.A., Via Morelli 2, 20090 Segrate, Milano, Italy
Упаковщик (вторичная/потребительская упаковка)
Рош С.п.А., Италия
Roche S.p.A., Via Morelli 2, 20090 Segrate, Milano, Italy
Выпускающий контроль качества
Рош С.п.А., Италия
Roche S.p.A., Via Morelli 2, 20090 Segrate, Milano, Italy
Претензии потребителей направлять в компанию ЗАО «Рош-Москва» по адресу:
107031, Россия, г. Москва, Трубная площадь, д. 2
Комментарии
(видны только специалистам, верифицированным редакцией МЕДИ РУ)
Фармакологическое действие
Алектиниб блокирует действие фермента — киназы апластической лимфомы (anaplastic lymphoma kinase — ALK-тирозинкиназа). Аномальные формы этого фермента(возникшие вследствие дефекта в гене, который образует этот фермент) способствует росту клеток опухоли. Препарат Алеценза® может замедлить или остановить рост клеток опухоли.
В доклинических исследованиях ингибирование активности тирозинкиназы ALK приводило к блокаде нисходящих сигнальных путей, включая STAT3 (переносчик сигнала и активатор транскрипции 3) и PI3K/AKT(фосфоинозитол-3-киназу/протеинкиназу В), и вызывало апоптоз опухолевых клеток.
Фармакокинетика
Фармакокинетические показатели алектиниба и М4 оценивали у пациентов с ALK-положительным НМРЛ (немелкоклеточным раком легкого) и у здоровых добровольцев. Средние геометрические (коэффициент вариации, %) показатели Cmax, Cmin и AUC0-12ч в равновесном состоянии для алектиниба составляли приблизительно 665 нг/мл (44.3%), 572 нг/мл (47.8%) и 7430 нг×ч/мл (45.7%) соответственно. Средние геометрические показатели Cmax, Cmin и AUC0-12ч в равновесном состоянии для М4 составляли приблизительно 246 нг/мл (45.4%), 222 нг/мл (46.6%) и 2810 нг×ч/мл (45.9%), соответственно.
Всасывание
После перорального приема алектиниба у пациентов с ALK-положительным НМРЛ в дозе 600 мг 2 раза/сут после приема пищи отмечалось быстрое всасывание препарата. Tmax — приблизительно от 4 до 6 ч.
При приеме алектиниба в дозе 600 мг 2 раза/сут равновесное состояние алектиниба достигается в течение 7 дней и остается неизменным, при этом согласно данным популяционного фармакокинетического анализа средний геометрический коэффициент накопления равен 5.6. Популяционный фармакокинетический анализ показал пропорциональность доз алектиниба в диапазоне от 300 мг до 900 мг после приема пищи.
У здоровых добровольцев абсолютная биодоступность при применении алектиниба после приема пищи составляла 36.9% (33.9-40.3%).
При однократном пероральном применении алектиниба в дозе 600 мг прием высококалорийной пищи с большим содержанием жиров увеличивал экспозицию препарата в 3 раза по сравнению с приемом натощак (среднее геометрическое соотношение комбинированных показателей для алектиниба и М4 — Cmax: 3.31 [2.79-3.93], AUCinf: 3.11 [2.73-3.55]).
Распределение
Алектиниб и M4 имеют высокий уровень связывания с белками плазмы крови человека (>99%) независимо от концентрации действующего вещества. В условиях in vitro среднее соотношение концентраций в крови и в плазме человека составляет 2.64 и 2.50 для алектиниба и М4 соответственно (при клинически значимых концентрациях).
Средний геометрический Vss алектиниба после в/в введения составлял 475 л, указывая на широкое распределение алектиниба в тканях.
Метаболизм
Исследования метаболизма в условиях in vitro показали, что изофермент CYP3A4 является основным изоферментом CYP (цитохрома P450), который опосредует метаболизм алектиниба и М4, при этом определено, что данный изофермент осуществляет 40-50% метаболизма в гепатоцитах человека. Согласно результатам исследования массового баланса алектиниб и М4 являлись основными циркулирующими веществами в плазме крови, их общее содержание составляло 76% от общей радиоактивности в плазме крови. Среднее геометрическое соотношение метаболит/препарат в равновесном состоянии – 0.399.
Выведение
У здоровых добровольцев после однократного перорального применения 14С-меченого алектиниба большая часть радиоактивности была выявлена в кале (средняя степень извлечения 97.8%, диапазон 95.6-100%), минимальная – в моче (средняя степень извлечения 0.46%, диапазон 0.30-0.60%). Доля неизмененного алектиниба в кале составляла 84% от введенной дозы, а доля М4 составила 5.8%.
Согласно данным популяционного фармакокинетического анализа CL/F (кажущийся клиренс) алектиниба составлял 81.9 л/ч, М4 – 217 л/ч. Индивидуальный расчетный средний геометрический Т1/2 алектиниба составлял 32.5 ч, M4 – 30.7 ч.
Фармакокинетика у особых групп пациентов
Пациенты детского возраста. Исследования фармакокинетики алектиниба у пациентов детского возраста (<18 лет) не проводились.
Пациенты пожилого возраста. Возраст не оказывает влияния на экспозицию алектиниба.
Пациенты с нарушением функции почек. Алектиниб и М4 выводятся почками в незначительных количествах в неизмененном виде (<0.2% дозы). Данные, полученные у пациентов с нарушением функции почек легкой и средней степени тяжести, показывают, что нарушение функции почек не оказывает значительного влияния на фармакокинетику алектиниба. Специальные исследования фармакокинетики не проводились, данные популяционной фармакокинетики для пациентов с нарушением функции почек тяжелой степени тяжести отсутствуют, однако коррекции дозы у таких пациентов не требуется, поскольку алектиниб выводится почками в незначительной степени.
Пациенты с нарушением функции печени. Основным путем выведения алектиниба является печеночный метаболизм. У пациентов с нарушением функции печени возможно увеличение концентрации алектиниба и/или М4 в плазме крови. Согласно данным популяционного фармакокинетического анализа экспозиции алектиниба и М4 у пациентов с нарушением функции печени легкой степени тяжести (общий билирубин на исходном уровне ≤ВГН и активность АСТ >ВГН или общий билирубин на исходном уровне >1.0-1.5×ВГН и любая активность АСТ на исходном уровне) соответствовали таковым у пациентов с нормальной функцией печени (общий билирубин ≤ВГН и активность АСТ ≤ВГН).
После однократного перорального приема алектиниба в дозе 300 мг пациентами с нарушением функции печени тяжелой степени тяжести (класс С по шкале Чайлд-Пью) Сmax алектиниба была такой же как и у здоровых добровольцев; AUCinf у пациентов с нарушением функции печени тяжелой степени тяжести (класс С по шкале Чайлд-Пью) была в 2.2 раза выше, чем у здоровых добровольцев, а Сmax и AUCinf для М4 были на 39% и 34% ниже соответственно. Таким образом, общая комбинированная экспозиция для алектиниба и М4 (AUCinf) была в 1.8 раз выше у пациентов с нарушением функции печени тяжелой степени тяжести по сравнению со здоровыми добровольцами.
В ходе изучения применения препарата у пациентов с нарушением функции печени средней степени тяжести (класс B по шкале Чайлд-Пью) наблюдалась умеренно повышенная экспозиция алектиниба по сравнению со здоровыми добровольцами. Однако у пациентов с нарушением функции печени средней степени тяжести (класс B по шкале Чайлд-Пью), в целом, не отмечалось отклонений в показателях билирубина, альбумина или протромбинового времени, что указывает на их возможное неполное соответствие пациентам с нарушением функции печени умеренной степени тяжести со сниженной метаболической способностью.
Показания препарата
Алеценза®
- монотерапия местно-распространенного или метастатического немелкоклеточного рака легкого с опухолевой экспрессией киназы анапластической лимфомы (ALK-положительный) у взрослых старше 18 лет.
Режим дозирования
Препарат принимают внутрь. Капсулы следует принимать одновременно с приемом пищи и проглатывать целиком. Открывать или растворять капсулы нельзя.
Перед применением препарата Алеценза® в первой линии терапии НМРЛ необходимо установить наличие опухолевой экспрессии киназы анапластической лимфомы (ALK). Для выявления пациентов с ALK-положительным НМРЛ необходимо использовать валидированный метод определения ALK.
Рекомендуемая доза препарата Алеценза® составляет 600 мг (4 капсулы по 150 мг) 2 раза/сут (суточная доза составляет 1200 мг).
Пациенты с исходным нарушением функции печени тяжелой степени тяжести (класс С по шкале Чайлд-Пью) должны получать препарат Алеценза® в начальной дозе 450 мг (3 капсулы по 150 мг) 2 раза/сут (суточная доза составляет 900 мг).
Продолжительность лечения
Лечение препаратом Алеценза® рекомендуется проводить до прогрессирования заболевания или до развития непереносимой токсичности.
Пациент не должен самостоятельно Алеценза® прекращать прием препарата без консультации врача.
Коррекция дозы
Для контроля нежелательных явлений может потребоваться временное прерывание лечения, уменьшение дозы или полное прекращение применения препарата Алеценза®.
Дозу препарата необходимо уменьшать пошагово, на 150 мг 2 раза/сут, в зависимости от переносимости.
Если применение препарата в дозе 300 мг 2 раза/сут невозможно из-за непереносимости, терапию препаратом Алеценза® следует полностью прекратить.
В таблицах ниже приведены общие рекомендации по коррекции дозы препарата Алеценза®.
Таблица 1. Схема снижения дозы
Таблица 2. Коррекция дозы при развитии отдельных нежелательных реакций
а ЧСС менее 60 уд./мин.
Дозирование в особых случаях
Безопасность и эффективность препарата Алеценза® у детей и подростков (<18 лет) не изучены.
Коррекции дозы препарата Алеценза® у пациентов пожилого/старческого возраста (≥65 лет) не требуется.
Коррекции дозы препарата Алеценза® у пациентов с нарушением функции почек легкой и средней степени тяжести не требуется. Применение препарата Алеценза® у пациентов с нарушением функции почек тяжелой степени не изучено, однако поскольку алектиниб выводится почками в незначительной степени, коррекции дозы у таких пациентов не требуется.
Пациентам с исходным нарушением функции печени легкой (класс А по шкале Чайлд-Пью) или средней степени тяжести (класс В по шкале Чайлд-Пью) коррекции начальной дозы не требуется. Пациенты с исходным нарушением функции печени тяжелой степени тяжести (класс С по шкале Чайлд-Пью) должны получать препарат Алеценза® в начальной дозе 450 мг 2 раза/сут (суточная доза составляет 900 мг). Рекомендуется соответствующее наблюдение (контроль показателей функции печени) для всех пациентов с нарушением функции печени.
Задержка или пропуск приема препарата
Если до приема следующей дозы препарата осталось более 6 ч, следует принять пропущенную дозу сразу, как только пациент вспомнит об этом.
Если до приема следующей дозы осталось менее 6 ч, не следует принимать пропущенную дозу. Необходимо принять следующую дозу в обычное время.
Не принимать двойную дозу, чтобы компенсировать пропущенную дозу.
Если возникла рвота после приема препарата
Не следует принимать дополнительную дозу препарата. Следует принять следующую дозу в обычное время.
Если принятая доза оказалась больше назначенной
Пациент должен немедленно обратиться к лечащему врачу или в больницу. Необходимо взять с собой упаковку препарата и листок-вкладыш.
Побочное действие
Безопасность препарата Алеценза® оценивали в клинических исследованиях (КИ) у пациентов с ALK-положительным НМРЛ, ранее получавших терапию кризотинибом, при применении в рекомендованной дозе 600 мг 2 раза/сут; медиана экспозиции составила 11 месяцев (0-35 месяцев). Безопасность препарата Алеценза® также оценивали в КИ у пациентов с ALK-положительным НМРЛ, ранее не получавших лечение, при применении в рекомендованной дозе 600 мг 2 раза/сут; медиана экспозиции составила 17.9 месяцев.
Наиболее частыми (≥20%) нежелательными реакциями были запор (36%), отеки (34%, включая периферический, генерализованный, отек век и периорбитальный отек); миалгия (31%, включая миалгию и скелетно-мышечную боль), тошнота (22%), повышение концентрации билирубина (21%, включая повышение концентрации билирубина в крови, гипербилирубинемию и повышение концентрации конъюгированного билирубина), анемия (20%, включая анемию и снижение уровня гемоглобина) и сыпь (20%, включая сыпь, макулопапулезную сыпь, акнеиформный дерматит, эритему, генерализованную сыпь, папулезную сыпь, зудящую сыпь и макулярную сыпь).
Ниже представлен перечень нежелательных реакций, отмечавшихся в КИ у пациентов с ALK-положительным НМРЛ, ранее получавших терапию кризотинибом, и у пациентов с ALK-положительным НМРЛ, ранее не получавших лечение. Нежелательные реакции сгруппированы в соответствии с классами систем органов медицинского словаря для нормативно-правовой деятельности MedDRA.
Для описания частоты нежелательных реакций используется следующая классификация: очень часто (≥10%), часто (≥1% и <10%), нечасто (≥0.1% и <1%), редко (≥0.01% и <0.1%), очень редко (<0.01%).
Со стороны системы кроветворения: очень часто – анемия1; гемолитическая анемия, которая при которой возможны повышенная утомляемость, слабость или одышка.
Со стороны нервной системы: часто – дисгевзия (нарушение вкусовых восприятий)#2.
Со стороны органа зрения: очень часто – расстройства зрения3.
Со стороны сердечно-сосудистой системы: очень часто – брадикардия4, вследствие которой возможны обморок, головокружение, артериальная гипотензия.
Со стороны дыхательной системы: часто – интерстициальная болезнь легких/пневмонит.
Со стороны пищеварительной системы: очень часто – запор, тошнота, диарея, рвота; часто – стоматит#5.
Со стороны печени и желчевыводящих путей: очень часто – повышение концентрации билирубина6, повышение активности АСТ, повышение активности АЛТ; нечасто – лекарственно-индуцированное поражение печени7.
Со стороны кожи и подкожных тканей: очень часто – сыпь8, реакция фоточувствительности.
Со стороны костно-мышечной системы: очень часто – миалгия9, повышение активности КФК в крови.
Со стороны мочевыделительной системы: часто – повышение концентрации креатинина в крови*, острая почечная недостаточность*#.
Прочие: очень часто – отеки10; часто – увеличение массы тела#.
* Включая одно явление с летальным исходом.
# Явление отмечено в клиническом исследовании у пациентов с ALK-положительным НМРЛ, ранее не получавших лечение.
1 Включая случаи анемии и снижения уровня гемоглобина.
2 Включая случаи дисгевзии и гипогевзии (снижение вкусовых восприятий).
3 Включая случаи нечеткости зрения, нарушения зрения, плавающих помутнений в стекловидном теле, снижения остроты зрения, астенопии и диплопии.
4 Включая случаи брадикардии и синусовой брадикардии.
5 Включая случаи стоматита и язвенного стоматита.
6 Включая случаи повышения концентрации билирубина в крови, гипербилирубинемии, повышения концентрации конъюгированного билирубина.
7 Включая одного пациента с лекарственно-индуцированным поражением печени (термин MedDRA) и одного пациента с повышением активности АСТ и АЛТ 4 степени и задокументированным лекарственно-индуцированным поражением печени, подтвержденным биопсией.
8 Включая случаи сыпи, макулопапулезной сыпи, акнеформного дерматита, эритемы, генерализованной сыпи, папулезной сыпи, зудящей сыпи и макулярной сыпи.
9 Включая случаи миалгии и скелетно-мышечной боли.
10 Включая случаи отека, отека век и периферического, генерализованного, периорбитального отека.
Описание отдельных нежелательных реакций
Профиль безопасности препарата Алеценза® в целом был сходным в КИ у пациентов с ALK-положительным НМРЛ, ранее получавших терапию кризотинибом, и у пациентов, ранее не получавших лечение.
Интерстициальная болезнь легких (ИБЛ)/пневмонит
Часто наблюдались следующие серьезные нежелательные реакции: признаки появления или ухудшения нарушений дыхания, в т.ч. затрудненное дыхание, одышка, кашель (сухой или с выделением мокроты), или повышение температуры. Эти симптомы могут быть признаками пневмонита.
При применении препарата Алеценза® отмечались тяжелые случаи интерстициальной болезни легких/пневмонита. В КИ у 0.4% пациентов с ALK-положительным НМРЛ, ранее получавших терапию кризотинибом, развивалась ИБЛ 3 степени тяжести, что привело к прекращению терапии препаратом Алеценза®. Явлений ИБЛ с летальным исходом ни в одном КИ отмечено не было.
Гепатотоксичность
Нечасто наблюдались следующие серьезные нежелательные реакции: желтушность кожи или белков глаз, боли в правом подреберье, темный цвет мочи, кожный зуд, снижение аппетита, тошнота или рвота, повышенная утомляемость, кровотечения или повышенная склонность к появлению синяков. При появлении таких реакций пациент должен немедленно обратиться к врачу.
Повышение активности АСТ/АЛТ 3-4 степени тяжести отмечалось в КИ у двух пациентов с ALK-положительным НМРЛ, ранее получавших терапию кризотинибом; при этом было задокументировано лекарственно-индуцированное поражение печени, подтвержденное биопсией.
Повышение активности АСТ и АЛТ отмечалось у 16% и 14% пациентов в КИ, соответственно. Большинство явлений были 1 и 2 степеней тяжести; явления ≥3 степени тяжести наблюдались у 2.8% и 3.2% пациентов, соответственно. Данные явления обычно развивались в течение первых 3 месяцев терапии, были, как правило, преходящими и разрешались после временной приостановки лечения (у 1.2% и 3.2% пациентов, соответственно) или снижения дозы препарата Алеценза® (у 1.6% и 0.8%, соответственно). Повышение активности АСТ и АЛТ приводило к прекращению терапии препаратом Алеценза® у 1.2% и 1.6% пациентов, соответственно.
Повышение концентрации общего билирубина отмечалось в КИ у 17% пациентов с ALK-положительным НМРЛ, ранее получавших терапию кризотинибом. Большинство явлений были 1 и 2 степеней тяжести; явления 3 степени тяжести наблюдались у 3.2% пациентов. Данные явления обычно отмечались в течение первых 3 месяцев терапии, были преходящими и разрешались после временной приостановки лечения (у 4.7% пациентов) или снижения дозы препарата Алеценза® (у 2.8% пациентов). Повышение концентрации билирубина приводило к прекращению терапии препаратом Алеценза® у 1.6% пациентов.
Одновременное повышение активности АЛТ или АСТ ≥3×ВГН и повышение концентрации общего билирубина ≥2×ВГН при нормальной активности ЩФ отмечалось у одного пациента в КИ препарата Алеценза®.
Брадикардия
Случаи брадикардии отмечались в КИ у 7.9% пациентов с ALK-положительным НМРЛ, ранее получавших терапию кризотинибом. Все явления были 1 или 2 степени тяжести. У 20% пациентов после приема препарата Алеценза® ЧСС составляла <50 уд/мин.
Тяжелая миалгия и повышение активности КФК
Сообщения о миалгии, включавшие миалгию (25%) и скелетно-мышечную боль (7.5%) регистрировались в КИ у пациентов с ALK-положительным НМРЛ, ранее получавших терапию кризотинибом, с общей частотой 31%. Большинство явлений были 1 или 2 степени тяжести, у 1.2% пациентов наблюдались явления 3 степени тяжести. Только 0.8% пациентов потребовалась коррекция дозы в связи с данными явлениями.
Повышение активности КФК отмечалось в КИ у 46% пациентов с доступными лабораторными данными. Частота повышения активности КФК 3 степени тяжести составила 5.0%; при этом медиана времени до возникновения явления составила 14 дней у пациентов с ALK-положительным НМРЛ, ранее получавших терапию кризотинибом, и 27.5 дней у пациентов с ALK-положительным НМРЛ, ранее не получавших лечение. Коррекция дозы препарата Алеценза® потребовалась у 4.0% пациентов с повышением активности КФК.
Отклонения со стороны лабораторных показателей
У пациентов с ALK-положительным НМРЛ, ранее получавших терапию кризотинибом, и у пациентов, ранее не получавших лечение, на фоне терапии препаратом Алеценза® наблюдались следующие отклонения со стороны лабораторных показателей:
Примечание: лабораторные отклонения определялись с учетом нормальных значений по классификации NCI CTCAE (Национального института рака National Cancer Institute Common Terminology Criteria of Adverse Events).
* Частота, отмечавшаяся в КИ у пациентов с ALK-положительным НМРЛ, ранее получавших терапию кризотинибом.
** Только пациенты с повышением концентрации креатинина на основании определения ВГН по классификации нежелательных явлений CTCAE.
# Частота, отмечавшаяся в КИ у пациентов с ALK-положительным НМРЛ, ранее не получавших лечение.
° Отклонений лабораторных показателей 5 степени тяжести не наблюдалось.
Пострегистрационный опыт применения
При пострегистрационном применении препарата Алеценза® отмечалось повышение активности ЩФ; при этом в КИ данное явление наблюдалось у 7.5% пациентов с ALK-положительным НМРЛ, ранее получавших терапию кризотинибом.
Противопоказания к применению
- повышенная чувствительность к алектинибу или к другим вспомогательным веществам препарата в анамнезе;
- беременность;
- период грудного вскармливания;
- возраст до 18 лет (эффективность и безопасность применения не изучались).
С осторожностью: нарушение функции почек тяжелой степени; редкая наследственная непереносимость галактозы, врожденная недостаточность лактазы или глюкозо-галактозная мальабсорбция.
Применение при беременности и кормлении грудью
Контрацепция
Пациентки или половые партнерши пациентов, а также пациенты мужчины, обладающие детородным потенциалом, должны использовать высокоэффективные методы контрацепции во время терапии препаратом Алеценза® и в течение не менее 3 месяцев после приема последней дозы препарата.
Беременность
Женщинам детородного возраста необходимо избегать беременности при приеме препарата Алеценза®. Клинические исследования алектиниба у беременных женщин не проводились. В силу своего механизма действия препарат Алеценза® может оказывать повреждающее воздействие на плод.
Если беременность наступила во время лечения препаратом или в течение 3 месяцев после приема последней дозы препарата, пациентка должна незамедлительно сообщить об этом лечащему врачу.
Если беременность наступила у женщины во время лечения препаратом ее полового партнера или в течение 3 месяцев после того, как он принял последнюю дозу препарата, женщина должна незамедлительно сообщить об этом лечащему врачу.
Доклинические данные
В исследованиях на животных алектиниб вызывал развитие эмбриофетальной токсичности.
Период грудного вскармливания
Неизвестно, выделяется ли алектиниб с грудным молоком. В период лечения следует воздержаться от грудного вскармливания, поскольку прием препарата может причинить вред грудному ребенку.
Применение при нарушениях функции печени
Пациентам с исходным нарушением функции печени легкой (класс А по шкале Чайлд-Пью) или средней степени тяжести (класс В по шкале Чайлд-Пью) коррекции начальной дозы не требуется.
У пациентов с исходным нарушением функции печени тяжелой степени тяжести (класс С по шкале Чайлд-Пью) суточная доза составляет 900 мг.
Применение при нарушениях функции почек
С осторожностью следует назначать при нарушении функции почек тяжелой степени тяжести.
Применение у детей
Противопоказано применение препарата в возрасте до 18 лет.
Применение у пожилых пациентов
Коррекции дозы у пациентов пожилого/старческого возраста (≥65 лет) не требуется.
Особые указания
Интерстициальная болезнь легких (ИБЛ)/пневмонит
В клинических исследованиях препарата Алеценза® отмечались случаи ИБЛ/пневмонита. Необходимо наблюдать за пациентами на предмет развития легочных симптомов пневмонита.
Следует незамедлительно прервать лечение препаратом Алеценза® у пациентов с диагностированным ИБЛ/пневмонитом и полностью прекратить терапию препаратом Алеценза®, если иные потенциальные причины возникновения ИБЛ/пневмонита не установлены.
Гепатотоксичность
В клинических исследованиях препарата Алеценза® отмечалось повышение активности АЛТ и АСТ >5×ВГН, а также повышение концентрации билирубина >3×ВГН. Большинство явлений возникало в ходе первых 3 месяцев терапии.
У трех пациентов с повышением активности АСТ/АЛТ 3-4 степени тяжести наблюдалось лекарственно-индуцированное поражение печени. У одного пациента зарегистрировано одновременное повышение активности АЛТ или АСТ ≥3×ВГН и повышение концентрации общего билирубина ≥2×ВГН при нормальной активности ЩФ.
Следует проводить мониторинг показателей функции печени, в т.ч. АЛТ, АСТ и общего билирубина, на исходном уровне и затем каждые 2 недели в течение первых 3 месяцев лечения. Далее необходимо продолжать периодический мониторинг, поскольку отклонения могут возникать и после 3 месяцев лечения. Пациентам, у которых отмечалось повышение активности трансаминаз и концентрации билирубина, необходим более частый мониторинг. В зависимости от степени тяжести нежелательной реакции следует или приостановить лечение с последующим возобновлением приема препарата в сниженной дозе, или полностью прекратить терапию препаратом Алеценза® (см. таблицу 2, раздел «Режим дозирования»).
Тяжелая миалгия и повышение активности КФК
В клинических исследованиях препарата Алеценза® отмечались миалгия или скелетно-мышечная боль, в т.ч. 3 степени тяжести.
В клинических исследованиях препарата Алеценза® отмечалось повышение активности КФК, в т.ч. 3 степени тяжести. Медиана времени до возникновения явлений 3 степени тяжести составила 14 дней у пациентов с ALK-положительным НМРЛ, ранее получавших терапию кризотинибом, и 27.5 дней у пациентов с ALK-положительным НМРЛ, ранее не получавших лечение.
Следует рекомендовать пациентам сообщать о любых случаях мышечной боли, болезненности или слабости неясной этиологии. Необходимо оценивать активность КФК каждые 2 недели в первый месяц терапии и в соответствии с клиническими показаниями у тех пациентов, которые сообщают о симптомах.
В зависимости от степени тяжести повышения активности КФК следует приостановить лечение препаратом Алеценза® с последующим возобновлением приема препарата в прежней дозе или снизить дозу (см. таблицу 2, раздел «Режим дозирования»).
Брадикардия
При применении препарата Алеценза® возможно возникновение симптоматической брадикардии.
Необходим мониторинг ЧСС и АД в соответствии с клиническими показаниями. В случае возникновения бессимптомной брадикардии коррекции дозы не требуется.
При развитии симптоматической брадикардии или жизнеугрожающих состояний следует оценить действие сопутствующих препаратов с известной способностью вызывать брадикардию, а также действие антигипертензивных препаратов. Дозу препарата Алеценза® корректируют (см. таблицу 2, раздел «Режим дозирования»).
Фоточувствительность
При применении препарата Алеценза® развивалась фоточувствительность к воздействию солнечных лучей. Необходимо рекомендовать пациентам избегать длительного пребывания на солнце во время приема препарата Алеценза® и в течение не менее 7 дней после прекращения терапии. Также следует использовать солнцезащитные средства и бальзам для губ с защитой от УФ лучей широкого спектра (от воздействия УФА (ультрафиолетового излучения диапазона А)/УФ-В (ультрафиолетового излучения диапазона В)) с SPF (солнцезащитным фактором) ≥50 для защиты от возможных солнечных ожогов.
Вспомогательные вещества
Препарат Алеценза® содержит лактозу. Следует соблюдать осторожность при применении препарата Алеценза® у пациентов с редкой наследственной непереносимостью галактозы, врожденной недостаточностью лактазы или глюкозо-галактозной мальабсорбцией.
Препарат Алеценза® содержит 2.09 ммоль (или 48 мг) натрия на суточную дозу препарата (1200 мг). Это необходимо учитывать, если пациент находится на диете с ограничением потребления натрия.
Инструкция по уничтожению неиспользованного препарата или препарата с истекшим сроком годности
Попадание лекарственного препарата в окружающую среду должно быть сведено к минимуму. Не следует утилизировать препарат с помощью сточных вод или вместе с бытовыми отходами. Уничтожение неиспользованного препарата или препарата с истекшим сроком годности должно проводиться в соответствии с локальными требованиями.
Влияние на способность к управлению транспортными средствами и механизмами
Влияние препарата Алеценза® на способность управлять автомобилем и работать с механизмами не изучалось.
Передозировка
В клинических исследованиях не наблюдалось случаев передозировки.
Лечение: при передозировке необходимо тщательно наблюдать за состоянием пациентов и проводить поддерживающую терапию. Специфического антидота, который можно было бы использовать при передозировке препарата Алеценза®, нет.
Лекарственное взаимодействие
Влияние алектиниба на сопутствующие лекарственные препараты
Субстраты изоферментов цитохрома P450
В условиях in vitro ни алектиниб, ни его основной активный метаболит — М4 — не оказывали ингибирующего действия на изоферменты CYP1A2, CYP2B6, CYP2C9, CYP2C19 или CYP2D6 в клинически значимых концентрациях. Алектиниб и M4 оказывают слабое, зависящее от времени, ингибирующее действие на CYP3A4. Кроме того, алектиниб является слабым индуктором изоферментов CYP3A4 и CYP2B6 в клинических концентрациях in vitro.
Согласно результатам исследования межлекарственного взаимодействия у пациентов с ALK-положительным НМРЛ, многократное применение алектиниба не оказывает влияния на экспозицию мидазолама, который является чувствительным субстратом CYP3A. Таким образом, при одновременном применении с субстратами CYP3A коррекции дозы не требуется.
Несмотря на то, что исследования в условиях in vitro показывают, что алектиниб является ингибитором изофермента CYP2C8, данные фармакокинетического моделирования, учитывающего физиологические процессы организма человека, свидетельствуют о том, что алектиниб в клинически значимых концентрациях не будет увеличивать плазменные концентрации одновременно применяемых препаратов-субстратов изофермента CYP2C8.
Субстраты P-гликопротеина и BCRP
В условиях in vitro алектиниб и М4 являются ингибиторами P-гликопротеина и BCRP. Таким образом, алектиниб и М4 могут способствовать увеличению плазменных концентраций субстратов Р-гликопротеина или BCRP при их одновременном применении (не ожидается, что увеличение экспозиции будет более чем двукратным). Рекомендуется соответствующее наблюдение пациентов, которые одновременно принимают алектиниб и субстраты Р-гликопротеина или BCRP с узким терапевтическим индексом (например, дигоксин, дабигатран, метотрексат).
Влияние сопутствующих лекарственных препаратов на алектиниб
Согласно данным, полученным in vitro, изофермент CYP3A4 является основным ферментом, ответственным за метаболизм алектиниба и М4, при этом предполагается, что посредством CYP3A осуществляется 40-50% общего печеночного метаболизма. М4 и алектиниб обладают сходной эффективностью и активностью в отношении ALK в условиях in vitro.
Индукторы изофермента CYP3A
При многократном пероральном применении рифампицина (мощного индуктора изофермента CYP3A) в дозе 600 мг 1 раз/сут одновременно с однократным пероральным приемом алектиниба в дозе 600 мг отмечалось небольшое влияние на общую экспозицию алектиниба и М4 (среднее геометрическое соотношение с/без рифампицина — Cmax: 0.96 [0.88-1.05], AUCinf: 0.82 [0.74-0.90]). Таким образом, при одновременном приеме препарата Алеценза® с индукторами изофермента CYP3A коррекции дозы не требуется.
Ингибиторы изофермента CYP3A
При многократном пероральном применении позаконазола (мощного ингибитора изофермента CYP3A) в дозе 400 мг 2 раза/сут одновременно с однократным пероральным приемом алектиниба в дозе 300 мг отмечалось небольшое влияние на общую экспозицию алектиниба и М4 (среднее геометрическое соотношение с/без позаконазола — Cmax: 0.93 [0.81-1.08], AUCinf: 1.36 [1.24-1.49]). Таким образом, при одновременном приеме препарата Алеценза® с ингибиторами изофермента CYP3A коррекции дозы не требуется.
Лекарственные препараты, повышающие рН желудка
Несмотря на то, что растворимость алектиниба в воде в условиях in vitro зависит от рН, специальное клиническое исследование межлекарственного взаимодействия показало, что применение эзомепразола (ингибитора протонового насоса) в дозе 40 мг 1 раз/сут не оказывает клинически значимого влияния на общую экспозицию алектиниба и М4. Таким образом, при одновременном применении препарата Алеценза® с ингибиторами протонового насоса или другими препаратами, повышающими рН желудка (например, антагонистами Н2-рецепторов или антацидами), коррекции дозы не требуется.
Влияние белков-переносчиков на распределение алектиниба
Алектиниб не является субстратом P-гликопротеина in vitro. Ни алектиниб, ни М4 не являются субстратами для BCRP или OATP 1B1/B3. При этом М4 является субстратом Р-гликопротеина. Алектиниб ингибирует Р-гликопротеин, таким образом, не ожидается, что одновременное применение ингибиторов Р-гликопротеина будет оказывать значимое влияние на экспозицию М4.
Условия хранения препарата Алеценза®
Препарат следует хранить в оригинальной упаковке для защиты от света и влаги, при температуре не выше 30°С.
Срок годности препарата Алеценза®
Срок годности — 3 года. Не применять по истечении срока годности, указанного на упаковке.
Условия реализации
Препарат отпускается по рецепту.
1 капсула содержит 150 мг алектиниба (в виде алектиниба гидрохлорида 161.33 мг).
Вспомогательные вещества с известными свойствами
Каждая капсула содержит 33.7 мг лактозы (в виде моногидрата) и 6 мг натрия (в виде натрия лаурилсульфата).
Полный перечень вспомогательных веществ приведен в разделе «Перечень вспомогательных веществ».
Твердые капсулы, размер 1, корпус и крышечка капсулы от белого до желтовато-белого цвета.
На корпусе капсулы нанесена надпись «150 mg» черного цвета. На крышечке капсулы нанесена надпись «ALE» черного цвета.
Содержимое капсулы: порошок или скомковавшийся порошок от белого до светло-желтого цвета.
Препарат Алеценза в качестве монотерапии первой линии показан для лечения взрослых пациентов с ALK-положительным распространенным немелкоклеточным раком лёгкого (НМРЛ).
Препарат Алеценза в качестве монотерапии показан для лечения взрослых пациентов с ALK-положительным распространенным немелкоклеточным раком лёгкого, которые ранее получали терапию кризотинибом.
Лечение препаратом Алеценза должно начинаться и проводиться врачами, имеющими опыт использования противоопухолевых лекарственных средств.
Для отбора пациентов с ALK-положительным НМРЛ необходимо провести валидированный анализ ALK. Статус ALK-положительного НМРЛ определяется до начала лечения препаратом Алеценза.
Режим дозирования
Рекомендуемая доза препарата Алеценза составляет 600 мг (четыре капсулы по 150 мг), принимаемая два раза в сутки во время приема пищи (общая суточная доза составляет 1200 мг).
Пациенты с тяжелой печеночной недостаточностью (Child-Pugh С (Чайлд-Пью С)) должны получать начальную дозу 450 мг, принимаемую дважды в день во время приема пищи (общая суточная доза 900 мг).
Продолжительность лечения
Лечение препаратом Алеценза необходимо продолжать до прогрессирования заболевания или развития неприемлемой токсичности.
Несвоевременно принятые или пропущенные дозы
Если запланированный прием дозы препарата Алеценза пропущен, пациенты могут компенсировать эту дозу за 6 часов до приема следующей дозы. Пациентам не следует принимать две дозы одновременно для компенсации пропущенной дозы. При возникновении рвоты после приема дозы препарата Алеценза пациентам следует принять следующую дозу в запланированное время.
Коррекция дозы
Для устранения нежелательных реакций может потребоваться снижение дозы, временный перерыв или прекращение лечения препаратом Алеценза. Дозу препарата Алеценза снижают поэтапно по 150 мг два раза в сутки в зависимости от переносимости. Лечение препаратом Алеценза следует полностью прекратить, если пациенты не могут переносить дозу 300 мг два раза в сутки.
Рекомендации по коррекции дозы представлены ниже в таблицах 1 и 2.
Таблица 1 Схема снижения дозы
| Схема снижения дозы | Величина дозы |
|---|---|
| Доза | 600 мг два раза в сутки |
| Первое снижение дозы | 450 мг два раза в сутки |
| Второе снижение дозы | 300 мг два раза в сутки |
Таблица 2 Рекомендации по коррекции дозы для установленных нежелательных реакций на лекарственное средство (см. разделы «Особые указания и меры предосторожности при применении» и «Нежелательные реакции»)
|
Степень на основании Общих критериев терминологии нежелательных явлений (СТСАЕ) |
Лечение препаратом Алеценза |
|
ИЗЛ / пневмонит любой степени тяжести |
Незамедлительно прервать и полностью прекратить прием препарата Алеценза, если не было обнаружено других потенциальных причин возникновения ИЗЛ/пневмонита. |
|
Повышение АЛТ или ACT до ≥ 3 степени (в >5 раз выше ВГН) с общим билирубином в ≤ 2 раза ВГН |
Временно прекратить прием до восстановления до начального уровня или ≤ 1 степени (в ≤ 3 раза выше ВГН), затем возобновить со сниженной дозой (см. таблицу 1). |
|
Повышение АЛТ или ACT до ≥ 2 степени (в > 3 раза выше ВГН) с увеличением общего билирубина в > 2 раза ВГН при отсутствии холестаза или гемолиза |
Полностью прекратить прием препарата Алеценза |
|
Брадикардияa 2 или 3 степени (симптоматическая, может быть тяжелая и медицински значимая, показано медицинское вмешательство) |
Временно прекратить прием до восстановления брадикардии (бессимптомной) до ≤ 1 степени или до частоты сердечных сокращений ≥ 60 уд./мин. Провести оценку сопутствующих лекарственных средств, которые способны вызывать брадикардию, а также противогипертонических лекарственных средств. Если сопутствующие лекарственные средства, оказывающие воздействие, установлены и их прием прекращен или их доза скорректирована, возобновить прием препарата Алеценза в предыдущей дозе до восстановления (бессимптомной) до брадикардии ≤ 1 степени или до частоты сердечных сокращений ≥ 60 уд./мин. Если сопутствующее лекарственное средство, оказывающее воздействие, не установлено, или если их прием не прекращен или не скорректирована их доза, возобновить прием препарата Алеценза в сниженной дозе (см. таблицу 1) до восстановления брадикардии (бессимптомной) до ≤ 1 степени или до частоты сердечных сокращений ≥ 60 уд./мин. |
|
Брадикардияa 4 степени (нежелательные явления, угрожающие жизни; показано неотложное медицинское вмешательство) |
Полностью прекратить прием, если не установлены сопутствующие лекарственные средства, оказывающие воздействие. Если сопутствующее лекарственное средство, оказывающее воздействие, установлено и его прием прекращен или его доза скорректирована, возобновить прием препарата Алеценза в сниженной дозе (см. таблицу 1) до восстановления брадикардии (бессимптомной) до ≤ 1 степени или до частоты сердечных сокращений ≥ 60 уд./мин с частым проведением контроля по клиническим показаниям. В случае повторного проявления полностью прекратить прием. |
| Повышение КФК в > 5 раз ВГН |
Временно прекратить прием до восстановления до начального уровня или до уровня в ≤ 2,5 раза ВГН, затем возобновить в прежней дозе. |
|
Повышение КФК в > 10 раз ВГН или повторное повышение КФК в > 5 раз ВГН |
Временно прекратить прием до восстановления до начального уровня или до уровня в ≤ 2,5 раза ВГН, затем возобновить в сниженной дозе в соответствии с таблицей 1. |
АЛТ = аланинаминотрансфераза; ACT = аспарагинаминотрансфераза; КФК = креатинфосфокиназа; СТСАЕ = Общие терминологические критерии оценки нежелательных явлений Национального института исследования рака; ИЗЛ = интерстициальное заболевание легких; ВГН = верхняя граница нормы
а Частота сердечных сокращений менее 60 ударов в минуту (уд./мин).
Особые группы
Печеночная недостаточность
Пациентам с легкой (Child-Pugh А) или умеренной (Child-Pugh В) печеночной недостаточностью, коррекция начальной дозы не требуется. Пациенты с тяжелой печеночной недостаточностью (Child- Pugh С) должны получать начальную дозу 450 мг, принимаемую два раза в день (общая доза 900 мг) (см. раздел «Фармакокинетические свойства»). Для всех пациентов с печеночной недостаточностью рекомендуется проводить соответствующий мониторинг (например, маркеры функции печени), см. раздел «Особые указания и меры предосторожности при применении».
Почечная недостаточность
Пациентам с почечной недостаточностью легкой или умеренной степени корректировать дозу не требуется. Применение препарата Алеценза не изучалось у пациентов с почечной недостаточностью тяжелой степени. Однако, поскольку алектиниб через почки выводится в незначительном количестве, то не требуется корректировать дозу у пациентов с почечной недостаточностью тяжелой степени (см. раздел «Фармакокинетические свойства»).
Пациенты пожилого возраста (≥ 65 лет)
Ограниченные данные по безопасности и эффективности применения препарата Алеценза у пациентов в возрасте 65 лет и старше не предполагают необходимости коррекции дозы у пациентов пожилого возраста (см. раздел «Фармакокинетические свойства»). Нет данных касательно пациентов в возрасте старше 80 лет.
Дети
Безопасность и эффективность применения препарата Алеценза у детей и подростков до 18 лет не установлена. Данные отсутствуют.
Чрезмерная масса тела (> 130 кг)
Несмотря на то, что фармакокинетическое моделирование для препарата Алеценза не указывает на снижение экспозиции у пациентов с чрезмерной массой тела (например, > 130 кг), алектиниб широко распределяется в организме, и в клинические исследования алектиниба включали пациентов с массой тела от 36,9 до 123 кг. Данные о пациентах с массой тела выше 130 кг отсутствуют.
Способ применения
Препарат Алеценза предназначен для приема внутрь. Твердые капсулы проглатываются целиком и не должны открываться или растворяться. Капсулы следует принимать во время приема пищи (см. раздел «Фармакокинетические свойства»).
Гиперчувствительность к алектинибу или к любым вспомогательным веществам, перечисленным в подразделе «Перечень вспомогательных веществ».
В клинических испытаниях препарата Алеценза сообщалось о случаях возникновения ИЗЛ/пневмонита (см. раздел «Нежелательные реакции»). Следует наблюдать за пациентами с целью выявления симптомов со стороны легких, указывающих на пневмонит. Прием препарата Алеценза должен быть незамедлительно прекращен у пациентов с диагнозом ИЗЛ/пневмонит и должен быть полностью отменен, если не было установлено других потенциальных причин развития ИЗЛ/пневмонита (см. раздел «Режим дозирования и способ применения»).
Гепатотоксичность
Повышение аланинаминотрансферазы (АЛТ) и аспарагинаминотрансферазы (ACT) свыше 5 раз выше ВГН, также повышение билирубина более чем в 3 раза выше ВГН возникало у пациентов в основных клинических исследованиях препарата Алеценза (см. раздел «Нежелательные реакции»). Большинство этих случаев возникло в течение первых 3 месяцев лечения. В основных клинических исследованиях лечения препаратом Алеценза было зарегистрировано, что у трех пациентов с повышением АЛТ/АСТ до степени 3-4 было лекарственное поражение печени. Сопутствующие повышения АЛТ или ACT в 3 или более раза выше ВГН и общего билирубина в 2 или более раза выше ВГН с нормальным показателем щелочной фосфатазы наблюдалось у одного пациента, получавшего лечение в клинических исследованиях препарата Алеценза.
Следует контролировать функцию печени, включая АЛТ, ACT, а также общий билирубин на момент включения в исследование и затем каждые две недели в течение первых 3 месяцев лечения. Впоследствии контроль следует проводить периодически, поскольку осложнения могут возникнуть позже, чем через три месяца, при этом у пациентов, у которых уровень аминотрансферазы и билирубина повысился, контроль следует проводить чаще. В зависимости от тяжести нежелательной лекарственной реакции следует приостановить прием препарата Алеценза и возобновить его в сниженной дозе или полностью прекратить его прием, как указано в таблице 2 (см. раздел «Режим дозирования и способ применения»).
Тяжелая миалгия и повышение уровня креатинфосфокиназы (КФК)
В основных исследованиях препарата Алеценза сообщалось о случаях возникновения миалгии или мышечно-скелетной боли у пациентов, включая явления 3 степени (см. раздел «Нежелательные реакции»).
В основных исследованиях препарата Алеценза были зарегистрированы случаи повышения уровня КФК, включая явления 3 степени (см. раздел «Нежелательные реакции»). В клинических испытаниях (NP28761, NP28673, ВO28984) медиана времени до повышения уровня КФК до 3 степени составляла 14 дней.
Пациентам следует рекомендовать сообщать о любой необъяснимой боли в мышцах, чувствительности или слабости. У пациентов, сообщающих о появлении симптомов, оценку уровня КФК следует проводить каждые две недели в течение первого месяца лечения и по клиническим показаниям. В зависимости от тяжести повышения КФК следует приостановить прием препарата Алеценза, затем возобновить его прием в сниженной дозе (см. раздел «Режим дозирования и способ применения»).
Брадикардия
При лечении препаратом Алеценза может возникнуть симптоматическая брадикардия (см. раздел «Нежелательные реакции»). В соответствии с клиническими показаниями следует проводить мониторинг частоты сердечных сокращений и артериального давления. В случае бессимптомной брадикардии коррекции дозы не требуется (см. раздел «Режим дозирования и способ применения»). Если у пациентов наблюдается симптоматическая брадикардия или угрожающие жизни явления, следует провести оценку сопутствующих лекарственных средств, вызывающих брадикардию, и противогипертонических лекарственных средств, а также скорректировать лечение препаратом Алеценза, как указано в таблице 2 (см. разделы «Режим дозирования и способ применения» и «Взаимодействие с другими лекарственными препаратами и другие формы взаимодействия» «Субстраты Р-гликопротеина» (P-gp substrates), и «Субстраты белка резистентности рака молочной железы» (BCRP substrates)).
Фоточувствительность
При приеме препарата Алеценза сообщалось о случаях фоточувствительности к солнечному свету (см. раздел «Нежелательные реакции»). Пациентам рекомендуют избегать длительного солнечного воздействия во время приема препарата Алеценза и, по крайней мере, в течение 7 дней после прекращения лечения. Для предотвращения солнечных ожогов пациентам также рекомендуют использовать солнцезащитные средства и против ультрафиолетового излучение типа A (UVA) / ультрафиолетового излучения типа В (UVB) и бальзам для губ (SPF ≥ 50).
Женщины детородного возраста
Прием препарата Алеценза во время беременности может оказать неблагоприятное воздействие на плод. Пациентки детородного возраста, получающие лечение препаратом Алеценза, должны использовать высокоэффективные методы контрацепции во время лечения и в течение не менее 3 месяцев после приема последней дозы препарата Алеценза (см. разделы «Фертильность, беременность и лактация» и «Данные доклинической безопасности»).
Непереносимость лактозы
Данное лекарственное средство содержит лактозу. Пациентам с редкими наследственными нарушениями переносимости лактозы, врожденной лактазной недостаточностью или нарушением всасывания глюкозы-галактозы не следует принимать данное лекарственное средство.
Содержание натрия
Этот лекарственный препарат содержит 48 мг натрия в суточной дозе (1200 мг), что эквивалентно 2,4% рекомендуемого ВОЗ максимального ежедневного потребления 2 г натрия для взрослых.
По данным in vitro, изофермент CYP3A4 является основным ферментом, посредством которого осуществляется метаболизм как алектиниба, так и его основного активного метаболита М4, при этом посредством CYP3A метаболизируется 40-50% препарата. М4 продемонстрировал схожую активность in vitro в отношении ALK.
Индукторы изофермента CYP3A
В результате приема внутрь рифампицина, мощного индуктора фермента CYP3A, многократно в дозе 600 мг один раз в сутки одновременно с приемом алектиниба внутрь однократно в дозе 600 мг Сmax и AUCinf алектиниба снижались соответственно на 51% и 73%, а Сmax и AUCinf метаболита М4 повышались соответственно в 2,20 и 1,79 раз. Влияние на комбинированную экспозицию алектиниба и М4 было незначительным со снижением Сmax и AUCinf соответственно на 4% и 18%. На основании изменений комбинированной экспозиции алектиниба и М4 корректировки дозы препарата не требуется, если Алеценза назначается одновременно с индукторами CYP3A. Рекомендуется проводить соответствующий контроль пациентов, одновременно принимающих мощные индукторы CYP3A (включая среди прочих карбамазепин, фенобарбитал, фенитоин, рифабутин, рифампицин и препараты зверобоя продырявленного (Hypericum perforatum)).
Ингибиторы изофермента CYP3A
В результате приема внутрь позаконазола, мощного ингибитора фермента CYP3A, многократно в дозе 400 мг два раза в сутки одновременно с приемом алектиниба внутрь однократно в дозе 300 мг экспозиция (Сmax и AUCinf) алектиниба повышалась соответственно в 1,18 и 1,75 раз, а Сmax и AUCinf метаболита М4 снижались соответственно на 71% и 25% раз. Влияние на комбинированную экспозицию алектиниба и М4 было незначительным со снижением Сmax на 7% и повышением AUCinf в 1,36 раза. На основании изменений комбинированной экспозиции алектиниба и М4 корректировки дозы препарата не требуется, если Алеценза назначается одновременно с ингибиторами CYP3A. Рекомендуется проводить соответствующий контроль пациентов, одновременно принимающих мощные ингибиторы CYP3A (включая среди прочих ритонавир, саквинавир, телитромицин, кетоконазол, итраконазол, вориконазол, позаконазол, нефазодон, грейпфруты и померанцы (Seville oranges).
Лекарственные средства, повышающие pH желудка
В результате приема внутрь эзомепразола, ингибитора протонного насоса, многократно в дозе 40 мг один раз в сутки не было отмечено клинически значимого влияния на комбинированную экспозицию алектиниба и метаболита М4. Следовательно, корректировать дозу не требуется, если препарат Алеценза назначается одновременно с ингибиторами протонного насоса или другими лекарственными средствами, повышающими pH желудочного сока (например, блокаторами Н2-гистаминовых рецепторов или антацидами).
Влияние транспортеров на фармакокинетические свойства алектиниба
Метаболит М4 является субстратом Р-гликопротеина. В связи с тем, что алектиниб ингибирует Р-гликопротеин, не ожидается, что одновременный прием препарата с ингибиторами Р-гликопротеина будет оказывать значительное влияние на экспозицию М4.
Влияние алектиниба на другие лекарственные средства
Субстраты Р-гликопротеина (Р-gр substrates)
В условиях in vitro алектиниб и его основной метаболит М4 являются ингибиторами эффлюксного белка-транспортера Р-гликопротеина (P-gp). Следовательно, алектиниб и М4, возможно, обладают потенциалом повышения концентрации в плазме одновременно принимаемых субстратов Р-гликопротеина. Рекомендуется проводить мониторинг пациентов при одновременном назначении с субстратами Р-гликопротеина (например, дигоксином, дабигатрана этексилатом, топотеканом, сиролимусом, эверолимусом, нилотинибом и лапатинибом).
Субстраты белка резистентности рака молочной железы (ВСRР (Breast Cancer Resistance Protein) substrates)
В условиях in vitro алектиниб и метаболит М4 являются ингибиторами эффлюксного транспортера белка резистентности рака молочной железы (BCRP). Следовательно, алектиниб и метаболит М4, возможно, обладают потенциалом повышения концентрации в плазме одновременно принимаемых субстратов белка резистентности рака молочной железы. При одновременном приеме препарата Алеценза с субстратами белка резистентности рака молочной железы (например, с метотрексатом, митоксантроном, топотеканом и лапатинибом), рекомендуется проводить соответствующий мониторинг пациентов.
Субстраты изоферментов CYP
В условиях in vitro алектиниб и метаболит М4 демонстрируют слабые зависимые от времени ингибирующие свойства в отношении фермента CYP3A4, при этом алектиниб в клинических концентрациях показывает наличие слабого потенциала индукции ферментов CYP3A4 и CYP2B6.
Прием алектиниба многократно в дозе 600 мг не оказывал влияния на экспозицию мидазолама (2 мг), чувствительного субстрата CYP3A. Следовательно, при одновременном приеме с субстратами CYP3A коррекции дозы не требуется.
Нельзя полностью исключить риск индукции CYP2B6 и ферментов, регулируемых прегнан-Х-рецептором (PXR – Pregnane X receptor). Эффективность одновременно принимаемых пероральных контрацептивов может быть снижена.
Женщинам детородного возраста следует рекомендовать избегать беременности в период терапии препаратом Алеценза. Пациентки детородного возраста, принимающие препарат Алеценза, должны использовать эффективные методы контрацепции в ходе лечения и в течение не менее 3 месяцев после приема последней дозы препарата Алеценза.
Беременность
Данные о применении препарата Алеценза беременными женщинами в период беременности отсутствуют, или имеются лишь ограниченные данные. Исходя из механизма действия препарата Алеценза, возможно, что при приеме беременными женщинами он может оказывать вредное воздействие на плод. Данные исследований, проведенных на животных, демонстрируют наличие репродуктивной токсичности (см. раздел «Данные доклинической безопасности»).
Пациентки, которые в ходе терапии препаратом Алеценза или в течение 3 месяцев после приема последней дозы препарата Алеценза беременеют, должны в обязательном порядке обратиться к врачу, который должен предупредить их о потенциальном вреде для плода.
Лактация
Неизвестно, выделяется ли алектиниб и его метаболиты с грудным молоком. Нельзя исключить риск для новорожденного/младенца. Матерям следует рекомендовать отказаться от грудного вскармливания в ходе терапии препаратом Алеценза.
Фертильность
Исследований фертильности на животных для оценки действия препарата Алеценза не проводилось. В исследованиях общей токсичности не наблюдалось нежелательных эффектов на мужские и женские репродуктивные органы (см. раздел «Данные доклинической безопасности»).
Препарат Алеценза оказывает незначительное влияние на способность управлять автотранспортом и использовать механизмы. Следует соблюдать осторожность при вождении автомобиля и использовании механизмов в связи с тем, что при приеме препарата Алеценза у пациентов может наблюдаться симптоматическая брадикардия (т. е. синкопе, головокружение, пониженное АД) или нарушение зрения (см. раздел «Нежелательные реакции»).
В приведенных ниже данных описываются результаты применения препарата Алеценза у 405 пациентов с распространенным ALK-положительным НМРЛ, принимавших участие в одном рандомизированном клиническом исследовании фазы III (ВО28984) и в двух клинических исследованиях фазы II с одной группой лечения (NP28761, NP28673). Данные пациенты получали препарат в рекомендуемой дозе 600 мг два раза в сутки. В клинических исследованиях фазы II (NP28761, NP28673; N=253) медиана продолжительности приема препарата Алеценза составила 11 месяцев. В исследовании ВО28984 (ALEX; N=152) медиана продолжительности приема препарата Алеценза составила 17,9 месяцев, в то время как медиана продолжительности приема кризотиниба составила 10,7 месяцев.
Наиболее частыми нежелательными явлениями (НЯ) (≥ 20%) были запор (35%), отек (30%, в том числе периферический отек, отек, генерализированный отек, отек век, периорбитальный отек, отек лица и локализованный отек) и миалгия (28%, в том числе миалгия и костно-мышечные боли).
Резюме в форме таблицы нежелательных реакций
В таблице 3 перечисляются НЯ, возникавшие у пациентов, получавших препарат Алеценза в ходе двух клинических исследований фазы II (NP28761, NP28673) и одного клинического исследования фазы III ВО28984 (ALEX), а также в течение пострегистрационного периода применения.
НЯ, перечисленные в таблице 3, представлены по системно-органным классам и категориям частоты возникновения, которые были определены по следующим критериям: очень часто (>1/10), часто (>1/100, но <1/10), нечасто (>1/1 000, но <1/100), редко (>1/10 000, но <1/1 000), очень редко (<1/10 000). В рамках каждого системно-органного класса нежелательные явления представлены в порядке убывания частоты.
Таблица 3 Нежелательные реакции, о которых сообщалось в ходе клинических исследований препарата Алеценза (NP28761, NP28673, ВO28984; N=405) и в течение пострегистрационного периода
|
Системно-органные классы Нежелательные реакции (словарь MedDRA) |
Алеценза N=405 |
||
|
Все степени (%) |
Категория частоты возникновения (все степени) |
Степени 3-4 (%) |
|
|
Нарушения со стороны крови и лимфатической системы |
|||
| Анемия1) | 17 | Очень часто | 3.0 |
|
Нарушения со стороны нервной системы |
|||
| Дисгевзия2) | 5.2 | Часто | 0.2 |
|
Нарушения со стороны органа зрения |
|||
| Нарушение зрения3) | 8.6 | Часто | 0 |
|
Нарушения со стороны сердца |
|||
| Брадикардия4) | 8.9 | Часто | 0 |
|
Нарушения со стороны дыхательной системы, органов грудной клетки и средостения |
|||
|
Интерстициальное заболевание легких / пневмонит |
0.7 | Нечасто | 0.2 |
|
Желудочно-кишечные нарушения |
|||
| Запор | 35 | Очень часто | 0 |
| Тошнота | 19 | Очень часто | 0.5 |
| Диарея | 16 | Очень часто | 0.7 |
| Рвота | 11 | Очень часто | 0.2 |
| Стоматит5) | 3.0 | Часто | 0 |
|
Нарушения со стороны печени и желчевыводящих путей |
|||
| Повышенный уровень билирубина6) | 18 | Очень часто | 3.2 |
| Повышенный уровень ACT | 15 | Очень часто | 3.7 |
| Повышенный уровень АЛТ | 14 | Очень часто | 3.7 |
|
Повышенный уровень щелочной фосфатазы** |
6.2 | Часто | 0.2 |
| Лекарственное поражение печени7) | 0.7 | Нечасто | 0.7 |
|
Нарушения со стороны кожи и подкожных тканей |
|||
Сыпь 
|
18 | Очень часто | 0.5 |
| Фоточувствительность | 9.1 | Часто | 0.2 |
|
Нарушения со стороны мышечной, скелетной и соединительной ткани |
|||
| Миалгия9) | 28 | Очень часто | 0.7 |
|
Повышенный уровень креатинфосфокиназы в крови |
10 | Очень часто | 3.2 |
|
Нарушения со стороны почек и мочевыводящих путей |
|||
|
Повышенный уровень креатинина в крови |
7.2 | Часто | 0.7* |
| Острое поражение почек | 1.0 | Часто | 1.0* |
| Общие нарушения и реакции в месте введения | |||
| Отек10) | 30 | Очень часто | 0.7 |
| Лабораторные и инструментальные данные | |||
| Увеличение массы тела | 12 | Очень часто | 0.7 |
* Включает в себя одно явление 5 степени
** Сообщалось о повышении уровня щелочной фосфатазы в пострегистрационный период и в ходе опорных клинических исследований фазы II и фазы III.
1) включает в себя случаи анемии и снижения гемоглобина
2) включает в себя случаи дисгевзии и гипогевзии
3) включает в себя случаи нечеткости зрения, нарушения зрения, плавающих помутнений стекловидного тела, снижения остроты зрения, астенопии и диплопии
4) включает в себя случаи брадикардии и синусовой брадикардии
5) включает в себя случаи стоматита и язвенного стоматита
6) включает в себя случаи повышения уровня билирубина, гипербилирубинемии и повышения связанного билирубина
7) включает в себя двух пациентов, у которых сообщалось MedDRA лекарственное поражение печени (термин по словарю MedDRA), а также у одного пациента сообщалось о повышении уровня ACT и АЛТ до 4 степени, у которого было зарегистрировано лекарственное поражение печени по результатам биопсии
включает в себя случаи сыпи, макулопапулезной сыпи, акнеформного дерматита, эритемы, генерализованной сыпи, папулезной сыпи, зудящей сыпи, макулезной сыпи и эксфолиативной сыпи
9) включает в себя случаи миалгии и мышечно-скелетной боли
10) включает в себя случаи периферического отека, отека, генерализованного отека, отека век, периорбитального отека, отека лица и локализованного отека.
Описание отдельных нежелательных лекарственных реакций
Профиль безопасности препарата Алеценза в целом оставался постоянным в основном клиническом исследовании фазы III ВO28984 (ALEX) и исследованиях фазы II (NP28761, NP28673).
Интерстициальное заболевание легких (ИЗЛ) / пневмонит
У пациентов, получавших лечение препаратом Алеценза, возникало тяжелое ИЗЛ/пневмонит. В клинических исследованиях (NP28761, NP28673, ВO28984) у 1 из 405 пациентов (0,2%), получавших лечение препаратом Алеценза, отмечалось ИЗЛ 3 степени. Данное явление послужило причиной прекращения лечения препаратом Алеценза. В клиническом исследовании III фазы ВO28984 3-4 степень ИЗЛ/пневмонита не наблюдалась у пациентов, получавших лечение препаратом Алеценза, по сравнению с 2,0% пациентов, получавших лечение кризотинибом. В клинических исследованиях случаев смертельного исхода в связи с ИЗЛ зарегистрировано не было. Следует проводить мониторинг пациентов на предмет появления симптомов со стороны легких, указывающих на пневмонит (см. разделы «Режим дозирования и способ применения» и «Особые указания и меры предосторожности при применении»).
Гепатотоксичность
В клинических исследованиях (NP28761, NP28673, ВO28984) у двух пациентов с повышением АСТ/АЛТ до 3-4 степени по результатам биопсии печени было подтверждено лекарственное поражение печени. Кроме того, у одного пациента было зарегистрировано лекарственное поражение печени 4 степени. В двух из этих случаев лечение препаратом Алеценза было прекращено. В клинических исследованиях (NP28761, NP28673, ВO28984) сообщалось о таких нежелательных реакциях, как повышение уровня ACT и АЛТ (15% и 14% соответственно) у пациентов, получавших лечение препаратом Алеценза. Большинство этих явлений были 1 и 2 степени интенсивности, а о явлениях со степенью ≥ 3 сообщалось соответственно у 3,7% и 3,7% пациентов. Явления, возникавшие в основном в течение первых 3 месяцев лечения, как правило, были преходящими и исчезали после временного прекращения приема препарата Алеценза (сообщалось соответственно для 1,5% и 3,0% пациентов) или снижении дозы (соответственно 2,2% и 1,2%). У 1,2% и 1,5% пациентов повышение уровня соответственно ACT и АЛТ приводило к прекращению лечения препаратом Алеценза. В клиническом исследовании III фазы ВO28984 повышение уровня АЛТ или ACT до 3 или 4 степени наблюдалось у 5% пациентов, получающих лечение препаратом Алеценза, по сравнению с 15% и 11% пациентов, получающих лечение кризотинибом.
В клинических исследованиях (NP28761, NP28673, ВO28984) сообщалось о повышении билирубина у 18% пациентов, получавших лечение препаратом Алеценза. Большинство этих явлений были 1 и 2 степени; явления 3 степени были зарегистрированы у 3,2% пациентов. Эти явления, возникавшие в основном в течение первых 3 месяцев лечения, обычно были преходящими, и большинство из них исчезало после изменения дозы. У 5,2% пациентов в связи с повышением уровня билирубина дозу изменяли, а у 1,5% пациентов в результате повышения уровня билирубина лечение препаратом Алеценза было прекращено. В клиническом исследовании III фазы ВO28984 повышение уровня билирубина 3 или 4 степени возникало у 3,3% пациентов, получавших лечение препаратом Алеценза, против пациентов, получавших лечение кризотинибом.
В клинических исследованиях сопутствующее повышение уровня АЛТ или ACT в 3 или более раза выше ВГН и общего билирубина в 2 или более раза выше ВГН с нормальным показателем щелочной фосфатазы возникло у одного пациента (0,2%), получавшего препарат Алеценза.
Следует наблюдать за функцией печени пациентов, включая АЛТ, ACT и общий билирубин, в соответствии с указаниями в разделе «Особые указания и меры предосторожности при применении», и при необходимости принимать соответствующие меры, рекомендуемые в разделе «Режим дозирования и способ применения».
Брадикардия
В клинических исследованиях (NP28761, NP28673, В028984) сообщалось о случаях брадикардии (8,9%) 1 и 2 степени у пациентов, принимавших препарат Алеценза. Ни у одного из пациентов не было зарегистрировано явлений ≥ 3 степени тяжести. У 66 из 365 пациентов (18%), принимавших препарат Алеценза, частота сердечных сокращений после приема дозы составляла менее 50 ударов в минуту (уд./мин). В клиническом исследовании III фазы ВO28984 частота сердечных сокращений после приема дозы составляла менее 50 уд./мин у 15% пациентов, получавших лечение препаратом Алеценза, и у 20% пациентов, получавших лечение кризотинибом. Лечение пациентов, у которых развиваются клинические симптомы брадикардии, следует проводить в соответствии с рекомендациями, изложенными в разделах «Режим дозирования и способ применения» и «Особые указания и меры предосторожности при применении». Случаев прекращения лечения препаратом Алеценза вследствие брадикардии зарегистрировано не было.
Тяжелая миалгия и повышение уровня КФК
В клинических исследованиях (NP28761, NP28673, ВO28984) сообщалось о случаях миалгии (28%), в том числе случаи миалгии (22%) и мышечно-скелетной боли (7,4%) у пациентов, получавших лечение препаратом Алеценза. Большинство явлений были 1 или 2 степени, а у трех пациентов (0,7%) была зарегистрирована 3 степень. Изменение дозы препарата Алеценза вследствие перечисленных нежелательных реакций потребовалось только двум пациентам (0,5%); лечение препаратом Алеценза не было прекращено вследствие данных явлений миалгии. В клинических исследованиях (NP28761, NP28673, ВO28984) препарата Алеценза повышение уровня КФК возникло у 43% из 362 пациентов с наличием лабораторных данных. Частота повышения уровня КФК 3 степени составляла 3,7%. В исследованиях (NP28761, NP28673, ВO28984) медиана времени повышения уровня КФК до 3 степени составляла 14 дней. Изменение дозы в связи с повышением уровня КФК потребовалось у 3,2% пациентов; прекращения лечения препаратом Алеценза вследствие повышения уровня КФК не требовалось. В клиническом исследовании ВO28984 не сообщалось о случаях тяжелой миалгии. Повышение уровня КФК до 3 степени было зарегистрировано у 2,6% пациентов, получавших лечение препаратом Алеценза, и у 1,3% пациентов, получавших лечение кризотинибом; в основном клиническом исследовании фазы III ВO28984 (ALEX) медиана времени повышения уровня КФК до 3 степени составляла соответственно 27,5 дней и 369 дней.
Воздействие на желудочно-кишечный тракт
Запор (35%), тошнота (19%), диарея (16%) и рвота (11%) являлись самыми частыми зарегистрированными реакциями со стороны желудочно-кишечного тракта (ЖКТ). Большинство этих явлений были легкой или умеренной степени тяжести; 3 степень тяжести была зарегистрирована у явлений диареи (0,7%), тошноты (0,5%) и рвоты (0,2%). Эти явления не требовали прекращения лечения препаратом Алеценза. Медиана времени до возникновения запора, тошноты, диареи и/или рвоты в клинических исследованиях (NP28761, NP28673, ВO28984) составляла 21 день. Частота возникновения явлений снизилась после первого месяца лечения. В клиническом исследовании фазы III ВO28984 у одного пациента (0,2%) была зарегистрирована тошнота 4 степени в группе приема препарата Алеценза, при этом в группе приема кризотиниба частота возникновения тошноты, рвоты и диареи 3 и 4 степени составляла соответственно 3,3%,3,3%, и 2,0%.
Сообщение о подозреваемых нежелательных реакциях
Важно сообщать о подозреваемых нежелательных реакциях после регистрации лекарственного препарата с целью обеспечения непрерывного мониторинга соотношения «польза — риск» лекарственного препарата. Медицинским работникам рекомендуется сообщать о любых подозреваемых нежелательных реакциях лекарственного препарата через национальную систему сообщения о нежелательных реакциях.
Пациенты с передозировкой должны находиться под тщательным наблюдением и общей поддерживающей терапией. Специальный антидот в случае передозировки препаратом Алеценза отсутствует.
Противоопухолевые средства, ингибитор протеинкиназы; код АТХ: L01XE36.
Фармакодинамические свойства
Механизм действия
Алектиниб является высокоселективным и мощным ингибитором ALK и RET тирозинкиназы. В доклинических исследованиях ингибирование активности тирозинкиназы ALK приводило к блокированию нисходящих сигнальных путей, в том числе STAT 3 и PI3K/AKT, а также индукции гибели опухолевых клеток (апоптоз).
Алектиниб продемонстрировал активность в условиях in vitro и in vivo в отношении мутантных форм фермента ALK, включая мутации, ответственные за резистентность к кризотинибу. Основной метаболит алектиниба (М4) продемонстрировал схожую активность в условиях in vitro.
По данным доклинических исследований, алектиниб не является субстратом Р-гликопротеина или белка резистентности рака молочной железы (BCRP), являющихся эффлюксными транспортерами через гематоэнцефалический барьер и, следовательно, способных поступать и распределяться в центральной нервной системе.
Клиническая эффективность и безопасность
ALK-положительный немелкоклеточный рак легких
Пациенты, ранее не получавшие лечения
Безопасность и эффективность препарата Алеценза изучалась в глобальном рандомизированном открытом клиническом исследовании фазы III (ВО28984, ALEX) с участием пациентов с ALK- положительным немелкоклеточным раком легких, ранее не получавших лечения. Перед рандомизацией в исследование требовалось проведение централизованного тестирования на определение положительного статуса экспрессии белка ALK. в образцах тканей, взятых у пациентов, методом иммуногистохимического (ИГХ) анализа Ventana anti-ALK (D5F3).
В исследование фазы III всего было включено 303 пациента, 151 из которых были рандомизированы в группу кризотиниба и 152 – в группу Алеценза, где пациенты получали препарат Алеценза перорально в рекомендуемой дозе по 600 мг два раза в сутки.
Стратификационными факторами при рандомизации являлись оценка по шкале ECOG PS (0/1 или 2), раса (азиатская или неазиатская) и наличие метастазов ЦНС на момент включения в исследование (да или нет). Первичной конечной точкой исследования являлось подтверждение большей эффективности препарата Алеценза по сравнению с кризотинибом по результатам выживаемости без прогрессирования заболевания (PFS) по оценке исследователя при помощи Критериев оценки ответа при солидных опухолях (RECIST 1.1). Исходные демографические характеристики и характеристики заболевания в группе приема препарата Алеценза: медиана возраста 58 лет (54 года в группе кризотиниба), 55% пациентов были женского пола (58% в группе кризотиниба), 55% пациентов неазиатской расы (54% в группе кризотиниба), 61% некурящие (65% в группе кризотиниба), у 93% пациентов оценка по шкале ECOG PS была от 0 до 1 (93% в группе кризотиниба), 97% пациентов имели IV стадию заболевания (96% в группе кризотиниба), у 90% пациентов была гистологически подтвержденная аденокарцинома (94% в группе кризотиниба), у 40% были метастазы в ЦНС на момент включения в исследование (38% в группе кризотиниба), у 17% ранее получали облучению ЦНС (14% в группе кризотиниба).
По результатам первичного анализа исследование достигло первичной конечной точки со статистически достоверным улучшением по выживаемости без прогрессирования заболевания по оценке исследователя. Данные по эффективности обобщены в таблице 4; на рисунке 1 представлена кривая Каплана-Мейера, демонстрирующая результаты по выживаемости без прогрессирования заболевания по оценке исследователя.
Таблица 4 Обзор результатов по эффективности из исследования ВО28984 (ALEX)
|
Кризотиниб N=151 |
Алеценза N=152 |
|
|
Медиана продолжительности периода наблюдения (месяцы) |
17.6 (диапазон 0.3-27.0) |
18.6 (диапазон 0.5-29.0) |
|
Первичный показатель эффективности |
||
|
Выживаемость без прогрессирования заболевания, PFS (иссл.) |
||
|
Количество пациентов с явлениями, n (%) Медиана (месяцы) [95% ДИ] |
102 (68%) 11.1 [9.1; 13.1] |
62 (41%) н.м.б.о. [17.7; н.м.б.о.] |
|
Отношение рисков [95% ДИ] Стратифицированное логранговое р-значение |
0.47 [0.34; 0.65] р <0. 0001 |
|
|
Вторичные показатели эффективности |
||
|
Выживаемость без прогрессирования заболевания, PFS (ННК) |
||
|
Количество пациентов с явлениями, n (%) Медиана (месяцы) [95% ДИ] |
92 (61%) 10.4 [7.7; 14.6] |
63 (41%) 25.7 [19.9; н.м.б.о.] |
|
Отношение рисков (HR) [95% ДИ] Стратифицированное логранговое р-значение |
0.50 [0.36; 0.70] р <0.0001 |
|
|
Время до прогрессирования в ЦНС (ННК)*, ** |
||
|
Количество пациентов с явлениями, n (%) |
68 (45%) | 18 (12%) |
|
Отношение рисков (HR) в зависимости от причины [95% ДИ] Стратифицированное логранговое р-значение |
0.16 [0.10; 0.28] p<0/0001 |
|
|
Совокупная частота случаев прогрессирования в ЦНС за 12 месяцев (ННК) [95% ДИ] |
41.4% [33.2; 49.4] |
9.4% [5.4; 14.7] |
| Частота объективного ответа (ORR) (иссл.)*, *** | ||
|
Количество пациентов, достигших ответа, n (%) [95% ДИ] |
114 (75.5%) [67.8; 82.1] |
126 (82.9%) [76.0; 88.5] |
| Общая выживаемость* | ||
|
Количество пациентов с явлениями, n (%) Медиана (месяцы) [95% ДИ] |
40 (27%) н.м.б.о. [н.м.б.о.] |
35 (23%) н.м.б.о. [н.м.б.о.] |
| Отношение рисков (HR) [95% ДИ] | 0.76 [0.48; 1.20] |
|
| Продолжительность ответа (иссл.) | N=114 | N=126 |
|
Медиана (месяцы) [95% ДИ] |
11.1 [7.9; 13.0] |
н.м.б.о. [н.м.б.о.] |
|
Частота объективного ответа (CNS-ORR) у пациентов с измеримыми метастазами в ЦНС на момент включения в исследование |
N=22 | N=21 |
|
Количество пациентов, достигших ответа, n (%) [95% ДИ] |
11 (50.0%) [28.2; 71.8] |
17 (81.0%) [58.1; 94.6] |
|
Полный ответ (CNS-CR), n (%) |
1 (5%) | 8 (38%) |
| Продолжительность ответа (CNS-DOR), медиана (месяцы) | 5.5 | 17.3 |
| [95% ДИ] | [2.1; 17.3] | [14.8; н.м.б.о.] |
|
Частота объективного ответа (CNS-ORR) у пациентов с измеримыми и неизмеримыми метастазами в ЦНС на момент включения в исследование (ННК) |
N=58 | N=64 |
|
Количество пациентов, достигших ответа, n (%) [95% ДИ] |
15 (25.9%) [15.3; 39.0] |
38 (59.4%) [46.4; 71.5] |
|
Полный ответ (CNS-CR), n (%) |
5 (9%) | н.м.б.о. |
| Продолжительность ответа (CNS-DOR), медиана (месяцы) | 3.7 | н.м.б.о. |
| [95% ДИ] | [3.2; 6.8] | [17.3; н.м.б.о.] |
* Ключевые вторичные конечные точки в рамках многоуровневого анализа
** Анализ конкурирующих рисков прогрессирования в ЦНС, системного прогрессирования и смерти в качестве конкурирующих явлений
*** 2 пациента в группе кризотиниба и 6 пациентов в группе приема алектиниба достигли полного ответа
ДИ = доверительный интервал; ЦНС = центральная нервная система; CR = полный ответ; DOR = продолжительность ответа; HR = отношение рисков; ННК = независимый наблюдательный комитет; иссл. = исследователь; н.м.б.о. = не может быть оценено; ORR = частота объективного ответа; PFS = выживаемость без прогрессирования
У пациентов, имеющих метастазы в ЦНС на момент включения в исследование (отношение рисков HR = 0,40, 95% ДИ: 0,25-0,64; медиана выживания без прогрессирования заболевания в группе Алеценза = н.м.б.о., 95% ДИ: 9,2-н.м.б.о., медиана выживания без прогрессирования заболевания в группе кризотиниба = 7,4 месяца, 95% ДИ: 6,6-9,6) и не имеющих метастазы в ЦНС на момент включения в исследование (отношение рисков HR = 0,51, 95% ДИ: 0,33-0,80, медиана выживания без прогрессирования заболевания в группе Алеценза = н.м.б.о., 95% ДИ: н.м.б.о.-н.м.б.о., медиана выживания без прогрессирования заболевания в группе кризотиниба = 14,8 месяцев, 95% ДИ: 10,8- 20,3) показатель выживаемости без прогрессирования заболевания был выше, что указывает на более высокую эффективность препарата Алеценза по сравнению с кризотинибом в обеих подгруппах.
Рисунок 1: Кривая Каплана-Мейера для выживаемости без прогрессирования заболевания по оценке исследователя в исследовании ВО28984 (ALEX)
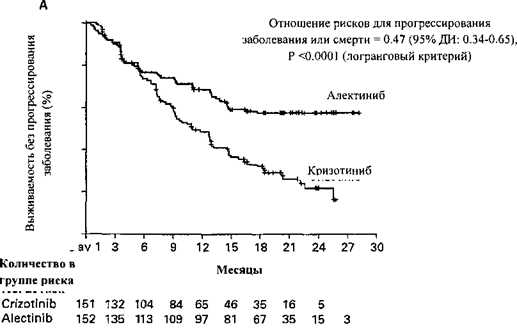
Пациенты, получавшие лечение кризотинибом
Безопасность и эффективность препарата Алеценза у пациентов с ALK-положительным НМРЛ, ранее получавших кризотиниб, исследовались в двух клинических исследованиях фазы I/II (NP28673 и NP28761).
NP28673
Исследование NP28673 являлось многоцентровым исследованием фазы I/II с одной группой лечения, проводившееся с участием пациентов с прогрессирующим ALK-положительным НМРЛ, у которых заболевание прогрессировало в ходе лечения кризотинибом. Помимо кризотиниба, пациенты могли проходить химиотерапию. Всего 138 пациентов были включены в фазу II исследования, в ходе которой они получали препарат Алеценза внутрь в рекомендуемой дозе 600 мг два раза в сутки.
Первичной конечной точкой являлась оценка эффективности препарата Алеценза на основании частоты объективного ответа (ORR) согласно централизованной оценке независимого наблюдательного комитета при помощи Критериев оценки ответа при солидных опухолях (RECIST 1.1) у пациентов, ранее проходивших цитотоксическую химиотерапию. При снижении доверительного интервала для оцененной частоты объективного ответа выше предварительно установленного предела 35% достигается статистически достоверный результат.
Демографические характеристики пациентов соответствовали популяции пациентов с ALK- положительным НМРЛ. Общие демографические характеристики в исследовании были следующими: 67% – европеоидная раса, 26% – монголоидная раса, 56% – женщины, медиана возраста – 52 года. Большинство пациентов ранее не курили (70%). На момент включения в исследование статус по шкале ECOG (Восточная объединенная группа онкологов) от 0 до 1 был зарегистрирован у 90,6% пациентов, а 2 – у 9,4% пациентов. На момент включения в исследование 99% пациентов имели IV стадию заболевания, у 61% были метастазы в головной мозг, а у 96% пациентов опухоли были классифицированы как аденокарцинома. Среди пациентов, включенных в исследование, у 20% наблюдалось значительное прогрессирование в ходе терапии кризотинибом, а у 80% заболевание ранее прогрессировало в ходе лечения кризотинибом и не менее одного курса химиотерапии.
NP28671
Исследование NP28671 являлось многоцентровым исследованием фазы I/II с одной группой, проводившимся с участием пациентов с ALK-положительным НМРЛ, у которых заболевание ранее прогрессировало в ходе лечения кризотинибом. Помимо кризотиниба, пациенты могли проходить химиотерапию. Всего 87 пациентов были включены в фазу II исследования, в ходе которой они получали препарат Алеценза внутрь в рекомендуемой дозе 600 мг два раза в сутки.
Первичной конечной точкой являлась оценка эффективности препарата Алеценза на основании частоты объективного ответа (ORR) согласно централизованной оценке независимого наблюдательного комитета при помощи Критериев оценки ответа при солидных опухолях (RECIST 1.1). При снижении доверительного интервала для оцененной частоты объективного ответа выше предварительно установленного предела 35% достигается статистически достоверный результат.
Демографические характеристики пациентов соответствовали популяции пациентов с ALK-положительным НМРЛ. Общие демографические характеристики в исследовании были следующими: 84% – европеоидная раса, 8% – монголоидная раса, 55% – женщины. Медиана возраста составила 54 года. Большинство пациентов ранее не курили (62%). На момент включения в исследование статус по шкале ECOG (Восточная объединенная группа онкологов) от 0 до 1 был зарегистрирован у 89,7% пациентов, а 2 – у 10,3% пациентов. На момент включения в исследование 99% пациентов имели IV стадию заболевания, у 60% были метастазы в головной мозг, а у 94% пациентов опухоли были классифицированы как аденокарцинома. Среди пациентов, включенных в исследование, у 26% наблюдалось значительное прогрессирование в ходе терапии кризотинибом, а у 74% заболевание ранее прогрессировало в ходе лечения кризотинибом и не менее одного курса химиотерапии.
Основные результаты по эффективности из исследований NP28673 и NP28671 обобщены в таблице 5. Обзор анализа объединенных данных по конечным точкам ЦНС представлен в таблице 6.
Таблица 5 Результаты по эффективности из исследований NP28673 и NP28671
|
NP28673 Алеценза 600 мг два раза в сутки |
NP28671 Алеценза 600 мг два раза в сутки |
|
|
Медиана продолжительности периода наблюдения (месяцы) |
21 (диапазон 1-30) |
17 (диапазон 1-29) |
|
Первичный показатель эффективности |
||
| Частота объективного ответа (ORR) в популяции RE | N=122а | N=67b |
|
Количество пациентов, N (%) [95% ДИ] |
62 (50.8%) [41.6%, 60.0%] |
35 (52.2%) [39.7%, 64.6%] |
|
Частота объективного ответа (ORR) у пациентов, ранее проходивших химиотерапию |
N=96 | |
|
Количество пациентов, N (%) [95% ДИ] |
43 (44.8%) [34.6%, 55.3%] |
|
|
Вторичные показатели эффективности |
||
| Продолжительность ответа (DOR) (ННК) | N=62 | N=35 |
|
Количество пациентов с явлениями, N (%) |
36 (58.1%) | 20 (57.1%) |
| Медиана (месяцы) | 15.2 | 14.9 |
| [95% ДИ] | [11.2, 24.9] | [6.9, н.м.б.о.] |
|
Выживаемость без прогрессирования заболевания (PFS) (ННК) |
N=138 | N=87 |
|
Количество пациентов с явлениями, N (%) |
98 (71.0%) | 58 (66.7%) |
| Медиана (месяцы) | 8.9 | 8.2 |
| [95% ДИ] | [5.6, 12.8] | [6.3, 12.6] |
ДИ = доверительный интервал; DOR = продолжительность ответа; ННК = независимый наблюдательный комитет; н.м.б.о. = не может быть оценено; ORR = частота объективного ответа; PFS = выживаемость без прогрессирования заболевания; RE = с ответом, который может быть оценен
а У 16 пациентов болезнь на момент включения в исследование была неизмерима согласно ННК, поэтому не была включена популяцию с ответом, который может быть оценен.
b У 20 пациентов болезнь на момент включения в исследование была неизмерима согласно ННК, поэтому не была включена популяцию с ответом, который может быть оценен.
В исследованиях NP28673 и NP28671 результаты по частоте объективного ответа были однородными по всем подгруппам исходных характеристик пациентов, таких как возраст, пол, раса, статус по шкале ECOG, наличие метастазов в ЦНС и прохождение химиотерапии, особенно если учитывать небольшое число пациентов в некоторых подгруппах.
Таблица 6. Обзор анализа объединенных данных по конечным точкам ЦНС из исследований NP28673 и NP28671
|
Показатели ЦНС (NP28673 и NP28671) |
Алеценза 600 мг два раза в сутки |
|
Пациенты с измеримыми поражениями ЦНС на момент включения в исследование |
N=50 |
|
Частота объективного ответа для ЦНС (CNS ORR) (ННК) |
|
|
Количество пациентов, достигших ответа (%) |
32 (64.0%) |
| [95% ДИ] | [49.2%, 77.1%] |
| Полный ответ | 11 (22.0%) |
| Частичный ответ | 21 (42.0%) |
|
Продолжительность ответа для ЦНС (CNS DOR) (ННК) |
N=32 |
|
Количество пациентов с явлениями (%) |
18(56.3%) |
| Медиана (месяцы) | 11.1 |
| [95% ДИ] | [7.6, н.м.б.о.] |
ДИ = доверительный интервал; DOR = продолжительность ответа; ННК = независимый наблюдательный комитет; ORR — частота объективного ответа; NE = не может быть оценено
Дети
Европейское агентство по лекарственным средствам отказалось от обязательства по предоставлению результатов исследований препарата Алеценза по всем подгруппам детской популяции пациентов с карциномой легких (мелкоклеточным и немелкоклеточным раком легких) (сведения о применении у детей см. в разделе «Режим дозирования и способ применения»).
Фармакокинетические свойства
Была дана характеристика фармакокинетических показателей алектиниба и его основного метаболита (М4) у пациентов с ALK-положительным НМРЛ и у здоровых участников. По результатам популяционного фармакокинетического анализа, среднее геометрическое значение (коэффициент вариации, %) равновесных Сmax, Сmin и AUC0-12ч алектиниба составляло соответственно приблизительно 665 нг/мл (44,3%), 572 нг/мл (47,8%) и 7430 нг*ч/мл (45,7%). среднее геометрическое значение (коэффициент вариации, %) равновесных Сmax, Сmin и AUC0-12ч метаболита М4 составляло соответственно приблизительно 246 нг/мл (45,4%), 222 нг/мл (46,6%) и 2810 нг*ч/мл (45,9%).
Абсорбция
После приема внутрь в дозе 600 мг два раза в сутки после еды у пациентов с ALK-положительным НМРЛ алектиниб всасывался с достижением Тmax через приблизительно 4-6 часов.
Равновесная концентрация алектиниба достигалась через 7 дней при непрерывном приеме в дозе 600 мг два раза в сутки. Коэффициент накопления при приеме в дозе 600 мг два раза в сутки составлял приблизительно 6. Популяционный фармакокинетический анализ подтверждает пропорциональность доз для всего диапазона доз от 300 до 900 мг при приеме после еды.
Абсолютная биодоступность алектиниба в капсулах у здоровых участников составляла 36,9% (90% ДИ: 33,9%, 40,3%) при приеме после еды.
После однократного приема внутрь в дозе 600 мг вместе с жирной высококалорийной пищей экспозиция алектиниба и метаболита М4 повышалась приблизительно в 3 по сравнению с приемом натощак (см. раздел «Режим дозирования и способ применения»).
Распределение
Алектиниб и его основной метаболит М4 активно связываются с белками плазмы человека (>99%) независимо от концентрации действующего вещества. Среднее соотношение концентраций алектиниба и М4 в крови и плазме in vitro соответственно составило 2,64 и 2,50 при приеме в клинически значимых дозах.
Среднее геометрическое значение объема распределения в равновесном состоянии (Vss) алектиниб после внутривенного введения составило 475 л, что свидетельствует о масштабном распределении в ткани.
По данным in vitro, алектиниб не является субстратом Р-гликопротеина. Алектиниб и метаболит М4 не являются субстратами белка резистентности рака молочной железы (BCRP) и полипептида транспортера органических анионов (ОАТР) 1В1/ВЗ.
Биотрансформация
Исследования метаболизма, проведенные в условиях in vitro, продемонстрировали, что CYP3A4 является основным ферментом, опосредующий метаболизм алектиниба и его основного метаболита М4. По оценкам, 40-50% метаболизма алектиниба приходится на долю данного фермента. По результатам исследования баланса масс у человека было продемонстрировано, что алектиниб и М4 являются основными компонентами, циркулирующими в плазме, и составляют 76% от всей радиоактивной метки в плазме. Среднее геометрическое значение соотношения метаболит/материнское соединение в равновесном состоянии составляет 0,399.
В условиях in vitro и в плазме крови здоровых участников был обнаружен второстепенный метаболит М1b. Вероятно, образование метаболита М1b катализируется комбинацией изоферментов CYP (включая другие изоферменты, помимо CYP3A) и ферментами альдегиддегидрогеназы (ALDH).
Исследования in vitro указывают на то, что ни алектиниб, ни его основной метаболит (М4) не ингибируют CYP1A2, CYP2B6, CYP2C9, CYP2C19 и CYP2D6 при клинически значимых концентрациях. Алектиниб не ингибирует ОАТР1В1/ОАТР1ВЗ, ОАТ1, ОАТЗ и ОСТ2 при клинически значимых концентрациях in vitro.
Элиминация
После однократного приема внутрь алектиниба, меченого 14С, у здоровых участников основная часть меченого 14С выводилась с калом (средний коэффициент извлечения 97,8%) с минимальной долей, выведенной с мочой (средний коэффициент извлечения 0,46%). С калом 84% и 5,8% дозы выводилось соответственно в виде неизмененного алектиниба или метаболита М4.
По результатам популяционного фармакокинетического анализа, кажущийся клиренс алектиниба (CL/F) составил 81,9 л/ч. Среднее геометрическое отдельных значений полувыведения алектиниба составило 32,5 ч. Соответствующие значения для метаболита М4 составили соответственно 217 л/ч и 30,7 ч.
Фармакокинетические характеристики у отдельных категорий пациентов
Почечная недостаточность
Незначительное количество алектиниба и активного метаболита М4 выводятся в неизмененном виде с мочой (<0,2% от дозы). По результатам популяционного фармакокинетического анализа, экспозиция алектиниба и метаболита М4 была схожей у пациентов с легкой и умеренной степенью почечной недостаточности и нормальной функцией почек. Фармакокинетические свойства алектиниба не изучались у пациентов с тяжелой почечной недостаточностью.
Печеночная недостаточность
Элиминация алектиниба происходит главным образом через печень, в связи с чем печеночная недостаточность может повысить концентрацию алектиниба и/или его основного метаболита в плазме. По результатам популяционного фармакокинетического анализа, экспозиция алектиниба и М4 были схожими у пациентов с умеренной печеночной недостаточностью и нормальной функцией печени.
Фармакокинетические свойства алектиниба не изучались у пациентов с тяжелой печеночной недостаточностью.
Влияние возраста, массы тела, расы и пола
Возраст, масса тела, раса и пол не оказывали клинически значимого влияния на системное воздействие алектиниба и метаболита М4. В клинические исследования были включены пациенты с массой тела в диапазоне от 36,9 до 123 кг. Данные о пациентах с крайне высокой массой тела (более 130 кг) отсутствуют (см. раздел «Режим дозирования и способ применения»).
Данные доклинической безопасности
Канцерогенность
Исследований канцерогенности по установлению канцерогенного потенциала препарата Алеценза не проводилось.
Мутагенность
Алектиниб не проявлял мутагенной активности in vitro в ходе испытания на обратные мутации у бактерий (тест Эймса), однако, он вызывал незначительное увеличение численных аберраций в цитогенетическом анализе in vitro, проводившемся на клетках легких китайских хомячков с метаболической активацией, а также в микроядрах спинного мозга крыс в микроядерном тесте. Механизм индукции микроядер представлял собой патологическую сегрегацию хромосом (анеугенность), но не кластогенное действие на хромосомы.
Снижение репродуктивной функции
Исследований по оценке эффекта препарата Алеценза на репродуктивную функцию на животных не проводились. В общих токсикологических исследованиях нежелательных эффектов на репродуктивные органы самцов и самок и не наблюдалось. Данные исследования проводились на крысах и обезьянах при экспозиции, соответственно в 2,6 и 0,5 или более раз превышающих экспозицию у человека, измерявшуюся в AUC, при применении в рекомендуемой дозе 600 мг два раза в сутки.
Тератогенность
Алектиниб вызывал токсическое воздействие на эмбрион и плод у беременных самок крыс и кроликов. У беременных крыс алектиниб вызывал потерю плода (выкидыш) при экспозиции, в 4,5 раз превышающей экспозицию у человека (на основании AUC), а также небольшой размер плода с задержкой формирования костей и незначительными патологиями органов при экспозиции, в 2,7 раз превышающей экспозицию у человека (на основании AUC). У беременных самок кроликов алектиниб вызывал потерю эмбриона или плода, небольшой размер плода и повышал частоту скелетных изменений при экспозиции, в 2,9 раз превышающей экспозицию у человека (на основании AUC) при приеме в рекомендуемой дозе.
Прочее
Алектиниб поглощает УФ-свет в диапазоне от 200 до 400 нм и демонстрировал наличие фототоксического потенциала в анализе фотобезопасности, проводившемся in vitro на культивированных фибробластах мыши после УФА-облучения.
В исследовании токсичности повторных доз целевыми органами, как у крыс, так и у обезьян при клинически значимой экспозиции были, среди прочего, эритроидная система, желудочно-кишечный тракт и гепатобилиарная система.
Патологическая морфология эритроцитов наблюдалась при экспозиции, в 10-60% превышающей экспозицию у человека (на основании AUC) при применении в рекомендуемых дозах. При экспозиции, в 20-120% превышающей экспозицию у человека (на основании AUC) при применении в рекомендуемых дозах, у обоих видов наблюдалось расширение пролиферативной зоны в слизистой ЖКТ. У крыс и/или обезьян при экспозиции, в 20-30% превышающей экспозицию у человека (на основании AUC) при применении в рекомендуемых дозах, повышался уровень печеночной щелочной фосфатазы (ЩФ) и прямого билирубина, а также наблюдалась вакуолизация/дегенерация/некроз эпителия желчных протоков и увеличение/очаговый некроз гепатоцитов.
У обезьян при клинически значимой экспозиции наблюдался слабый гипотензивный эффект.
Перечень вспомогательных веществ
Содержание капсулы
Лактоза моногидрат
Гидроксипропилцеллюлоза
Натрия лаурилсульфат
Магния стеарат
Карбоксиметилцеллюлоза кальция
Оболочка капсулы
Гипромеллоза
Каррагинан
Калия хлорид
Титана диоксид (Е171)
Кукурузный крахмал
Карнаубский воск
Чернила для печати
Железа оксид красный (Е172)
Железа оксид желтый (Е172)
Алюминиевый лак на основе индигокармина (Е132)
Карнаубский воск
Белый шеллак
Глицерил моноолеат
Совместимость
Неприменимо.
Хранить при температуре не выше 30 °C в оригинальной упаковке для защиты от света и влаги. Хранить в недоступном для детей месте.
По 8 капсул в контурную ячейковую упаковку (блистер) из композиционного материала (полиамид/алюминий/ поливинилхлорид) и фольги алюминиевой композиционной (алюминий/сополимер винил-хлорида/ винилацетата полибутилметакрилат).
7 контурных ячейковых упаковок (блистеров) вместе с инструкцией по применению помещают в картонную пачку (промежуточная упаковка), на которую с целью контроля первого вскрытия наклеивают голограммы.
4 промежуточных упаковки помещают в картонную пачку (потребительская упаковка) с контролем первого вскрытия (в виде перфорации).
Особые меры предосторожности при уничтожении использованного лекарственного препарата или отходов, полученных после применения лекарственного препарата или работы с ним
Неиспользованное лекарственное средство или отходы утилизируют в соответствии с местными требованиями.
Владелец регистрационного удостоверения
Ф. Хоффманн-Ля Рош Лтд., Швейцария
F. Hoffmann-La Roche Ltd, Grenzacherstrasse 124, 4070 Basel, Switzerland
Производитель
Экселла ГмбХ и Ко. КГ, Германия
Excella GmbH & Со. KG, Nurnberger Strasse 12, 90537 Feucht, Germany
В случае поставок и реализации на территории Республики Беларусь претензии потребителей направлять на адрес ИООО «Рош Продактс Лимитед»:
220073, г. Минск, 1-й Загородный пер., 20, 8-й этаж, кабинет 20.
Тел. (017) 256 23 08; факс (017) 256 23 06.
Email: belarus.safety@roche.com
Алектиниб
Alectinib
Фармакологическое действие
Алектиниб является ингибитором тирозинкиназы, таргетированной к ALK и RET. В неклинических исследованиях алектиниб ингибировал ALK-фосфорилирование и ALK-опосредованную активацию сигнальных белков STAT3 и AKT и уменьшал выживаемость опухолевых клеток в множественных клеточных линиях, содержащих слияния ALK, амплификации или активирующие мутации. Основной активный метаболит алектиниба М4 показал сходную активность in vitro.
Фармакодинамика
У пациентов с распространённым ALK-позитивным немелкоклеточным раком лёгкого средняя геометрическая (коэффициент вариации%) установившаяся максимальная концентрация (Cmax,ss) для алектиниба составляла 665 н /мл (44 %), а для М4 — 246 нг/мл (45 %). Средняя геометрическая стационарная площадь под кривой AUC0–12 h,ss для алектиниба составила 7 430 нг × ч/мл (46 %), а для М4 — 2 810 нг × ч/мл (46 %). Воздействие алектинибом пропорционально дозе в диапазоне доз от 460 до 900 мг (то есть в 0,75–1,5 раза выше рекомендованной рекомендуемой дозы) в условиях приёма пищи. Алектиниб и M4 достигли стабильных концентраций к седьмому дню.
Фармакокинетика
Абсорбция
Алектиниб достигал максимальных концентраций через 4 часа после приёма 600 мг два раза в день.
Абсолютная биодоступность алектиниба была 37 % (90 % ДИ: 34 %, 40 %) в условиях приёма пищи.
Жирная высококалорийная пища увеличивала комбинированное воздействие (AUC0–inf) алектиниба и M4 в 3,1 раза (90 % CI: 2,7, 3,6) после перорального введения одной дозы 600 мг алектиниба.
Распределение
Кажущийся объём распределения (Vd) составляет 4 016 л для алектиниба и 10 093 л для М4.
Алектиниб и M4 связываются с белками плазмы более 99 %, независимо от концентрации лекарственного средства.
Концентрации алектиниба в спинномозговой жидкости у пациентов с ALK-позитивным немелкоклеточным раком лёгкого приблизительно на уровне свободной концентрации алектиниба в плазме крови.
Исследования in vitro показывают, что алектиниба не является субстратом P-гликопротеина (P-gp), но M4 является субстратом P-gp. Алектиниб и M4 не являются субстратами белка устойчивости к раку молочной железы (BCRP), органического анионтранспортирующего полипептида OATP1B1 или OATP1B3.
Метаболизм
Алектиниб метаболизируется CYP3A4 до его основного активного метаболита M4. Среднее геометрическое соотношение M4/алектиниб при стабильной концентрации составляет 0,40. M4 затем метаболизируется CYP3A4. Алектиниб и M4 были основными циркулирующими компонентами в плазме, составляя 76 % от общей радиоактивности.
Выведение
Клиренс (CL/F) составляет 81,9 л/час для алектиниба и 217 л/час для M4. Среднее геометрическое время полувыведения составляет 33 часа для алектиниба и 31 час для M4 у пациентов с ALK-позитивным немелкоклеточным раком лёгкого.
98 % выводится с фекалиями. 84 % дозы выводится с фекалиями в виде неизменённого алектиниба, 6 % — М4. Выделение с мочой составляло менее 0,5 % введённой меченной дозы алектиниба.
Особые группы популяции
Возраст, масса тела, печёночная недостаточность лёгкой степени, слабая или умеренная почечная недостаточность (клиренс креатинина 30–89 мл/мин), раса и пол не имели клинически значимого эффекта при системном воздействии алектиниба и M4. Фармакокинетика лёгкая не изучалась у пациентов с тяжёлой почечной недостаточностью, терминальной стадией почечной недостаточности, умеренной или тяжёлой печёночной недостаточностью.
Показания
Алектиниб показан для лечения пациентов с распространённым анапластическим ALK-позитивным немелкоклеточным раком лёгкого при неэффективности или непереносимости кризотиниба.
Беременность и грудное вскармливание
Категория действия на плод по FDA — N.
Основываясь на исследованиях на животных и механизме действия алектиниба, препарат противопоказан при беременности.
Женщины репродуктивного возраста должны использовать эффективные методы контрацепции во время лечения и по крайней мере 1 неделю после последней дозы алектиниба.
Грудное вскармливание противопоказано во время лечения и по крайней мере 1 неделю после последней дозы алектиниба.
Способ применения и дозы
Рекомендуемая доза алектиниба составляет 600 мг перорально два раза в день во время приёма пищи.
Побочные действия
Серьёзные побочные реакции алектиниба наблюдались у 19 % пациентов, наиболее частыми серьёзными побочными реакциями были лёгочная эмболия (1,2 %), одышка (1,2 %) и гипербилирубинемия (1,2 %).
Фатальные побочные реакции наблюдались у 2,8 % пациентов и включали кровотечения (0,8 %), перфорацию кишечника (0,4 %), одышку (0,4 %), лёгочную эмболию (0,4 %) и эндокардит (0,4 %).
Прекращение приёма алектиниба из-за побочных реакций произошло у 6 % пациентов. Наиболее частыми побочными реакциями, которые привели к окончательному прекращению приёма, были гипербилирубинемия (1,6 %), повышение уровня АЛТ (1,6 %) и повышение уровня АСТ (1,2 %). В целом, 23 % пациентов, начавших лечение в рекомендуемой дозе, требовали по крайней мере одного снижения дозы. Среднее время до первого снижения дозы составляло 48 дней. Наиболее частыми побочными реакциями, которые привели к снижению или прерыванию дозы, были повышение уровня билирубина (6 %), КФК (4,3 %), АЛТ (4,0 %) и АСТ (2,8 %) и рвота (2,8 %).
Светочувствительность наблюдалась у 9,9 % пациентов, пациентам рекомендовалось избегать воздействия солнечных лучей и использовать солнцезащитные крема.
Меры предосторожности
Гепатотоксичность
Повышение АСТ более чем в 5 раз от верхнего предела нормы отмечалось у 3,6 % пациентов, а повышение АЛТ более чем в 5 раз от верхнего предела нормы у 4,8 % пациентов. Трёхкратное превышение уровня билирубина наблюдалось у 2,8 % пациентов. Большинство отклонений имели место в течение первых 2 месяцев применения алектиниба.
Необходим мониторинг функций печени, включая показатели АЛТ, АСТ и общего билирубина каждые 2 недели в течение первых 2 месяцев применения алектиниба, затем периодически с более частым тестированием пациентов, у которых развивается подъем трансаминаз и билирубина.
Интерстициальная болезнь лёгких/пневмонит
Тяжёлая интерстициальная болезнь лёгких/пневмонит произошла у одного (0,4 %) из 253 пациентов, принимавших алектиниб в ходе клинических исследований.
Необходимо немедленно отменить алектиниб, если не выявлено других потенциальных причин развития осложнений со стороны лёгких.
Брадикардия
Симптоматическая брадикардия может возникать при использовании алектиниба. Сообщалось о случаях брадикардии (7,5 %) у пациентов, получавших алектиниб. 20 % из 221 пациентов, получавших алектиниб, имели частоту сердечных сокращений менее 50 ударов в минуту (уд / мин).
Регулярно контролируйте частоту сердечных сокращений и артериальное давление. Модификация дозы не требуется в случаях бессимптомной брадикардии. В случаях симптоматической брадикардии, которая не угрожает жизни, приостановите приём алектиниба до восстановления до бессимптомной брадикардии или частоты сердечных сокращений до 60 уд/мин или выше.
Тяжёлая миалгия и повышение КФК
Миалгия или мышечно-скелетная боль наблюдались у 29 % пациентов в исследовании 1 и в исследовании 2. Частота миалгии/мышечно-скелетной боли 3-й степени тяжести составляла 1,2 %. Для 0,8 % пациентов потребовались изменение дозы алектиниба.
Повышения КФК произошли у 43 % из 218 пациентов. Выраженный подъём КФК отмечался у 4,6 %. пациентов. Изменения дозы алектиниба потребовались 5,0 % пациентов.
Классификация
-
АТХ
L01XE36, L01ED03
-
Фармакологическая группа
-
Код МКБ 10
-
Категория при беременности по FDA
N
(не классифицировано FDA)
Информация о действующем веществе Алектиниб предназначена для медицинских и фармацевтических специалистов, исключительно в справочных целях. Инструкция не предназначена для замены профессиональной медицинской консультации, диагностики или лечения. Содержащаяся здесь информация может меняться с течением времени. Наиболее точные сведения о применении препаратов, содержащих активное вещество Алектиниб, содержатся в инструкции производителя, прилагаемой к упаковке.